

Abstract
Single-stranded DNA binding proteins (SSBs) play central roles in cellular and viral processes involving the generation of single-stranded DNA. These include DNA replication, homologous recombination and DNA repair pathways. SSBs bind DNA using four ‘OB-fold’ (oligonucleotide/oligosaccharide binding fold) domains that can be organised in a variety of overall quaternary structures. Thus eubacterial SSBs are homotetrameric whilst the eucaryal RPA protein is a heterotrimer and euryarchaeal proteins vary significantly in their subunit compositions. We demonstrate that the crenarchaeal SSB protein is an abundant protein with a unique structural organisation, existing as a monomer in solution and multimerising on DNA binding. The protein binds single-stranded DNA distributively with a binding site size of ~5 nt per monomer. Sulfolobus SSB lacks the zinc finger motif found in the eucaryal and euryarchaeal proteins, possessing instead a flexible C-terminal tail, sensitive to trypsin digestion, that is not required for DNA binding. In comparison with Escherichia coli SSB, the tail may play a role in protein–protein interactions during DNA replication and repair.
INTRODUCTION
Single-stranded DNA binding proteins (SSB in eubacteria; RPA in eucarya and euryarchaea) are required by all cellular life forms, as well as many viruses, in order to bind, sequester and protect DNA that has had its natural double-stranded structure disrupted. Such disruptions occur during vital information processing pathways such as DNA replication, recombination and repair, as a result of DNA damage sustained from exposure to cellular or environmental factors, or simply due to DNA ‘breathing’ (1). Although there is little conservation at sequence level, the crystal structures of human RPA (2,3), Escherichia coli SSB and human mitochondrial SSB (4–6) indicate the presence of a core ‘OB-fold’ (oligonucleotide/oligosaccharide binding fold) (7). This domain comprises approximately 100 amino acids and is responsible for the major interactions with single-stranded DNA (ssDNA). The universality of OB-fold domains in SSB proteins suggests that an ancestral SSB existed in the Last Universal Cellular Ancestor (LUCA) (8) and therefore that all extant SSB and RPA proteins are derived from a common ancestral SSB. In the eubacteria, SSBs exist as homotetramers, whilst eucaryal RPA is a heterotrimer and euryarchaeal SSBs exist as monomers, heterodimers and heterotrimers (9).
Both E.coli SSB and the yeast and human RPA proteins have been characterised extensively, both biochemically and at a structural level. Comparisons of amino acid sequence and protein domain organisation suggest strongly that the eucaryal and euryarchaeal RPA proteins are more closely related to one another than either is to the eubacterial SSBs (9,10). By way of an illustration, the large subunits of both eucaryal and euryarchaeal RPA contain a putative zinc finger domain that is absent in the eubacterial and mitochondrial SSBs. This tallies with an extensive body of evidence that suggests that the archaea and eucarya share information processing pathways, including DNA replication and transcription, in common (reviewed in 11). Protein domains outwith the core OB-fold regions are thought to function as recognition surfaces for protein–protein interactions, for example RPA70 has an extensive N-terminal sequence that has also been postulated to interact with other repair proteins (Discussion).
We are investigating DNA recombination and repair pathways in the crenarchaeote Sulfolobus solfataricus. The crenarchaeal lineage is highly diverged from that of the euryarchaea, and has some distinct features, notably the lack of true histone proteins for double-stranded DNA (dsDNA) binding and compaction (12). To date, only one crenarchaeal genome sequence, that of Aeropyrum pernix, has been published (13), though several others are nearing completion. As there is no annotated open reading frame (ORF) encoding an SSB protein in the A.pernix genome, we undertook the identification of the Sulfolobus SSB using an assay for ssDNA binding activity. We report here the purification, identification and characterisation of Sulfolobus SSB, and its relationship with other members of this important class of proteins.
MATERIALS AND METHODS
Oligonucleotides
The following oligonucleotides were used for binding and cross-linking studies, and were radioactively 5′-32P-labelled using polynucleotide kinase where appropriate: 18mer, 5′-CGTCGGATCCCCATGGCC; 69mer, 5′-GGCGGAAAATGAGAAAATTCGACCTATCCTTGCGCAGCTCGAGAAGCTCTTACTTTGCGACCTTTCGCC.
Purification and identification of SSB from Sulfolobus
The S.solfataricus P2 biomass was supplied by the Centre for Extremophile Research, Porton Down, UK. Cell lysis, centrifugation and chromatography steps were carried out at 4°C. Cells (50 g) were thawed in 150 ml lysis buffer and immediately sonicated for 5 × 1 min with cooling. The lysate was centrifuged at 40 000 g for 30 min. The supernatant was diluted 4-fold with buffer A and applied to an SP-Sepharose High Performance 26/10 column (Hi-Load, Amersham Pharmacia) equilibrated with buffer A. A 500 ml linear gradient comprising 0–1000 mM NaCl was used to elute cationic proteins. Fractions were assayed for the ability to retard a 32P-labelled ssDNA oligonucleotide in an electrophoretic mobility shift assay (EMSA). The relevant fractions were pooled, concentrated and loaded onto a 26/70 gel filtration column (Superdex 200 Hi Load, Amersham Pharmacia) and developed with buffer A containing 300 mM NaCl. Fractions containing ssDNA binding activity were assessed by EMSA, pooled and applied to a 1 ml Mono-S column. A 50 ml linear gradient comprising 200–750 mM NaCl was used to elute cationic proteins. Active fractions identified by the EMSA coincided with a single, essentially homogeneous polypeptide observed by SDS–PAGE. The protein was identified by MALDI-TOF mass spectroscopy following in-gel tryptic digestion and interrogation of the public domain release of the Sulfolobus genome.
