


Abstract
The protein subunit of RNase P from a thermophilic bacterium, Thermotoga maritima, was overexpressed in and purified from Escherichia coli. The cloned protein was reconstituted with the RNA subunit transcribed in vitro. The temperature optimum of the holoenzyme is near 50°C, with no enzymatic activity at 65°C or above. This finding is in sharp contrast to the optimal growth temperature of T.maritima, which is near 80°C. However, in heterologous reconstitution experiments in vitro with RNase P subunits from other species, we found that the protein subunit from T.maritima was responsible for the comparative thermal stability of such complexes.
INTRODUCTION
The extreme thermophile Thermotoga maritima has a growth range between 55 and 90°C with an optimum temperature of 80°C (1). Evolutionary studies have placed this bacterium in one of the deepest and most slowly evolving lineages of the Eubacteria (2,3). It is, therefore, of some interest to understand how the enzymatic machinery of T.maritima functions at high temperatures. We report here the biochemical characterization of RNase P, a ubiquitous enzyme essential for tRNA biosynthesis (4), from T.maritima. We also report the function of the subunits of this enzyme in heterologous reconstitution experiments with the overexpressed subunits from various other Eubacteria, including one thermophile. Although RNase P from T.maritima has an optimal temperature for function in vitro lower than that observed for growth of the intact organism, its properties, nevertheless, make it better suited for structural studies than its analogs isolated from mesophilic bacteria.
MATERIALS AND METHODS
Materials
Reagents used in this study were obtained as follows: restriction enzymes, T4 DNA polymerase and T4 DNA ligase from New England Biolabs; T7 RNA polymerase, a gift from Dr R. Breaker (Yale University); RNasin from Promega; nucleoside triphosphates from Pharmacia Biotech; protein assay reagent from Bio-Rad; QuickSpin Sephadex columns from Roche Biochemicals; and [α-32P]GTP from Amersham Biochemicals. Oligonucleotides were synthesized by the DNA Synthesis Facility at Yale University (Keck Center) or by MWG-Biotech.
Cloning of the RNA and protein subunits of RNase P from T.maritima
DNA encoding the protein subunit (rnpA) was generated by PCR amplification using genomic DNA as template [kindly provided by Dr Wolfgang Liebl (Technische Universität München, Germany)]. The PCR reaction mixture contained 25 ng genomic DNA, 25 pmol of each primer (5′-TmC5-1: TGCATGCCATGGGAACAGAGAGTTTCACCCGCA; 3′-TmC5-2: TGCATGCTCGAGCTAATGATGATGATGATGATGACTTCCTTCTATCCTCTTCAAGAG), 1× PCR reaction buffer, 200 nM dNTP mix and 2.5 U Expand High Fidelity DNA polymerase mix (all from Roche Biochemicals) in a total volume of 50 µl, all overlaid with mineral oil (Perkin Elmer). After initial denaturation for 2 min at 92°C, the DNA was amplified in 30 cycles (1 min denaturation at 92°C, 1 min annealing at 60°C and 1 min extension at 72°C), followed by a final extension step of 7 min at 72°C. The PCR product was purified with the PCR Purification Kit (Qiagen) and cloned into the NcoI and XhoI sites of the pBAD-A vector (Invitrogen). The sequence of the insert was confirmed using Sequenase 2.0 (USB) for dideoxy sequencing of both strands.
DNA encoding the RNA subunit (rnpB ) was PCR amplified from plasmid DNA (clone Tm-gp-52) (5) as described above using the primer pair 5′-TmM1-1: GCCGAAT TCTAATACGACTCACTATAGGAGAGGAGCAGGCGGTCGC and 3′-TmM1-2: CGCGGATCCGGATGACGTAAATCCACGGGAGAGGAGCATAAGCCGG. The PCR product was purified and cloned into the EcoRI and BamHI sites of the pUC19 vector. The T7 promoter for transcription in vitro of RNA was provided by the 5′ primer.
In vitro transcription of RNase P RNA and substrate RNA
The plasmid harboring the gene encoding the T.maritima RNase P RNA subunit (rnpB) was linearized with FokI, and RNA was transcribed by T7 RNA polymerase as described (6). The RNA was then isolated using a QuickSpin column procedure (6).
