


Abstract
A detailed computational analysis of 32 protein–RNA complexes is presented. A number of physical and chemical properties of the intermolecular interfaces are calculated and compared with those observed in protein–double-stranded DNA and protein–single-stranded DNA complexes. The interface properties of the protein–RNA complexes reveal the diverse nature of the binding sites. van der Waals contacts played a more prevalent role than hydrogen bond contacts, and preferential binding to guanine and uracil was observed. The positively charged residue, arginine, and the single aromatic residues, phenylalanine and tyrosine, all played key roles in the RNA binding sites. A comparison between protein–RNA and protein–DNA complexes showed that whilst base and backbone contacts (both hydrogen bonding and van der Waals) were observed with equal frequency in the protein–RNA complexes, backbone contacts were more dominant in the protein–DNA complexes. Although similar modes of secondary structure interactions have been observed in RNA and DNA binding proteins, the current analysis emphasises the differences that exist between the two types of nucleic acid binding protein at the atomic contact level.
INTRODUCTION
RNA performs essential and diverse functions within the cell. It forms part of the ribosome (1,2) and the spliceosome (3) and also exhibits catalytic activity (4–6). A common thread to many of these functions is the interaction of RNA with proteins. For example, specific tRNAs are bound to aminoacyl-tRNA synthetases for the translation of the genetic code during protein synthesis (7), and ribonucleoprotein particles (RNPs) bind RNA in post-transcriptional regulation of gene expression (8). However, despite their obvious functional importance, the specific mechanisms of protein–RNA interactions are still poorly understood. This is in contrast to the much clearer picture of interactions in protein–DNA complexes (9–11). The lack of information about protein–RNA complexes reflects the smaller number of structures that have been solved by crystallography and NMR. The current work was completed prior to the structure of the ribosomal subunits being solved (12–14). When the analysis was conducted there were a total of 330 known protein–DNA complex structures compared with just 35 protein–RNA complexes [Nucleic Acid Database (NDB) (15)]. With the coordinates of the ribsomal structures now available the number of protein–RNA complexes with known structures has risen to 89, and whilst this number is still small, these new structures provide important new data for analysis.
In contrast to the regular double helical structure of B-DNA commonly found in protein–DNA complexes, RNAs display structures almost as diverse as their function. RNA structures are flexible molecules that display complex secondary and tertiary structures. RNAs are commonly single-stranded but structures also include short lengths of double helices (A-form), hairpin loops, bulges and pseudoknots. Proteins tend to interact with RNA where it forms complex secondary structure elements such as stem–loops and bulges (16). In addition non-Watson–Crick base pairing can occur in loop regions of RNA structures and such features can also be preferentially identified by proteins (17).
Work in this field has primarily centred on the identification of recurring RNA recognition motifs such as the RNP and arginine-rich motifs (16,18,19) and interactions within individual complexes (20–22). A large amount of data has also been derived from aminoacyl-tRNA synthetases for which a number of complexes have been solved (7,23).
Now that an increasing number of protein–RNA structures are known, there is a need to draw together the structural data to look for common features that might characterise the intermolecular interactions within them. A comprehensive review of protein–RNA structures has most recently been published by Draper (24). This work goes further than previous reviews on the subject, by dividing complexes into two main classes based on the mode of RNA recognition: (i) groove binding and (ii) β-sheet binding. In the former, proteins position a secondary structure element, such as an α-helix or loop, into the groove of an RNA helix. In the latter, proteins use β-sheet surfaces to create binding pockets that bind unpaired RNA bases. These two recognition themes are adopted in the current analysis (Table 1).