Cloning and heterologous expression of the Sulfolobus SSB gene
The gene encoding S.solfataricus SSB was amplified from S.solfataricus strain P2 genomic DNA, using proofreading polymerase Pfu (Promega), with the following oligonucleotides: forward primer, 5′-CGTCGGATCCCCATGGAAGAAAAAGTAGGTAATCTAAAACC; reverse primer, 5′-CCGGGGATCCGTCGACTCACTCCTCTTCACCTTCTTCGTTTTC. The oligonucleotides introduced restriction sites at either end of the amplified gene to facilitate subcloning. Amplified SSB was subcloned into the BamHI site of vector pUC119 (Clontech), creating the plasmid pUC119-SSB. The SSB gene was sequenced completely to ensure that no errors had been introduced in the amplification process; the sequence was identical to that in the database (ORF bac19_023 at http://niji.imb.nrc.ca/sulfhome/). The SSB gene was subcloned from pUC119 into the BamHI and NcoI sites of the expression vector pET19b (Novagen), allowing expression of SSB with a native N-terminus in BL21 CodonPlus (DE3) RIL cells (Stratagene).
Purification of recombinant SSB protein
Protein expression was induced by addition of 0.2 mM IPTG at 37°C for 3 h, after which cells were pelleted and frozen until required. Cell lysis, centrifugation and chromatography steps were carried out at 4°C. Cells (20 g) were thawed in 50 ml lysis buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM benzamidine, 1 mM AEBSF) and immediately sonicated for 5 × 1 min with cooling. The lysate was centrifuged at 40 000 g for 30 min. The supernatant was heated to 70°C for 30 min in a water bath, and denatured proteins were precipitated by centrifugation at 40 000 g at 4°C. The supernatant was analysed by SDS–PAGE, and shown to contain recombinant SSB, which migrated as a band of ~16 kDa as expected. The supernatant was diluted 4-fold with buffer A (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 1 mM DTT) and applied to an SP-Sepharose High Performance 26/10 column equilibrated with buffer A. A 500 ml linear gradient comprising 0–1000 mM NaCl was used to elute cationic proteins. Fractions corresponding to a distinct absorbance peak were analysed by SDS–PAGE, pooled, concentrated and loaded onto a 26/70 gel filtration column and developed with buffer A containing 300 mM NaCl. This step removes endogenous E.coli SSB, which is tetrameric in solution (Mr ~75 kDa). Active fractions were pooled and shown to contain essentially homogeneous SSB protein (Fig. 3). This protein was used for all subsequent analyses.
Figure 3.
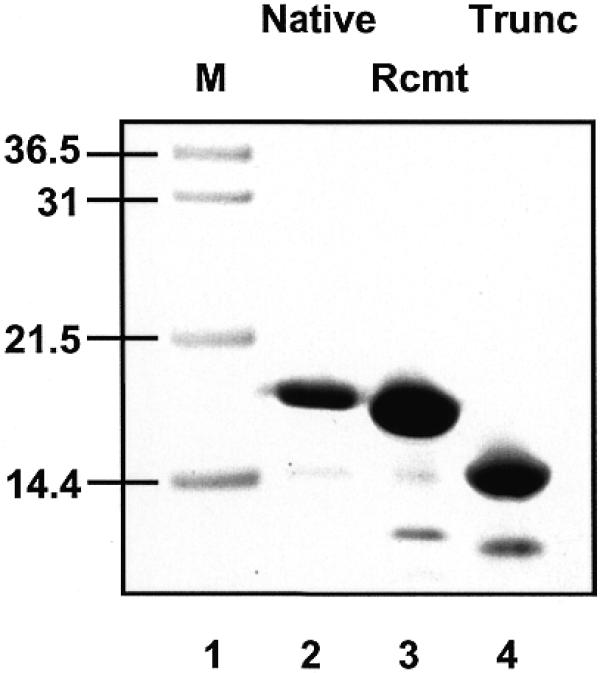
SDS–PAGE analysis of purified native SSB protein, purified recombinant SSB protein, and recombinant SSB truncated by limited trypsin digestion. Lane 1, markers; lane 2, SSB purified from S.solfataricus; lane 3, recombinant Sulfolobus SSB purified from E.coli; lane 4, recombinant SSB after trypsin digestion.
Digestion of recombinant SSB with proteolytic enzymes
Recombinant SSB was incubated in digest buffer (50 mM Tris–HCl pH 8.0, 2 mM CaCl2) at 37°C for 1 h in the presence of a 1:250 (w/w) ratio of porcine trypsin (Promega), or a 1:250 (w/w) ratio of Glu-C (Promega). Following digestion, products were analysed by SDS–PAGE and MALDI-TOF. Trypsin was inhibited irreversibly by the addition of 1 mM AEBSF.
Antibody production and immunodetection
Purified recombinant SSB was used as an antigen to raise polyclonal antibodies in sheep (antibody production by Diagnostics Scotland, Law Hospital, Carluke, Scotland). The specificity of the resulting antibodies was tested by western- and dot-blot analyses, using an anti-goat HRP-conjugated secondary antibody and chemiluminescent detection with ECL-Plus reagents (Amersham Pharmacia). The immune serum at a dilution of 1:1000 allowed detection of 2 ng SSB in a dot-blot, and 10 ng SSB in a western blot. SSB concentrations in Sulfolobus cell extracts were estimated by western blotting, comparing the signal obtained from a set of known concentrations of the pure recombinant protein with that from a known volume of cell extract. Total protein in the extracts was estimated by Bradford analysis.