After purification from the transcription reaction mixture, the RNA was dissolved in 1× PA buffer (50 mM Tris, pH 7.5, 100 mM NH4Cl, 10 mM MgCl2), heated for 5 min at 65°C and then allowed to cool slowly to room temperature.
The plasmid encoding substrate RNA (precursor to tRNATyr from Escherichia coli, i.e. pTyr) was linearized with FokI. During transcription in vitro with T7 RNA polymerase the RNA was internally labeled with [α-32P]GTP and then purified on a 5% (w/v) polyacrylamide/7 M urea gel.
Overexpression and purification of the RNase P protein subunit
As a consequence of the procedure used for cloning the rnpA gene into the NcoI site of the pBAD-A expression vector, a codon encoding a glycine residue (GGA) was inserted into the gene product immediately following the AUG initiator methionine codon. At the C-terminus of the open reading frame, a single serine residue followed by six histidine residues was added to facilitate the purification of the recombinant protein by metal ion affinity chromatography.
The pBAD-rnpA expression plasmid was transformed into E.coli TOP10 (Invitrogen). To enrich the host bacterium with genes for certain low abundance tRNAs, this bacterial strain was also transformed with the plasmid pSJS1240 (7) lacking the PvuI/ScaI fragment (unpublished results).
For overexpression, the bacteria were diluted from an overnight culture and grown in LB with 100 µg/ml ampicillin at 37°C to OD600 of 0.5–0.6. After induction by addition of arabinose to a final concentration of 0.02%, the temperature was shifted to 30°C. After 3 h, the bacterial cultures were spiked with an additional 0.02% arabinose and 100 µg/ml ampicillin. Six hours after the start of induction the bacteria were harvested by centrifugation at 6000 g for 15 min. Bacterial pellets were stored at –70°C.
Bacteria were lysed by suspension in ice cold lysis buffer [50 mM Tris, pH 8.0, 8 mM CDTA (1,2-cyclohexanediamine-N,N,N′N′-tetraacetic acid), 5 mM DTT, 4% Triton X100, 100 µg/ml polymyxin-B-sulfate, 1 mM Pefabloc] and immediately centrifuged at 27 200 g for 15 min (8). The pellets were washed once with solubilization buffer (50 mM Tris, pH 8.0, 2% Triton X100, 100 µg/ml polymyxin-B-sulfate, 1 mM Pefabloc) to remove CDTA and DTT. The washed pellet was again suspended in solubilization buffer, and freshly prepared egg white lysozyme (Sigma) and DNase A (Worthington Biochemical Corporation) were added to a final concentration of 200 and 0.2 µg/ml, respectively, according to a protocol supplied by Pierce Chemical Co. After 5 min incubation on ice, 600 ml of 0.1× solubilization buffer was added, and the solution was centrifuged for 20 min at 27 200 g. In small scale preparations, the supernatant of this latter centrifugation step contained the T.maritima RNase P protein, but >50% of the total recombinant protein remained in the pellet after large scale purification.
Purification of RNase P protein under non-denaturing conditions
The soluble recombinant protein could be precipitated with 2.5 M, but not with 1 M ammonium sulfate (pH 7.9). Two successive ammonium sulfate precipitations were, therefore, performed: first with 1 M ammonium sulfate during which most of the recombinant protein stayed in the supernatant, and then with 2.5 M ammonium sulfate. After centrifugation (20 min at 27 200 g) the ammonium sulfate pellet was dissolved in ~3 ml of 1× His-binding buffer (500 mM NaCl, 20 mM Tris, 5 mM imidazole, pH 7.9), and loaded on a His-Resin column (5 ml; Novagen) equilibrated with the same buffer. The column was washed with ∼30 vol 1× His-binding buffer, and then a gradient of imidazole (from 5 mM to 1 M) was applied to elute the recombinant protein. Fractions containing protein [as determined by the Bradford method (9)] were dialyzed against 20 mM Tris, 100 mM ammonium acetate, pH 7.9. However, recombinant protein precipitated out of the solution both before and after dialysis, an indication that this protein was insoluble at high concentrations. Efforts to solubilize the precipitated protein in the absence of urea or guanidinium hydrochloride have been unsuccessful to date.