Table 1. Dataset of 32 protein–RNA complexes selected from the NDB (December 7, 1999).
| |
Family |
PDB |
NDB |
Protein name |
Protein recognition |
RNA type |
RNA structure |
Res. |
| A | 1 | 1A34 | PRV020 | Coat protein STMV | β-sheet | vRNA | Double-stranded | 1.81 |
| 2 | 1BMV | PRV001 | Coat protein BPMV | β-sheet | vRNA | Single-stranded | 3.00 | |
| 3 | 2BBV | PRV004 | Coat protein BBV | Groove binding | vRNA | Double-stranded | 2.80 | |
| 4 | 1ZDI | PRV006 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 2.70 | |
| 1ZDH | PRV003 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 2.70 | ||
| 1ZDJ | PRV021 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 2.70 | ||
| 1ZDK | PR0003 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 2.86 | ||
| 1AQ3 | PRV008 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 2.80 | ||
| 1AQ4 | PRV009 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 3.00 | ||
| 5MSF | PR0001 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 2.80 | ||
| 6MSF | PRV010 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 2.80 | ||
| 7MSF | PR0002 | Coat protein, Bacteriophage MS2 | β-sheet | vRNA | Single-stranded, single loop | 2.80 | ||
| 5 | 1A1T | N/A | Nucleocapsid protein HIV-1 | Groove binding | vRNA | Single-stranded, single loop | NMR | |
| B | 6 | 1ASY | PTR005 | Aspartyl-tRNA synthetase | Groove binding + β-sheet | Asp-tRNA | Single-stranded, multiple loops | 3.00 |
| 1ASZ | PTR008 | Aspartyl-tRNA synthetase (+ATP) | Groove binding + β-sheet | Asp-tRNA | Single-stranded, multiple loops | 3.00 | ||
| 7 | 1QTQ | PTE003 | Glutaminyl-tRNA synthetase | Groove binding + β-sheet | Gln-tRNA | Single-stranded, multiple loops | 2.40 | |
| 1GTS | PTR002 | Glutaminyl-tRNA synthetase (+AMP) | Groove binding + β-sheet | Gln-tRNA | Single-stranded, multiple loops | 2.80 | ||
| 1GSG | PTR001 | Glutaminyl-tRNA synthetase (+ATP) | Groove binding + β-sheet | Gln-tRNA | Single-stranded, multiple loops | 2.80 | ||
| 1GTR | PTR003 | Glutaminyl-tRNA synthetase (+ATP) | Groove binding + β-sheet | Gln-tRNA | Single-stranded, multiple loops | 2.50 | ||
| 1QRS | PTR009 | Glutaminyl-tRNA synthetase | Groove binding + β-sheet | Gln-tRNA | Single-stranded, multiple loops | 2.60 | ||
| 1QRT | PTR010 | Glutaminyl-tRNA synthetase | Groove binding + β-sheet | Gln-tRNA | Single-stranded, multiple loops | 2.70 | ||
| 1QRU | PTR011 | Glutaminyl-tRNA synthetase | Groove binding + β-sheet | Gln-tRNA | Single-stranded, multiple loops | 3.00 | ||
| 8 | 1SER | PTR004 | Seryl-tRNA synthetase | Groove binding | Ser-tRNA | Single-stranded, multiple loops | 2.90 | |
| 9 | 1QF6 | N/A | Threonyl-tRNA synthetase | Groove binding + β-sheet | Thr-tRNA | Single-stranded, multiple loops | 2.90 | |
| 10 | 1TTT | PTR012 | Elongation factor Tu | Groove binding + β-sheet | Phe-tRNA | Single-stranded, multiple loops | 2.70 | |
| 1B23 | PDR0004 | Elongation factor Tu | Groove binding + β-sheet | Cys-tRNA | Single-stranded, multiple loops | 2.60 | ||
| 11 | 1QA6 | N/A | L11 | Groove binding | 23S rRNA | Single-stranded, multiple loops | 2.60 | |
| C | 12 | 1AV6 | PRV007 | Methyltransferase VP39 | Groove binding | MRNA | Single-stranded | 2.70 |
| 13 | 1A9N | PTR016 | Spliceosomal U2B″/U2A′ complex | β-sheet | snRNA | Single-stranded, single loop | 2.38 | |
| 1URN | PRV002 | U1A spliceosomal protein | β-sheet | snRNA | Single-stranded, single loop | 1.92 | ||
| 1AUD | N/A | U1A spiceosomal protein | β-sheet | snRNA | Single-stranded, multiple loops | NMR | ||
| 14 | 1B7F | N/A | Sex-lethal protein | β-sheet | mRNA | Single-stranded | 2.60 |
The structures have been divided into those that bind viral RNA (A), those involved in protein synthesis (B) and those involved in RNA modification (C). STMV, Satellite tobacco mosaic virus; BPMV, bean pod mosaic virus; BBV, black beetle virus.