Agarose gel electrophoretic retardation analysis
A range of concentrations of purified SSB protein was incubated with a known concentration of single-stranded phage phiX174 DNA in binding buffer (20 mM Tris pH 7.5, 50 mM NaCl, 1 mM EDTA and 0.1 mg/ml bovine serum albumin), in 10 µl total volume. After 15 min at 20°C, one-sixth vol loading buffer (0.25% bromophenol blue, 0.25% xylene cyanol FF, 35% Ficoll type 400) was added, and samples were loaded onto 0.7% agarose gels and electrophoresed in 1× TBE buffer for 90 min. After electrophoresis, gels were stained in ethidium bromide and visualised under UV light.
Acrylamide gel electrophoretic retardation analysis
During protein purification, fractions eluting from chromatography columns were tested for the ability to retard a ssDNA oligonucleotide. An aliquot (1 µl) of each fraction was incubated with a known concentration of a radioactively 5′-32P-labelled oligonucleotide in binding buffer (20 mM Tris pH 7.5, 50 mM NaCl, 1 mM EDTA and 0.1 mg/ml bovine serum albumin) in 10 µl total volume. After 15 min at 20°C, one-sixth vol loading buffer was added, and samples were loaded onto 8% polyacrylamide gels (19:1 acrylamide to bisacrylamide) and electrophoresed in 1× TBE buffer. After electrophoresis, gels were dried and exposed to X-ray film for documentation or phosphor screens for quantitation as described previously (14).
Fluorescence titrations
The fluorimetric titration experiments were performed on a Perkin-Elmer LS50B Luminescence Spectrometer. The tryptophan emission spectra of Sulfolobus SSB were obtained by excitation at 290 nm (excitation and emission slit widths 5 nm) in reaction buffer (20 mM Tris–HCl pH 7.5, 50 mM NaCl) at 20°C. Fluorescence quenching at the emission peak of 345 nm was monitored whilst titrating ssDNA species into a fixed concentration of SSB protein. Relative fluorescence values were corrected for dilution effects and subtraction of fluorescence due to buffer and DNA components.
Glutaraldehyde cross-linking
Chemical cross-linking of SSB protein with glutaraldehyde was carried out according to the method of Jaenicke and Rudolph (15). SSB protein (50 µg) was incubated in the presence or absence of 1 µM 18mer oligonucleotide in a 500 µl final volume in X-link buffer (50 mM NaPO4 pH 7.6, 50 mM NaCl) for 10 min at 20°C. After addition of 20 µl glutaraldehyde (25%, w/v) and incubation for 2 min at 20°C, the reaction was quenched by the addition of 25 µl NaBH4 (2 M in 0.1 M NaOH, freshly prepared), and incubation at 20°C for a further 20 min. Protein was precipitated by the addition of 1.5 µl 10% sodium deoxycholate followed by 22.5 µl 78% (w/v) TCA and incubation on ice for 5 min, followed by centrifugation. The supernatant was discarded, and the pellet resuspended in 600 µl cold acetone, followed by a final centrifugation step (13 000 g at 4°C for 10 min). The pellet was drained and allowed to air dry. Cross-linked proteins were solubilised by the addition of 20 µl 1× SDS–PAGE sample loading buffer and heating to 80°C for 10 min, followed by SDS–PAGE.
RESULTS
Purification, identification and cloning of the Sulfolobus SSB protein
Using a single-stranded oligonucleotide probe in an EMSA, we detected a ssDNA binding activity in S.solfataricus protein extracts fractionated by anion exchange chromatography (Fig. 1). The protein responsible for this binding activity was purified through two further columns until it was essentially homogeneous as assessed by SDS–PAGE (Fig. 3). The purified protein was identified by MALDI-TOF mass spectroscopy of tryptic peptides, and interrogation of the public release of the Sulfolobus genome sequence. This resulted in the unambiguous identification of a single ORF (bac19_023) encoding a 16 184 Da polypeptide. The protein eluted from a calibrated gel filtration column with an estimated size of 20 ± 4 kDa (data not shown), consistent with a monomeric subunit composition in solution. Analysis of the primary structure revealed the presence of a putative OB-fold domain within the first 100 amino acids that is common to all known SSB proteins, together with an approximately 40 amino acid C-terminal extension. No N-terminal extension was apparent, and the sequence does not include the putative zinc finger found in RPA70 and several euryarchaeal RPA sequences (9). Polyclonal antibodies raised against the purified recombinant protein were used to estimate the in vivo concentration of SSB in Sulfolobus cells as ~2–5% of total soluble protein. This apparent abundance is consistent with the important role of SSB in many DNA transactions in cells, and with previous estimates of the abundance of human RPA (1,16). It may also reflect an important role for SSB proteins in ssDNA stabilisation in hyperthermophilic organisms.
Figure 1.
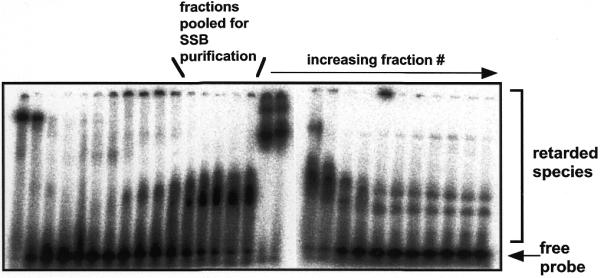
Detection of a ssDNA binding activity in extracts of S.solfataricus fractionated by anion-exchange chromatography. Aliquots (1 µl) of protein eluting from a SP-Sepharose column developed with a gradient of increasing NaCl were incubated with a ssDNA probe (radioactively labelled 18mer oligonucleotide) in binding buffer containing a large excess of unlabelled calf thymus duplex DNA, prior to detection of retarded species by EMSA. The fractions indicated were pooled and purified through a further two chromatography steps, yielding essentially homogeneous native Sulfolobus SSB protein (Fig. 3). The strongly retarded species apparent in fractions later than those collected are due to the presence of an abundant dsDNA binding protein, Sso10b (data not shown).