Purification under denaturing conditions
Protein which remained insoluble after the lysozyme treatment of the lysed cell pellet (see above) could be dissolved by addition of guanidinium–HCl as a dry powder to a final concentration of 6 M in 500 mM NaCl, 20 mM Tris, 5 mM imidazole, pH 7.9. His-Resin (5 ml equilibrated in the same buffer) was then added to the solution. After stirring on ice for 1 h, the His-Resin was recovered by centrifugation (5 min at 1000 g), and poured into a column. The column was washed successively with 5 vol 6 M guanidinium–HCl (500 mM NaCl, 20 mM Tris, 5 mM imidazole, pH 7.9) and 25 vol 6 M urea (500 mM NaCl, 20 mM Tris, 5 mM imidazole, pH 7.9). The recombinant protein was eluted with an imidazole gradient (from 5 mM to 1 M). After elution, the fractions containing the protein, as determined by Bradford assays (9), were immediately dialyzed against 6 M urea, 20 mM Tris and 100 mM ammonium acetate, pH 7.9. The identity of the purified recombinant protein was confirmed by mass spectroscopy analysis at the Keck Center of Yale University.
Assays of RNase P activity and reconstitution of the RNase P holoenzyme
Freshly diluted (to 100 nM) recombinant protein was added in 10-fold molar excess to the RNA subunit of RNase P that had been transcribed in vitro. After incubation for 5 min in the RNase P reaction buffer (e.g., 400 mM NH4Cl, 50 mM HEPES, pH 7.5, 5 mM MgCl2) at the assay temperature, the reaction was started by the addition of labeled substrate
To calculate the initial velocity of the reactions, aliquots were withdrawn at regular time intervals (1 or 2 min) and quenched with 9 M urea/10% phenol. The reaction products were analyzed by electrophoresis on 8% polyacrylamide/7 M urea gels and the extent of cleavage was calculated by quantifying the intensity of the various radioactive species using a phosphoimager (Fuji). Measurements of the extent of cleavage were always restricted to the linear range of product formation.
Determination of kinetic constants of T.maritima RNase P
Initial velocities were determined by incubation of 1 nM RNA and 10 nM protein subunits in buffer that contained 400 mM NH4Cl, 50 mM HEPES, pH 7.5, and 5 mM MgCl2 at 50°C. The reactions were started by addition of various concentrations (35, 50, 75, 100, 150 or 180 nM) of 32P-labeled substrate. Aliquots were taken at 1 min intervals and analyzed as described above for RNase P assays. The numbers cited in the text reflect the results of four independent experiments and were determined using a Lineweaver–Burk plot.
RESULTS AND DISCUSSION
Purification of the T.maritima RNase P protein subunit
The T.maritima RNase P protein subunit was purified as described in Materials and Methods. Overexpressed recombinant protein was analyzed on SDS–polyacrylamide gels to show that it was purified to apparent homogeneity. The purification procedure under denaturing conditions proved to be somewhat more efficient in removing contaminating host cell proteins (Fig. 1). RNase P protein samples purified both under denaturing and non-denaturing conditions were also analyzed by mass spectroscopy. The analysis revealed the initiator methionine at the N-terminus to be missing in both protein samples, and the appropriate C-termini, as well as the rest of the sequence, to be present.
Figure 1.
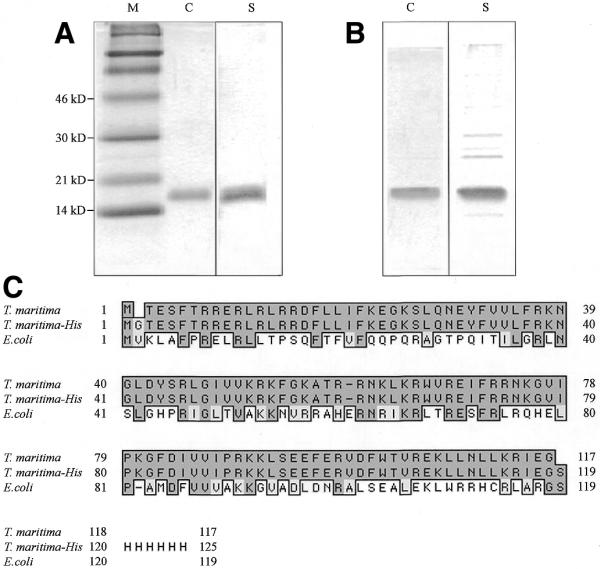
The T.maritima RNase P protein subunit. His-tagged T.maritima RNase P protein subunit was purified on His-Resin columns under (A) denaturing and (B) non-denaturing conditions. After dialysis of the proteins eluted from the columns, 0.5 µg of each protein preparation was separated on a 15% SDS–PA gel and stained with either Coomassie dye (C) or silver (S). (C) Clustal W alignment of the wild-type T.maritima RNase P protein subunit with its recombinant homolog (T.maritima–His) and with the E.coli RNase P protein subunit.