The PDB codes shown in bold are those structures included in the non-homologous dataset used for the interface parameter calculations (see Materials and Methods).
Here we present a comprehensive analysis of protein–RNA interactions at the residue and atom level, and compare them with interactions observed in protein–double-stranded DNA (dsDNA) complexes (N.M.Luscombe and J.M.Thornton, manuscript in preparation; 25) and protein–single-stranded DNA (ssDNA) complexes. Data for this analysis has been drawn from both the NDB (15) and the Protein Data Bank (PDB) (26). A computational analysis of chemical and physical properties of nucleic acid binding sites on proteins, including the size, polarity and packing is described. In addition the distribution of observed atom–atom contacts in the protein–nucleic acid complexes have been calculated and compared to expected values.
MATERIALS AND METHODS
Datasets
For this analysis a total of 35 protein–RNA complexes were extracted from the NDB (on December 7, 1999) with resolutions of 3.0 Å or better and the full coordinates of all atoms (by December 12, 2000 there were 59 protein–RNA complexes in the NDB with a resolution of 3.0 Å or better) (15). Of these, 27 involved at least five RNA bases. To this initial dataset two protein–RNA complexes solved by NMR and a further three structures very recently solved by X-ray crystallography (22) were added, to produce a dataset of 32 protein–RNA complexes (Table 1). The proteins in each complex were classified into structural families using the structural alignment program SSAP (27). A SSAP score of ≥80 (and a sequence identity of >20%) between a pair of protein chains indicates that the two are structurally related and hence they were clustered into the same structural family. A representative complex (with the best resolution) from each family was selected to be included in a non-homologous dataset. Additional proteins were included from a family if the sequence of the bound RNA was different, hence the same protein could be included in the non-homologous dataset but only if the RNA sequences were different in each case. This resulted in a non-homologous dataset of 20 protein–RNA complexes (Table 1). The protein–RNA complexes were also divided into three subsets dependant upon their function: (A) proteins binding viral RNA (vRNA), (B) those involved in protein synthesis, binding transfer RNA (tRNA) and ribosomal RNA (rRNA) and (C) those involved in RNA modification, binding messenger RNA (mRNA) and small nuclear RNA (snRNA) (Table 1).
In Table 1, the RNA molecules bound to the proteins are classified into double-stranded (A-type double helix), single-stranded (elongated structures with no tertiary structure elements), single-stranded with single loop (commonly forming a hairpin loop), single-stranded with multiple loops (commonly forming the classic cloverleaf structures observed in the tRNAs). The type of recognition used by the protein is additionally included in Table 1, using the two classes identified by Draper (21). One structure from each family is shown in a Molscript diagram in Figure 1.
Figure 1.