The closest homolog to Sulfolobus SSB was found in an unannotated ORF (accession no. Q9YCD3) present in A.pernix, the only other crenarchaeote for which substantial sequence data is in the public domain. Neither Sulfolobus nor Aeropyrum contain any other ORFs that are recognisable as SSB domains. BLAST searches suggest that the OB-fold in the crenarchaeal SSBs is most similar to those in euryarchaeal SSB proteins, supporting the monophyly of the archaea. The next closest matches were to hypothetical ORFs in the genome sequences of the higher plant Arabidopsis thaliana and the metazoan Caenorhabditis elegans (Fig. 2A). In contrast, alignment of the eucaryal, euryarchaeal and newly discovered crenarchaeal SSB proteins with eubacterial sequences required the insertion of several gaps in the OB-fold domain.
Figure 2.
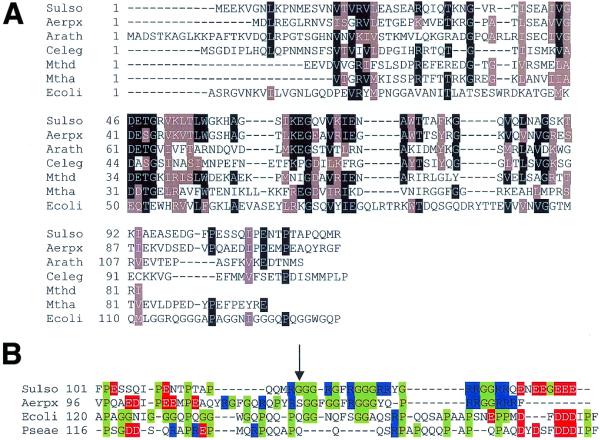
Sequence alignment of the OB-fold domain of a variety of SSB proteins. (A) Comparison of the protein sequence of Sulfolobus SSB with the closest matches identified using a PSI-BLAST search. Homologues include predicted proteins from crenarchaeal, euryarchaeal, higher plant and metazoan organisms. The comparison is restricted to the first 119 residues of Sulfolobus SSB, which is generated by trypsin digestion (see Fig. 3). The sequence of the eubacterial SSB from E.coli is included for comparative purposes, and demonstrates several insertions in the OB-fold DNA binding domain with respect to the other sequences. (Sulso: S.solfataricus SSB, bac19_023; Aerpx: A.pernix SSB, accession no. Q9YCD3; Arath: A.thaliana Q9ZUC0; Celeg: C.elegans Q17749; Mthd and Mtha: Methanobacterium thermoautotrophicum SSB domains d and a, respectively, O27438; Ecoli: E.coli SSB P02339). (B) Comparison of the C-terminal extension sequences of crenarchaeal and eubacterial SSBs. The C-terminal sequences of crenarchaeal (Sulfolobus and Aeropyrum) and eubacterial (E.coli and Pseudomonas aeruginosa, accession no. P40947) SSBs are aligned, with glycine and proline residues highlighted in green, arginine in blue and acidic residues in red. The highly flexible nature of the sequence in all four proteins is apparent. The crenarchaeal sequences appear to have a positively charged region that is not found in the eubacterial SSBs, and Sulfolobus SSB has an acidic region at the extreme C-terminus. The C-terminus of Sulfolobus SSB is clipped by trypsin proteolysis at Arg-119 (arrow).
Outwith the OB-fold, the crenarchaeal SSB sequences contain surprising similarities to the equivalent C-termini of the eubacterial SSBs. This includes the presence of a large number of glycine and proline residues that are expected to confer flexibility on the C-terminal ‘tail’ of the protein (shown in green in Fig. 2B), together with areas of positively and negatively charged residues (blue and red, respectively, in Fig. 2B).
The gene encoding the putative SSB was cloned from S.solfataricus P2 genomic DNA by PCR amplification, and cloned into a plasmid allowing heterologous expression in E.coli. The recombinant protein was purified by a heat denaturation and two column chromatography steps as described in Materials and Methods (Fig. 3). In order to test the predicted flexibility of the C-terminal tail of Sulfolobus SSB, the pure recombinant protein was subjected to partial digestion with a 250:1 ratio of SSB:porcine trypsin. SDS–PAGE and MALDI-TOF analysis of the digested protein showed a single major peak with a molecular mass of 12 933 Da. This allowed the unambiguous identification of the site of cleavage by trypsin as Arg-119, the first arginine present in the Gly/Pro-rich C-terminal region, suggesting that the C-terminal 30 amino acids of Sulfolobus SSB exist in a flexible conformation. Digestion of intact recombinant SSB with the enzyme Glu-C, which cleaves C-terminal to glutamate residues, did not result in the formation of any further-truncated species, despite the presence of several potential target sites (data not shown), suggesting that the remainder of the protein has a defined folded structure.
DNA binding characteristics of native, recombinant and truncated SSBs
Sulfolobus SSB was identified on the basis of its ability to bind and retard a ssDNA oligonucleotide during acrylamide gel electrophoresis (Fig. 1). However, for the experimental determination of binding affinities and stoichiometries, the EMSA technique was not ideal for this protein, probably due to DNA–protein dissociation over the lifetime of the electrophoretic separation. Accordingly, we investigated alternative procedures for the characterisation of the interaction of SSB with nucleic acids. Using a circular ssDNA molecule, phage phiX174, composed of 5386 nt, we demonstrated DNA binding using an agarose gel electrophoretic assay (Fig. 4). As greater numbers of protein molecules bind to the ssDNA circles, their mobility is gradually reduced in the gel, until an end-point is reached where all available ssDNA binding sites are saturated by protein. This end-point coincided with a dramatic reduction in fluorescence intensity after staining with ethidium bromide. We interpret this effect as being due to the occlusion of ethidium bromide from potential binding sites on the DNA when it is fully bound by the protein, suggesting that Sulfolobus SSB can intercalate side chains between DNA bases.