Characterization of the enzymatic activity of the T.maritima RNase P holoenzyme
We compared the activities of the RNase P RNA subunit alone, the holoenzyme (consisting of the RNA and protein subunits) and the protein subunit alone (Fig. 2). The RNA subunit alone was active only in buffer that contained high concentrations of MgCl2 (e.g., 100 mM) as has been found for the RNA subunits of RNase P from other bacterial sources. In contrast, the holoenzyme was optimally active in a buffer that contained 10 mM MgCl2. The protein subunit alone showed no activity at all. These results are in accordance with experiments performed with the RNase P holoenzymes and RNase P RNA subunits of other Eubacteria and show that the RNA is catalytic, while the protein subunit has an auxiliary function (10).
Figure 2.
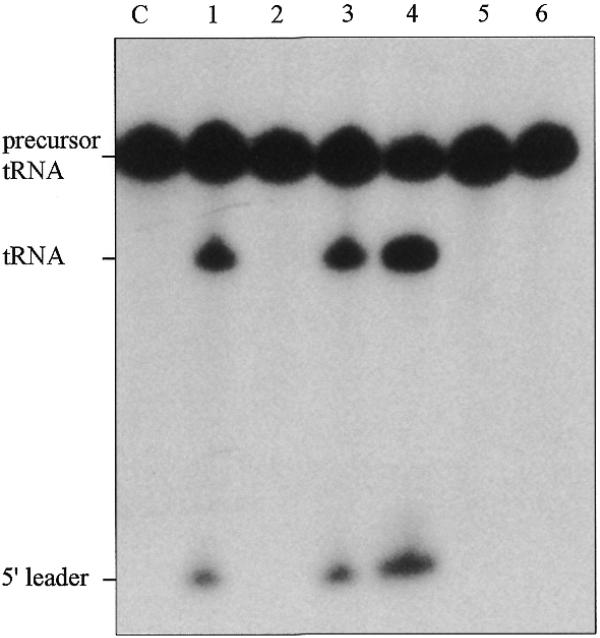
Cleavage of precursor tRNATyr by T.maritima RNase P RNA and RNase P holoenzyme. The activities of the T.maritima RNase P holoenzyme and its RNA and protein subunits alone were assayed with precursor tRNATyr (E.coli). The assay mixtures were incubated for 15 min at 50°C with 1 nM RNA alone and/or with 10 nM protein and contained either 100 or 10 mM MgCl2 and 100 nM of the substrate RNA. Lane C, control, substrate RNA alone incubated for 15 min in reaction buffer; lane 1, RNA alone in 100 mM MgCl2; lane 2, RNA alone in 10 mM MgCl2; lane 3, holoenzyme in 100 mM MgCl2; lane 4, holoenzyme in 10 mM MgCl2; lane 5, protein alone in 100 mM MgCl2; lane 6, protein alone in 10 mM MgCl2.
We compared the activity of T.maritima RNase P holoenzyme reconstituted with the protein subunit purified under both denaturing and non-denaturing conditions (data not shown). No difference in the specificity or amount of specific activity for protein purified under denaturing or non-denaturing conditions was found. In both cases, maximal activity was observed when the protein subunit was added in 10-fold molar excess to the RNA subunit. We conclude that protein stored in 6 M urea buffer is able to refold and reconstitute functional holoenzyme after its dilution into buffer lacking urea and addition of the RNA subunit.
Further characterization of the T.maritima RNase P holoenzyme was undertaken with a 10-fold molar excess of the RNase P protein subunit purified under denaturing conditions with respect to the concentration of the RNA subunit. This resulted in a urea concentration of 4 mM in the reaction mixtures (urea in a concentration up to 40 mM had little effect on the activity of the holoenzyme; data not shown).