MOLSCRIPT diagrams depicting protein–RNA complexes. One complex from each of the 14 families in Table 1 is presented. The sizes of the proteins are not comparable between diagrams and each is viewed from an angle that best depicts both the protein and RNA. In each diagram the RNA molecule is shown in ball-and-stick format and the proteins in ribbon format. Different subunits of the same protein are differentiated by colour. (1) Coat protein from Satellite tobacco mosaic virus (1A34); (2) bean pod mottle virus (middle component) (1BMV); (3) black beetle virus capsid protein (2BBV); (4) MS2 protein capsid (1ZDI); (5) HIV-1 nucleocapsid protein (1A1T); (6) aspartyl tRNA synthetase (1ASY); (7) glutaminyl tRNA synthetase (1QTQ); (8) seryl tRNA synthetase (1SER); (9) threonyl tRNA synthetase (1QF6); (10) elongation factor EF-TU (1TTT); (11) ribosomal protein L11 (1QA6); (12) methyltransferase VP39 (1AV6); (13) spliceosomal U2B″/U2A′ complex (1A9N); (14) sex-lethal protein (1B7F).
A second dataset of proteins bound to ssDNA structures was also selected from the NDB (on December 7, 1999). There were 16 protein–ssDNA complexes in the NDB with full coordinates available, that bind between 3 and 16 nucleic acid bases (by December 12, 2000 there were 29 protein–ssDNA complexes in the NDB). These 16 proteins were clustered into eight structural families using SSAP (27) as described above (Table 2). One complex with the best resolution was selected as a representative from each family if it included at least five DNA bases. As before, additional proteins were included from a family if the DNA sequence bound was different. This resulted in a non-homologous dataset of just three protein–ssDNA complexes (Table 2).
Table 2. Dataset of 16 protein–ssDNA complexes selected from the NDB (December 7, 1999).
| Family |
PDB |
NDB |
Protein name |
Res. |
No. bases |
| 1 | 2KFN | PD0014 | Exonuclease: Klenow fragment | 2.03 | 3 |
| 2KFZ | PD0015 | Exonuclease: Klenow fragment | 2.03 | 3 | |
| 1KFS | PDE0137 | Exonuclease: Klenow fragment | 2.10 | 3 | |
| 1KRP | PDE0138 | Exonuclease: Klenow fragment | 2.20 | 3 | |
| 1KSP | PDE0136 | Exonuclease: Klenow fragment | 2.30 | 3 | |
| 2 | 4DPV | PDV006 | Coat protein CPV | 2.90 | 11 |
| 1IJS | PDV005 | Coat protein | 3.25 | 11 | |
| 1MVM | PDV007 | Coat protein MMV | 3.50 | 16 | |
| 3 | 1RTA | PDE0116 | Ribonuclease A | 2.50 | 4 |
| 1RCN | PDE0117 | Ribonuclease A | 2.50 | 4 | |
| 1RBJ | PDE023 | Ribonuclease B | 2.70 | 4 | |
| 4 | 1JMC | PDO001 | Replication protein A | 2.40 | 8 |
| 5 | 1CBV | PDA001 | FAB antibody | 2.66 | 3 |
| 6 | 1LAU | PDE026 | Uracil DNA glycosylase | 1.80 | 3 |
| 7 | 1NOY | PDE090 | DNA polymerase fragment | 2.20 | 3 |
| 8 | 2BPA | PDV002 | Capsid proteins GPF, GPG, GPJ | 3.00 | 5 |
The PDB codes shown in bold are those structures included in the non-homologous dataset used for the interface parameter calculations (see Materials and Methods).
Analysis of nucleic acid binding site properties
As described in our previous analysis (28), an amino acid was defined as an interface residue if it lost >1 Å2 of accessible surface area (ASA) when passing from the uncomplexed state (protein only) to the complexed state (protein–RNA). The ASA of the protein complexed with RNA and the protein molecule without the RNA present was calculated using the computer program Naccess (http://wolf.bms.umist.ac.uk/naccess). With these two ASA calculations it is possible to identify those protein residues whose ASA is reduced by >1 Å2 on complex formation with RNA, termed the interface residues. The total number of interface residues in a single protein defines its nucleic acid binding site.