Figure 4.
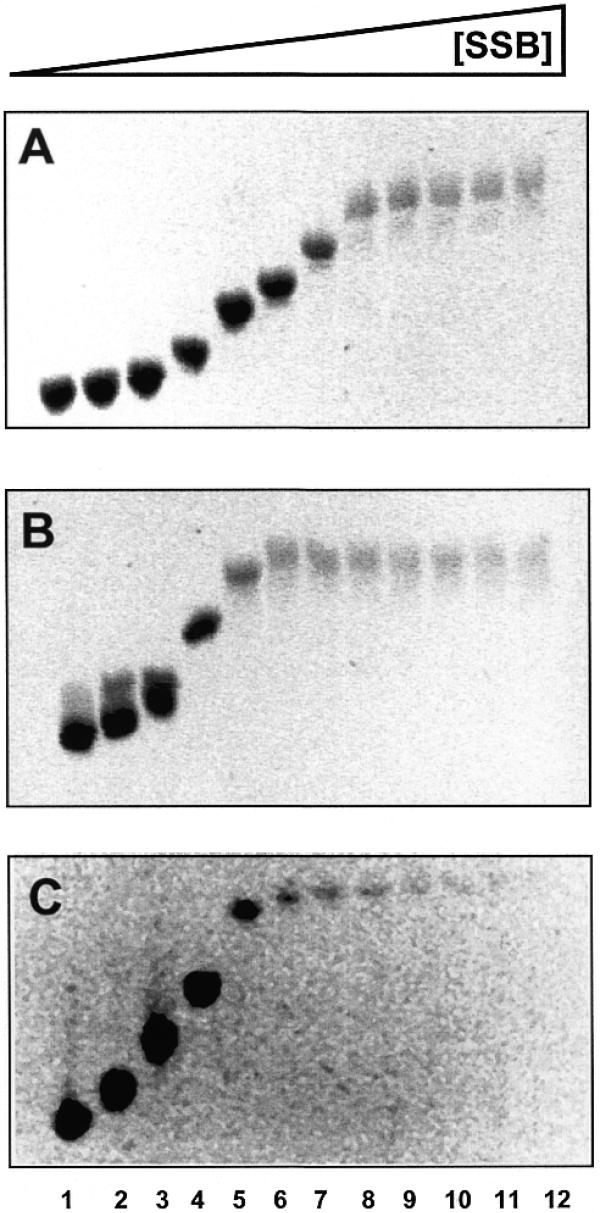
Agarose gel electrophoretic analysis of ssDNA binding by native (A), recombinant (B) and truncated (C) SSB. PhiX174 single-stranded virion DNA (250 ng) was incubated with the indicated amount of SSB protein in binding buffer, in a total volume of 10 µl for 10 min at 20°C, prior to addition of Ficoll loading buffer. DNA species were fractionated in 0.7% agarose gels in 1× TBE buffer and DNA visualised after electrophoresis by staining with ethidium bromide. Saturation of the viral DNA with bound SSB protein coincided with strongly reduced ethidium bromide fluorescence, suggesting that intercalation sites for ethidium bromide are occluded by the bound protein. Lanes 1–12 contain 250 ng phiX174 and 0.05, 0.125, 0.25, 0.5, 1, 1.5, 2, 3, 4.5, 6, 7.5 and 10 ng SSB, respectively.
The agarose gel retardation assay was repeated for the purified native SSB protein, the recombinant SSB protein and the recombinant protein after truncation by trypsin. By calculating the molar ratios of DNA nucleotides:SSB molecules at the point of saturation, we estimated the binding site size of Sulfolobus SSB as 4–6 nt per SSB monomer for the native protein, ~8–12 nt per monomer for the recombinant protein and 7–10 nt per monomer for the truncated protein. Thus the truncated protein retains the ability to bind DNA, as seen for E.coli SSB (17,18). The slight differences in binding site size calculated for the native, recombinant and truncated species are more likely to be due to experimental error rather than having a functional significance, particularly in light of the fluorescence quenching data described below. This experiment also demonstrated that DNA binding is not highly co-operative, but has some distributive character, otherwise we would not expect to see a gradual reduction in the mobility of the bulk of the DNA, but rather an interconversion between non-retarded and fully-retarded species.
DNA binding monitored by quenching of intrinsic tryptophan fluorescence
Sulfolobus SSB has two tryptophan residues in the OB-fold domain, allowing analysis of ssDNA binding by tryptophan fluorescence quenching (Fig. 5). The protein displayed strong intrinsic fluorescence with a peak wavelength of 345 nm when excited at 290 nm. As ssDNA was titrated into the cuvette, the intrinsic fluorescence of the protein was progressively quenched, reaching an end-point at ~20–40% of the unquenched value. The binding isotherms suggest a dissociation constant in the low nanomolar concentration range. Regardless of the length of ssDNA strand added (18, 69 or 5386 nt), binding was saturated at a ratio of ~4–5 nt ssDNA per monomer of Sulfolobus SSB, or 20–25 nt per tetramer. This ratio was unaffected by the presence or absence of the C-terminal tail and is in broad agreement with the values estimated by agarose gel electrophoresis. In comparison, Methanococcus jannaschii RPA binds 20 nt ssDNA (10) and human RPA binds DNA non-cooperatively, with a binding site size of 30 nt per heterotrimer under most conditions (19). Escherichia coli SSB displays multiple binding modes, with 35 or 65 nt bound per tetramer (20–22).