Optimal conditions for the specific cleavage of precursor tRNATyr by T.maritima RNase P were determined at 50°C (see below). The dependence of the reaction on monovalent cations was determined in a buffer that contained 50 mM Tris, pH 7.5, and 10 mM MgCl2. Potassium chloride yielded optimal activity at a concentration of 200 mM (Fig. 3A), sodium chloride at 50 mM (Fig. 3B) and ammonium chloride between 200 and 400 mM (Fig. 3C).
Figure 3.

Characterization of the T.maritima RNase P holoenzyme. Initial velocities of cleavage of precursor tRNATyr were determined as follows: 1 nM RNA and 10 nM protein were preincubated in their respective reaction buffers for 5 min at 50°C (see text). The reactions were started by the addition of 100 nM 32P-labeled substrate. Aliquots were taken at 1 min intervals and separated on 8% polyacrylamide/7 M urea denaturing gels. The amount of cleavage was determined using a phosphoimager, and is indicated as nmol product/nmol RNase P RNA (min). Thermotoga maritima RNase P holoenzyme dependence on (A) KCl (in 50 mM Tris, pH 7.5, 10 mM MgCl2 in this and other cases that follow), (B) NaCl, (C) NH4Cl, (D) MgCl2 (in 50 mM Tris, pH 7.5, 400 mM NH4Cl).
The dependence of T.maritima RNase P holoenzyme on the presence of magnesium was determined in a buffer that contained 50 mM Tris, pH 7.5, and 400 mM NH4Cl. The peak of activity was at 5 mM MgCl2 (Fig. 3D).
The pH dependence of the enzyme was determined in buffers containing 5 or 10 mM MgCl2 and 400 mM NH4Cl. Tris–HCl (50 mM) was used as buffer for the pH range 7.0–9.0, and 50 mM Bis–Tris–propane buffer for the pH range 6.5–9.5 [(Bis–Tris–propane was toxic to RNase P at concentrations >50 mM) (data not shown)]. The maximum of specific activity occurred near pH 7.5 in both buffer systems. HEPES buffer at pH 7.5 showed no significant inhibition of holoenzyme activity at various concentrations (data not shown). HEPES buffer was, therefore, used in further experiments with the holoenzyme.
Determination of kcat and Km
The kcat and Km of the T.maritima holoenzyme were determined at 50°C with 1 nM RNase P RNA and 10 nM RNase P protein in a buffer that contained 400 mM NH4Cl, 50 mM HEPES, pH 7.5, and 5 mM MgCl2 as further described in Materials and Methods. The Km was 223 nM and the kcat 8.2 nmol product/nmol RNase P RNA (min) (data not shown).
The temperature dependence of the T.maritima RNase P holoenzyme
Temperature dependence of the RNase P holoenzyme was assayed in buffer systems that contained 50 mM HEPES, pH 7.5, 5 mM MgCl2 and 400 mM NH4Cl, in Tris-based buffers with 5 or 10 mM MgCl2, and in a buffer containing 10 mM HEPES, pH 7.5, 5 mM Mg(OAc)2 and 1 M ammonium acetate. In all these buffer systems, the optimal temperature for activity was 50°C (Fig. 4). This finding is in marked contrast to the optimal growth temperature of T.maritima, which is ∼80°C (1). Of course, we cannot exclude the possibility that the holoenzyme would manifest greater thermal stability in buffers of composition yet to be determined.
Figure 4.

Temperature dependence of T.maritima RNase P holoenzyme activity. Initial velocities were determined in 5 mM MgCl2 (in 50 mM HEPES, pH 7.5, 400 mM NH4Cl) at each respective reaction temperature. The reactions were started by addition of 100 nM of 32P-labeled substrate RNA. Aliquots were taken at 1 min intervals and separated on 8% denaturing polyacrylamide gels. The amount of cleavage was determined using a phosphoimager.
We note that measurements of activity versus temperature were also carried out with precursor tRNA substrates from T.maritima: no differences in temperature optima compared to those measured with substrate from E.coli were found but the absolute activities of the enzyme variations listed in Table 1 were somewhat higher with the T.maritima substrates at higher temperatures (data not shown).