An algorithm was used to calculate a series of parameters summarising the characteristics of the RNA and ssDNA binding sites of the protein. This was a modified version of the algorithm used to calculate the same parameters for protein–dsDNA complexes (25). The parameters calculated for each binding site included the size, polarity, interface sequence segmentation, numbers of intermolecular hydrogen bonds, the gap volume between the protein and the nucleic acid chain, and the number of water molecules forming hydrogen bond bridges between the protein and the nucleic acid. The definitions of these parameters are given in the legend to Table 3. The means and standard deviations for these parameters are shown for the protein–RNA non-homologous dataset, the protein–ssDNA non-homologous dataset and, for comparison, a dataset of 26 non-homologous protein–dsDNA complexes taken from our previous analysis (25) (Table 3). The means and standard deviations of the same parameters have also been calculated for the three protein–RNA subsets viral proteins, proteins involved in protein synthesis and proteins involved in RNA modification (Table 4).
Table 3. Protein interface properties for datasets of protein–RNA, protein–ssDNA and protein–dsDNA complexes.
| |
Protein–RNA |
Protein–ssDNA |
Protein–dsDNA |
| Number of examples | 20 | 3 | 26 |
| ΔASA | 1128.9 (554.3) | 906.7 (106.4) | 1586.3 (499.4) |
| Segments | 8.5 (5.4) | 7.3 (0.6) | 7.31 (3.5) |
| Gap volume index | 3.3 (1.8) | 2.9 (1.3) | 2.6 (0.87) |
| Hydrogen bonds | 1.2 (0.5) | 0.6 (0.4) | 1.4 (0.4) |
| Bridging waters | 0.3 (0.4) | 0.1 (0.1) | 0.6 (0.6) |
| % Polarity | 45.7 (7.1) | 45.1 (4.2) | 48.1 (9.1) |
The data for the protein–dsDNA complexes is taken from our previous analysis (25).
The parameter definitions are as follows:
ΔASA: for the protein–DNA complexes this is the ASA of the protein that is buried on complex formation with the DNA. For the protein–protein complexes this is the ASA of one protomer that is buried on complex formation. For hetero-complexes the mean ASA buried by each protomer was calculated. The ASAs were calculated with Naccess (http://wolf.bms umist.ac.uk/naccess).
Segments: the number of sequence segments in the protein interface was defined such that interface residues separated by more than five residues in sequence were defined in different segments.
Gap volume index: the gap volume between protein and DNA, or two protein protomers was calculated using the algorithm SURFNET (http://www.biochem.ucl.ac.uk/~roman/surfnet/surfnet.html). The index is defined as Gap Index (Å) = gap volume between molecules (Å3)/interface ASA (Å2) per complex.
Hydrogen bonds: the number of intermolecular hydrogen bonds per 100Å2 ΔASA were calculated using HBPLUS (29), in which hydrogen bonds are defined according to standard geometric criteria.
Bridging waters: the number of water molecules that form hydrogen bonds with both parts of a complex were calculated using HBPLUS (29).
% Polarity: this is defined as [ΔASA(polar)/ΔASA(p)] × 100 where ΔASA(polar) is the ASA of polar atoms buried on complexation and ΔASA(p) is the ASA of protein buried on complexation with DNA.
Table 4. Protein interface properties for three functional subsets of protein–RNA complexes.
| |
Virus proteins |
Protein synthesis |
RNA modification |
| Number of examples | 8 | 7 | 5 |
| Families (see Table 1) | (A) 1–5 | (B) 6–11 | (C) 12–14 |
| ΔASA | 826.9 (321.9) | 1614.2 (577.3) | 932.7 (368.0) |
| Segments | 8.6 (6.7) | 11.0 (4.5) | 11.2 (6.1) |
| Gap volume index | 3.9 (2.0) | 3.2 (1.8) | 2.3 (1.1) |
| Hydrogen bonds | 1.3 (0.6) | 1.0 (0.2) | 1.2 (0.4) |
| Bridging waters | 0.2 (0.2) | 0.3 (0.3) | 0.4 (0.7) |
| % Polarity | 48.5 (9.7) | 45.2 (3.5) | 42.0 (4.8) |
See legend to Table 3 for definition of properties.