Figure 5.

DNA binding by Sulfolobus SSB monitored by intrinsic fluorescence quenching. Reaction mixtures (0.5 ml) containing 400 nM SSB monomer were excited at 290 nm, and fluorescence emission with a maximum at 345 nm was recorded. Scans were repeated after addition of aliquots of ssDNA species, the data were corrected for dilution effects and normalised to the initial fluorescence in the absence of DNA. Fluorescence quenching of the intact SSB protein was monitored in response to binding of an 18mer oligo (open circles), 69mer oligo (closed circles) and phiX174 ssDNA (closed squares). Binding of the 69mer was also monitored for the SSB truncated by trypsin (open squares). For all four conditions, we observed saturation of SSB binding at a ratio of ~5 nt DNA per SSB monomer.
Glutaraldehyde cross-linking
To address the question of the subunit composition of Sulfolobus SSB, we incubated the protein with glutaraldehyde in order to cross-link lysine residues in the presence and absence of an 18mer oligonucleotide. Conditions were chosen in order to maximise inter-subunit cross-links whilst minimising cross-links between non-associated protein subunits. Cross-linked species were precipitated and analysed by SDS–PAGE (Fig. 6). Whilst there was a suggestion of the presence of higher molecular species in the absence of DNA (lanes 3 and 4), addition of a roughly equimolar concentration of a short oligonucleotide resulted in a significant increase in the amount of cross-linked species, which may correspond to the presence of dimers and tetramers of SSB. Taken together with the gel filtration data, these studies suggest that Sulfolobus SSB is a monomer in solution, and that multimers (probably tetramers) are formed, or at least significantly stabilised, only in the presence of DNA.
Figure 6.
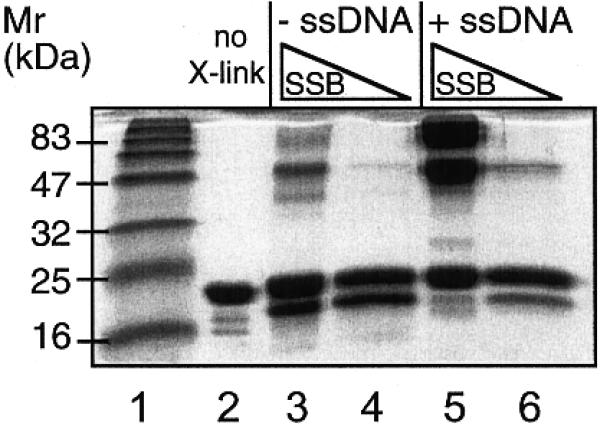
Analysis of the subunit structure of SSB assessed by glutaraldehyde chemical cross-linking in the presence and absence of ssDNA (1 µM 18mer oligonucleotide). The presence of DNA gives rise to a significant increase in the proportion of cross-linked species evident after SDS–PAGE (lane 5). Retarded species have mobilities consistent with the formation of cross-linked dimers and tetramers of SSB. No further-retarded complexes were observed. Lane 1, markers; lane 2, 5 µM recombinant SSB, no glutaraldehyde; lane 3, 5 µM SSB without DNA; lane 4, 1 µM SSB without DNA; lane 5, 5 µM SSB with DNA; lane 6, 1 µM SSB with DNA.
DISCUSSION
DNA is subjected to a continual assault by cellular and environmental factors, and must be repaired efficiently to preserve the genetic integrity of all DNA-based life forms. Many of these stresses encountered by cells are likely to be highly exacerbated by adaptation to life in extreme conditions, and this has led to the evolution of highly efficient DNA repair pathways in extremophiles. For example, the extreme radiation-resistance of the bacterium Deinococcus radiodurans probably arose in response to the high levels of DNA damage suffered upon desiccation (23). Adaptation to growth at elevated temperatures presents a unique set of challenges, as the rates of chemical reactions resulting in DNA modification are correspondingly higher, and the structural integrity of all cellular components, including nucleic acids and proteins, must be preserved. Most of the hyperthermophilic organisms known at present are archaeal, and we still have only a very patchy knowledge of DNA repair pathways in these organisms. The tantalising suggestion that some archaeal DNA repair pathways may, like those of DNA replication and transcription, resemble their eucaryal equivalents deserves further study. Accordingly, we have undertaken the identification and characterisation of archaeal DNA repair proteins, using genomic information where possible, and biochemical alternatives when necessary. The latter approach has led to the identification of the first crenarchaeal ssDNA binding protein, reported here.
Analysis of the domain and subunit composition of Sulfolobus SSB has yielded some surprises. Whilst the Sulfolobus OB-fold domain responsible for DNA binding is clearly related most closely to other archaeal SSB domains, the C-terminal region of the protein has unexpected similarities to eubacterial SSBs. The role of the C-terminal tail of E.coli SSB, which consists of a flexible spacer region and a highly acidic extreme C-terminal α-helix, has been investigated in some detail. Deletions in the C-terminal α-helix result in severe repair-deficient phenotypes that are not caused by alterations in DNA binding characteristics, suggesting the tail has a role in protein–protein interactions. Escherichia coli SSB interacts directly with the base excision repair protein exonuclease I via the C-terminal helix (24,25). It is tempting to speculate that the C-terminal tail of Sulfolobus SSB plays a similar role in vivo. If so, the clear differences apparent in the DNA binding domains of Sulfolobus and E.coli SSB suggest that the evolution of similar C-terminal domains is an example of convergent rather than divergent evolution. In the eucaryal and euryarchaeal RPA proteins, other domains probably fulfil similar roles, and it is notable that these proteins have N-terminal extensions and a zinc binding domain that are not present in the eubacterial and crenarchaeal proteins. The crenarchaeota possess some unique features in dsDNA as well as ssDNA binding proteins. They lack the histones found in most of the euryarchaeota, and instead rely on small non-histone dsDNA binding proteins such as Sso7 and Sso10 in Sulfolobus. The roles of all of these proteins in DNA stability, replication and repair in hyperthermophilic archaea are currently under scrutiny.