Table 1. Heterologous reconstitution of RNase P protein and RNA subunits at (A) 37°C and (B) 55°C.
| Protein
subunit |
RNA
subunit |
nmol product/ nmol RNA
(min) |
% cleavage by homologous T.maritima complex |
| A | |||
| T.maritima | T.maritima | 6.6 ± 0.6 | 100 |
| T.maritima | E.coli | 5.1 ± 1 | 77 |
| T.maritima | T.thermophilus | 2.2 ± 0.7 | 33 |
| E.coli | T.maritima | 6.1 ± 0.3 | 93 |
| E.coli | E.coli | 5.6 ± 1.4 | 85 |
| E.coli | T.thermophilus | 1.0 ± 0.4 | 15 |
| B | |||
| T.maritima | T.maritima | 6.5 ± 1.6 | 100 |
| T.maritima | E.coli | 3.8 ± 1.4 | 58 |
| T.maritima | T.thermophilus | 0.7 ± 0.04 | 11 |
| E.coli | T.maritima | 0 | – |
| E.coli | E.coli | 0 | – |
| E.coli | T.thermophilus | 0 | – |
The initial velocities of cleavage of precursor tRNATyr were determined by incubation of 1 nM of the RNA subunit and 10 nM of the protein subunit in reaction buffer (400 mM NH4Cl, 50 mM HEPES, pH 7.5, 5 mM MgCl2) for 5 min at 50°C. The reactions were started by addition of 100 nM of 32P-labeled substrate (see Materials and Methods). Aliquots were taken at 1 min intervals and separated on 8% polyacrylamide/7 M urea denaturing gels. The amount of cleavage was determined with a phosphoimager. A ‘homologous’ complex refers to a complex that contains protein and RNA subunits from the same organism.
The sequence of the recombinant protein is not identical to the native amino acid sequence since alterations were introduced at both the N-terminus (an additional glycine residue) and at the C-terminus (the poly-histidine tag; Fig. 1). However, another recombinant T.maritima RNase P protein subunit with an N-terminal His-tag, but no additional glycine residue after the initiator methionine, also showed optimal activity in reconstitution experiments near 50°C (data not shown).
The comparison of the amino acid sequences of bacterial RNase P protein subunits shows homology mainly between internal portions of the proteins and less conservation near the N- and C-termini (11). We would expect, therefore, that the recombinant proteins we used would function in an equivalent manner to the wild-type protein.
Heterologous reconstitution of the RNase P protein subunits of T.maritima and E.coli with RNase P RNA subunits
Heterologous reconstitution in vitro of the RNase P protein subunits from T.maritima and E.coli with the RNase P RNA subunits from T.maritima, E.coli and Thermus thermophilus were carried out to determine which of the two subunits might be responsible for comparative thermal stability, if any, of these complexes. At 37°C, both the T.maritima and E.coli proteins were able to reconstitute activity with each of the three RNase P RNA subunits (Table 1). The heterologous reconstitution of the two proteins with the RNA subunits from E.coli and T.maritima RNase P yielded about the same specific activities as the respective homologous reconstitution. Both the T.maritima and E.coli RNase P protein subunits were also able to reconstitute with the T.thermophilus RNA subunit at 37°C, although with less efficiency compared to the T.maritima and E.coli RNase P RNA subunits. (A clone for the T.thermophilus RNase P protein subunit is not yet available.)
When the heterologous reconstitution of protein and RNA subunits was repeated at 55°C, only the T.maritima RNase P protein subunit was able to reconstitute activity with the RNase P RNA subunits: the E.coli RNase P protein subunit could not reconstitute activity with any of the catalytic RNAs at this temperature (Table 1). These results indicate that the recombinant T.maritima RNase P protein can participate in the formation of a complex more stable to thermal denaturation than the E.coli C5 protein.
We also note that T.maritima RNase P protein (N-terminal His-tagged) can sustain growth of an E.coli strain that harbors a temperature sensitive mutation in the rnpA gene (C5 protein) at the restrictive temperature (data not shown).