Residue interface propensities were calculated for the non-homologous dataset of protein–RNA complexes. These propensities give a measure of the relative importance of different amino acid residues in the RNA binding site of the protein. Residue interface propensities were calculated for each amino acid type (AAj) as the fraction of ASA that AAj contributed to the RNA binding site compared with the fraction of ASA contributed to the remainder of the surface of the protein (equation 1).
Interface residue propensity AAj =
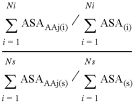 1
1
where ASAAAj(i) is the sum of the ASA (in the protein) of the amino acid residues of type j in the interface (the ASA of each type of residue is calculated without the RNA present); ASA(i) is the sum of the ASA in the protein of all amino acid residues of all types in the interface (the ASA of each type of residue is calculated without the RNA present); ASAAAj(s) is the sum of the ASA (in the protein) of the amino acid residues of type j on the protein surface (the surface being defined as those residues with >5% relative ASA in isolation); ASA(s) is the sum of the ASA in the protein of all amino acid residues of all types on the protein surface. Ni is the number of residues in the interface and Ns is the number of residues on the protein surface, excluding the interface residues.
A propensity of >1 indicates that a residue occurs more frequently in the interface than on the protein surface. Propensities for the protein–RNA dataset are shown compared with those of protein–dsDNA dataset (25) (Fig. 2). Propensities were not calculated for the protein–ssDNA as the dataset of non-homologous structures was too small.
Figure 2.

Histogram of the interface residue propensities calculated for the protein–RNA complexes and compared to a dataset of protein–dsDNA complexes (25). A propensity of more than one denotes that a residue occurs more frequently in the protein–nucleic acid interface than in the remainder of the protein surface. The amino acid residues on the x-axis are ordered according to the Fauchere and Pliska (39) hydrophobicity scale, moving from the most hydrophilic residues on the left-hand side to the most hydrophobic on the right.
An internet resource
The protein–RNA interface parameters calculated here can be calculated for any protein–RNA complex using the protein–nucleic acid server on the World Wide Web (http://www.biochem.ucl.ac.uk/bsm/DNA/server). This tool allows the user to upload the three-dimensional coordinates of any protein–nucleic acid complex and receive back a report of its interface parameters. This server provides a simple means of comparing new complexes with those already known.
Analysis of atom–atom contacts
The non-homologous datasets of protein–RNA and protein–ssDNA complexes each contain relatively few members (20 and 3, respectively) (Tables 1 and 2). Hence for the atom–atom contact analysis, all the structures were used to extract a dataset of non-homologous intermolecular contacts. This method also ensures that if a complex contains interactions that are unique within a family, these interactions were not lost.
Intermolecular hydrogen bonds and van der Waals contacts were calculated for each protein–nucleic acid complex using HBPLUS (29). This algorithm locates proximal donor (D) and acceptor (A) atom pairs and calculates theoretical hydrogen atom (H) positions that fit geometrical criteria. The criteria used to define a hydrogen bond were H–A distance <2.7 Å, D–A distance <3.35 Å, D–H–A angle >90°. van der Waals contacts were defined as all contacts between atoms not involved in hydrogen bonds that were <3.9 Å apart. The algorithm GROW (30) was used to extract all the intermolecular protein–nucleic acid contacts from each complex.
For each family in each dataset a structural alignment was generated using CORA (31). Then from each alignment a set of non-homologous contacts (hydrogen bonds and van der Waals interactions) were extracted from the total set of interactions, using a method designed by N.M.Luscombe and J.M.Thornton (manuscript in preparation). In this process, if more than two structures used the same atoms from the same residue to contact the same atoms in the same nucleic acid base or backbone, only the contact from the highest resolution structure was retained. Not every contact was included as this would mean that a specific type of contact would occur multiple times in the dataset just because it was present in proteins that are members of a large family. When a protein was the only member of a family all its protein–nucleic acid interactions were included. In addition, a second filter was used in the case of van der Waals contacts. If a residue was involved in an intermolecular hydrogen bond, all the contacts from the atoms in that single residue were excluded from the set of van der Waals contacts. However, when nucleic acid bases were involved in intermolecular hydrogen bonds, contacts from atoms within the bases were included in the set of van der Waals contacts.