The DNA binding characteristics of Sulfolobus SSB appear similar to most other SSB proteins. The analyses described here have all been undertaken at 20°C, some 60°C below the optimal growth temperature of Sulfolobus. Our studies of other Sulfolobus proteins such as the Holliday junction resolving enzyme Hjc (26) and the dsDNA binding protein Sso10b (B.N.Wardleworth and M.F.White, unpublished data) suggest that DNA binding by thermostable proteins is not highly temperature dependent in general, whereas enzymatic activity clearly obeys the Arrhenius equation. Nevertheless, more detailed studies of the interaction of Sulfolobus SSB with ssDNA at more physiological temperatures may identify some significant effects. The binding site footprint of 5 nt ssDNA per monomer most likely reflects the amount of DNA accommodated by each OB-fold domain, which is known to be 4 nt for the co-crystal structure of RPA70 (i.e. 8 nt ssDNA are bound by the two OB-folds of RPA70) (2). However, Sulfolobus SSB appears unique amongst cellular SSB proteins in being monomeric in solution. Chemical cross-linking experiments suggest that the protein multimerises on DNA binding, and by analogy with other SSB proteins we would expect a tetramer to represent the functional subunit structure. Further analysis of the subunit composition of Sulfolobus SSB using analytical ultracentrifugation is currently underway.
The central role of SSB proteins in DNA replication and repair is emphasised by the large number of publications reporting functional interactions of RPA with other proteins. For example, RPA has been shown to interact with the nucleotide excision repair protein XPA (27–29), the Bloom’s syndrome helicase BLM (30), the strand exchange proteins Rad51 (31) and Rad52 (32–34) and P53 (35). The identification of the crenarchaeal ssDNA binding protein reported here brings us a step closer to the reconstitution of crenarchaeal DNA replication and repair pathways in vitro, and may aid the identification of further archaeal repair proteins that are not detectable solely on the basis of their protein sequence.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Neil Raven for advice and supply of the biomass and Mamuka Kvaratskhelia, Derek Richard and Ben Wardleworth for helpful advice and discussion. M.F.W. is a Royal Society University Research Fellow. This work was funded by the BBSRC.
References
- 1.Wold M.S. (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem., 66, 61–92. [DOI] [PubMed] [Google Scholar]
- 2.Bochkarev A., Bochkareva,E., Frappier,L. and Edwards,A.M. (1999) The crystal structure of the complex of replication protein A subunits RPA32 and RPA14 reveals a mechanism for single-stranded DNA binding. EMBO J., 18, 4498–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bochkarev A., Pfuetzner,R.A., Edwards,A.M. and Frappier,L. (1997) Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature, 385, 176–181. [DOI] [PubMed] [Google Scholar]
- 4.Webster G., Genschel,J., Curth,U., Urbanke,C., Kang,C. and Hilgenfeld,R. (1997) A common core for binding single-stranded DNA: structural comparison of the single-stranded DNA-binding proteins (SSB) from E. coli and human mitochondria [published errata appear in FEBS Lett. (1997) 415, 351 and (1997) 416, 387]. FEBS Lett., 411, 313–316. [DOI] [PubMed] [Google Scholar]
- 5.Raghunathan S., Kozlov,A.G., Lohman,T.M. and Waksman,G. (2000) Structure of the DNA binding domain of E.coli SSB bound to ssDNA. Nat. Struct. Biol., 7, 648–652. [DOI] [PubMed] [Google Scholar]
- 6.Yang C., Curth,U., Urbanke,C. and Kang,C. (1997) Crystal structure of human mitochondrial single-stranded DNA binding protein at 2.4 Å resolution. Nat. Struct. Biol., 4, 153–157. [DOI] [PubMed] [Google Scholar]
- 7.Murzin A.G. (1993) OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J., 12, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiRuggiero J., Brown,J.R., Bogert,A.P. and Robb,F.T. (1999) DNA repair systems in archaea: mementos from the Last Universal Common Ancestor? J. Mol. Evol., 49, 474–484. [DOI] [PubMed] [Google Scholar]
- 9.Chedin F., Seitz,E.M. and Kowalczykowski,S.C. (1998) Novel homologs of replication protein A in archaea: implications for the evolution of ssDNA-binding proteins. Trends Biochem. Sci., 23, 273–277. [DOI] [PubMed] [Google Scholar]
- 10.Kelly T.J., Simancek,P. and Brush,G.S. (1998) Identification and characterization of a single-stranded DNA-binding protein from the archaeon Methanococcus jannaschii. Proc. Natl Acad. Sci. USA, 95, 14634–14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keeling P.J. and Doolittle,W.F. (1995) Archaea: narrowing the gap between prokaryotes and eukaryotes. Proc. Natl Acad. Sci. USA, 92, 5761–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reeve J.N., Sandman,K. and Daniels,C.J. (1997) Archaeal histones, nucleosomes and transcription initiation. Cell, 89, 999–1002. [DOI] [PubMed] [Google Scholar]