Basic and aromatic residues make up 30 and 12%, respectively, of the amino acid content of the protein subunit of T.maritima RNase P (Table 2). These residues might contribute to the high thermal stability of this protein as well as to that of the corresponding T.maritima RNase P holoenzyme in vivo through salt bridges and hydrophobic interactions (12). High-resolution structural approaches are required to investigate this proposal.
Table 2. Comparison of the amino acid composition of T.maritima RNase P protein and homologous proteins from other bacteria.
| Organism |
Cysteine (C) |
Proline (P) |
Aromatic (F,W,Y) |
Acidic (D,E) |
Basic (R,K) |
Histidine (H) |
Total residues |
| T.maritima | 0 | 2 | 14 | 14 | 33 | 0 | 117 |
| Bacillus subtilis | 0 | 2 | 9 | 13 | 30 | 5 | 119 |
| Staphylococcus aureus | 1 | 1 | 8 | 8 | 27 | 5 | 117 |
| E.coli | 1 | 6 | 7 | 9 | 25 | 4 | 119 |
CONCLUSION
Overexpression and purification of RNase P protein from an extreme thermophile, T.maritima have allowed the biochemical characterization of the holoenzyme complex. The RNase P protein from T.maritima may be more useful than its counterparts from mesophilic bacteria for structural studies of RNase P.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs R. Biswas and V. Gopalan (Ohio State University) and our colleagues at Yale University, in particular Dr Cecilia Guerrier-Takada, for many helpful discussions. This research was supported by grants from the Human Frontiers of Science Program and the National Institutes of Health to S.A.
References
- 1.Huber R., Langworthy,T.A., Koenig,H., Thomm,M., Woese,C.R., Sleytr,U.B. and Stetter,K.O. (1986) Thermotoga maritima sp. nov. represents a new genus of unique extremely thermophilic eubacteria growing up to 90°C. Arch. Microbiol., 144, 324–333. [Google Scholar]
- 2.Achenbach-Richter L., Gupta,R., Stetter,K.O. and Woese,C.R. (1987) Were the original eubacteria thermophiles? Syst. Appl. Microbiol., 9, 34–39. [DOI] [PubMed] [Google Scholar]
- 3.Bocchetta M., Gribaldo,S., Sanangelantoni,A. and Cammarano,P. (2000) Phylogenetic depth of the bacterial genera Aquifex and Thermotoga inferred from analysis of ribosomal protein, elongation factor, and RNA polymerase subunit sequences, Mol. Evol., 50, 366–380. [DOI] [PubMed] [Google Scholar]
- 4.Frank D.N. and Pace,N.R. (1998) Ribonuclease P: unity and diversity in a tRNA processing ribozyme. Annu. Rev. Biochem., 67, 153–180. [DOI] [PubMed] [Google Scholar]
- 5.Nelson K.E., Clayton,R.A., Gill,S.R., Gwinn,M.L., Dodson,R.J., Haft,D.H., Hickey,E.K., Peterson,J.D., Nelson,W.C., Ketchum,K.A. et al. (1999) Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature, 399, 323–329. [DOI] [PubMed] [Google Scholar]
- 6.Vioque A., Arnez,J. and Altman,S. (1988) Protein–RNA interactions in the RNase P holoenzyme from Escherichia coli. J. Mol. Biol., 202, 835–848. [DOI] [PubMed] [Google Scholar]
- 7.Kim R., Sandler,S.J., Goldman,S., Yokota,H., Clark,A.J. and Kim,S.-H. (1998) Overexpression of archael proteins in Escherichia coli. Biotechnol. Lett., 20, 207–210. [Google Scholar]
- 8.Schupp J.M., Travis,S.E., Price,L.B., Shand,R.F. and Keim,P. (1995) Rapid bacterial permeabilization reagent useful for enzyme assays. Biotechniques, 19, 18–20. [PubMed] [Google Scholar]
- 9.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 10.Guerrier-Takada C., Gardiner,K., Marsh,T., Pace,N. and Altman,S. (1983) The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell, 35, 849–857. [DOI] [PubMed] [Google Scholar]
- 11.Brown J.W. (1998), The ribonuclease P database. Nucleic Acids Res., 26, 351–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaenicke R. and Bohm,G. (1998), The stability of proteins in extreme environments. Curr. Opin. Struct. Biol., 8, 738–748. [DOI] [PubMed] [Google Scholar]