These observed contact distributions cannot be used to detect possible preferential contacts without the calculation of expected contact distributions. Such expected values can be generated by assessing the availability of protein residues and nucleic acid groups to make potential contacts, by calculating the average solvent accessibility of such groups. All the solvent accessibilities were calculated using Naccess (http://wolf.bms.umist.ac.uk/naccess). For the expected distribution of hydrogen bond contacts the average contribution to the accessible surface area made by polar atoms was calculated for each of the 20 amino acids from a dataset of 119 non-homologous monomeric proteins as used by N.M.Luscombe and J.M.Thornton (manuscript in preparation). The average accessible surface area contribution made by polar atoms in the bases and backbone components of the nucleic acids were also calculated from RNA molecules in the non-homologous dataset of protein–RNA complexes, and from the ssDNA molecules in the complete dataset of protein–ssDNA complexes. For the expected distributions of the van der Waals contacts average solvent accessibilities were calculated using all the atoms in the dataset of proteins, and in the bases and backbone of the nucleic acids in the RNA and ssDNA molecules, as before. All these values are given as additional material at http://www.biochem.ucl.ac.uk/bsm/RNA. Using the percentage ASA contributions for the protein residues and the nucleic acid components, expected contact distributions were calculated.
The numbers of observed and expected contacts made by each type of base and each type of amino acid residue in the protein–RNA complexes are shown in Table 5. The contacts for the protein–ssDNA complexes and for a dataset of 131 protein–DNA complexes (N.M.Luscombe and J.M.Thornton, manuscript in preparation) are included as additional material at http://www.biochem.ucl.ac.uk/bsm/RNA.
Table 5. Observed frequency distributions of (A) hydrogen bond contacts and (B) van der Waals contacts between the 20 amino acid residues and the components of RNA.
| |
|
A |
G |
U |
C |
Sugar |
Phosphate |
Total |
| A | ALA | 1 (0.6) | 1 (0.6) | 0 (0.5) | 0 (0.5) | 0 (2.2) | 0 (3.7) | 2 (8.1) |
| ARG | 2 (0.6) | 3 (0.6) | 4 (0.5) | 2 (0.5) | 2 (2.2) | 23 (3.7) | 36 (8.1) | |
| ASN | 1 (0.9) | 7 (0.8) | 6 (0.7) | 3 (0.8) | 2 (3.1) | 3 (5.2) | 22 (11.5) | |