- 13.Kawarabayasi Y., Hino,Y., Horikawa,H., Yamazaki,S., Haikawa,Y., Jin-no,K., Takahashi,M., Sekine,M., Baba,S., Ankai,A. et al. (1999) Complete genome sequence of an aerobic hyper-thermophilic crenarchaeon, Aeropyrum pernix K1. DNA Res., 6, 83–101, 145–152. [DOI] [PubMed] [Google Scholar]
- 14.White M.F. and Lilley,D.M.J. (1997) Characterisation of a Holliday junction-resolving enzyme from Schizosaccharomyces pombe. Mol. Cell. Biol., 17, 6465–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaenicke R. and Rudolph,R. (1986) Refolding and association of oligomeric proteins. Methods Enzymol., 131, 218–250. [DOI] [PubMed] [Google Scholar]
- 16.Seroussi E. and Lavi,S. (1993) Replication protein A is the major single-stranded DNA binding protein detected in mammalian cell extracts by gel retardation assays and UV cross-linking of long and short single-stranded DNA molecules. J. Biol. Chem., 268, 7147–7154. [PubMed] [Google Scholar]
- 17.Curth U., Genschel,J., Urbanke,C. and Greipel,J. (1996) In vitro and in vivo function of the C-terminus of Escherichia coli single-stranded DNA binding protein. Nucleic Acids Res., 24, 2706–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams K.R., Spicer,E.K., LoPresti,M.B., Guggenheimer,R.A. and Chase,J.W. (1983) Limited proteolysis studies on the Escherichia coli single-stranded DNA binding protein. Evidence for a functionally homologous domain in both the Escherichia coli and T4 DNA binding proteins. J. Biol. Chem., 258, 3346–3355. [PubMed] [Google Scholar]
- 19.Kim C., Snyder,R.O. and Wold,M.S. (1992) Binding properties of replication protein A from human and yeast cells. Mol. Cell. Biol., 12, 3050–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffith J.D., Harris,L.D. and Register,J.D. (1984) Visualization of SSB–ssDNA complexes active in the assembly of stable RecA–DNA filaments. Cold Spring Harbor Symp. Quant. Biol., 49, 553–559. [DOI] [PubMed] [Google Scholar]
- 21.Bujalowski W. and Lohman,T.M. (1986) Escherichia coli single-strand binding protein forms multiple, distinct complexes with single-stranded DNA. Biochemistry, 25, 7799–7802. [DOI] [PubMed] [Google Scholar]
- 22.Lohman T.M. and Overman,L.B. (1985) Two binding modes in Escherichia coli single strand binding protein–single stranded DNA complexes. Modulation by NaCl concentration. J. Biol. Chem., 260, 3594–3603. [PubMed] [Google Scholar]
- 23.Mattimore V. and Battista,J.R. (1996) Radioresistance of Deinococcus radiodurans: functions necessary to survive ionizing radiation are also necessary to survive prolonged desiccation. J. Bacteriol ., 178, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Genschel J., Curth,U. and Urbanke,C. (2000) Interaction of E. coli single-stranded DNA binding protein (SSB) with exonuclease I. The carboxy-terminus of SSB is the recognition site for the nuclease. Biol. Chem., 381, 183–192. [DOI] [PubMed] [Google Scholar]
- 25.Sandigursky M., Mendez,F., Bases,R.E., Matsumoto,T. and Franklin,W.A. (1996) Protein–protein interactions between the Escherichia coli single-stranded DNA-binding protein and exonuclease I. Radiat. Res., 145, 619–623. [PubMed] [Google Scholar]
- 26.Kvaratskhelia M. and White,M.F. (2000) Two Holliday junction resolving enzymes in Sulfolobus solfataricus. J. Mol. Biol., 297, 923–932. [DOI] [PubMed] [Google Scholar]
- 27.Li L., Lu,X., Peterson,C.A. and Legerski,R.J. (1995) An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol. Cell. Biol., 15, 5396–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stigger E., Drissi,R. and Lee,S.H. (1998) Functional analysis of human replication protein A in nucleotide excision repair. J. Biol. Chem., 273, 9337–9343. [DOI] [PubMed] [Google Scholar]
- 29.Wang M., Mahrenholz,A. and Lee,S.H. (2000) RPA stabilizes the XPA-damaged DNA complex through protein–protein interaction. Biochemistry, 39, 6433–6439. [DOI] [PubMed] [Google Scholar]
- 30.Brosh R.M.,Jr, Li,J.L., Kenny,M.K., Karow,J.K., Cooper,M.P., Kureekattil,R.P., Hickson,I.D. and Bohr,V.A. (2000) Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J. Biol. Chem., 275, 23500–23508. [DOI] [PubMed] [Google Scholar]
- 31.Golub E.I., Gupta,R.C., Haaf,T., Wold,M.S. and Radding,C.M. (1998) Interaction of human rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res., 26, 5388–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugiyama T., New,J.H. and Kowalczykowski,S.C. (1998) DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc. Natl Acad. Sci. USA, 95, 6049–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hays S.L., Firmenich,A.A., Massey,P., Banerjee,R. and Berg,P. (1998) Studies of the interaction between Rad52 protein and the yeast single- stranded DNA binding protein RPA. Mol. Cell. Biol., 18, 4400–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shinohara A., Shinohara,M., Ohta,T., Matsuda,S. and Ogawa,T. (1998) Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells, 3, 145–156. [DOI] [PubMed] [Google Scholar]
- 35.Abramova N.A., Russell,J., Botchan,M. and Li,R. (1997) Interaction between replication protein A and p53 is disrupted after UV damage in a DNA repair-dependent manner. Proc. Natl Acad. Sci. USA, 94, 7186–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]