| ASP | 0 (1.1) | 1 (1.0) | 2 (0.8) | 2 (1.0) | 2 (3.9) | 1 (6.5) | 8 (14.4) | |
| CYS | 0 (0.1) | 0 (0.1) | 0 (0.1) | 0 (0.1) | 0 (0.3) | 0 (0.5) | 0 (1.1) | |
| GLN | 0 (0.5) | 0 (0.5) | 1 (0.5) | 2 (0.5) | 1 (2.0) | 3 (3.3) | 7 (7.3) | |
| GLU | 0 (0.9) | 5 (0.8) | 1 (0.8) | 0 (0.8) | 0 (3.4) | 0 (5.6) | 6 (12.4) | |
| GLY | 0 (0.8) | 4 (0.7) | 0 (0.7) | 2 (0.7) | 0 (2.8) | 1 (4.7) | 7 (10.3) | |
| HIS | 0 (0.2) | 0 (0.2) | 0 (0.2) | 1 (0.2) | 2 (0.8) | 0 (1.3) | 3 (3.0) | |
| ILE | 0 (0.2) | 0 (0.2) | 0 (0.2) | 0 (0.2) | 1 (0.7) | 0 (1.2) | 1 (2.6) | |
| LEU | 0 (0.4) | 0 (0.3) | 0 (0.3) | 1 (0.3) | 0 (1.4) | 1 (2.3) | 2 (5.0) | |
| LYS | 0 (1.1) | 2 (1.0) | 2 (0.9) | 1 (1.0) | 1 (3.9) | 3 (6.4) | 9 (14.1) | |
| MET | 0 (0.1) | 0 (0.1) | 0 (0.1) | 0 (0.1) | 1 (0.4) | 0 (0.7) | 1 (1.5) | |
| PHE | 0 (0.2) | 1 (0.2) | 0 (0.1) | 0 (0.2) | 0 (0.6) | 0 (1.1) | 1 (2.3) | |
| PRO | 0 (0.9) | 1 (0.8) | 0 (0.7) | 0 (0.8) | 0 (3.1) | 0 (5.2) | 1 (11.4) | |
| SER | 3 (0.7) | 0 (0.6) | 0 (0.6) | 1 (0.6) | 0 (2.6) | 3 (4.2) | 7 (9.3) | |
| THR | 4 (0.6) | 0 (0.5) | 0 (0.5) | 0 (0.5) | 4 (2.2) | 5 (3.6) | 13 (8.0) | |
| TRP | 0 (0.1) | 1 (0.1) | 0 (0.1) | 0 (0.1) | 0 (0.4) | 0 (0.6) | 1 (1.4) | |
| TYR | 0 (0.3) | 1 (0.3) | 0 (0.3) | 1 (0.3) | 6 (1.1) | 4 (1.9) | 12 (4.2) | |
| VAL | 0 (0.3) | 0 (0.3) | 0 (0.3) | 0 (0.3) | 1 (1.1) | 0 (1.8) | 1 (4.0) | |
| Total | 11 (10.5) | 27 (9.6) | 16 (8.8) | 16 (9.4) | 23 (38.3) | 47 (63.3) | 140 (140) | |
| B | ALA | 13 (4.9) | 7 (4.9) | 1 (4.1) | 3 (4.0) | 12 (23.3) | 1 (37.7) | 37 (79.0) |
| ARG | 32 (8.6) | 19 (8.5) | 24 (7.1) | 13 (6.9) | 44 (40.5) | 93 (65.5) | 225 (137.1) | |
| ASN | 5 (6.6) | 25 (6.6) | 58 (5.5) | 10 (5.3) | 16 (31.2) | 31 (50.4) | 145 (105.6) | |
| ASP | 2 (8.5) | 6 (8.5) | 6 (7.1) | 15 (6.9) | 11 (40.3) | 4 (65.1) | 44 (136.3) | |
| CYS | 3 (0.5) | 9 (0.5) | 0 (0.4) | 0 (0.4) | 0 (2.5) | 0 (4.0) | 12 (8.3) | |
| GLN | 3 (5.9) | 32 (5.8) | 9 (4.9) | 7 (4.8) | 33 (27.8) | 16 (44.9) | 100 (94.0) | |
| GLU | 0 (10.0) | 16 (9.9) | 23 (8.3) | 1 (8.1) | 20 (47.1) | 1 (76.1) | 61 (159.4) | |
| GLY | 0 (4.8) | 31 (4.7) | 2 (3.9) | 12 (3.8) | 9 (22.5) | 13 (36.3) | 67 (76.1) | |
| HIS | 1 (2.7) | 0 (2.7) | 0 (2.2) | 12 (2.2) | 2 (12.7) | 0 (20.6) | 15 (43.1) | |
| ILE | 0 (2.4) | 23 (2.4) | 3 (2.0) | 2 (1.9) | 16 (11.4) | 3 (18.4) | 47 (38.5) | |
| LEU | 16 (4.4) | 11 (4.4) | 2 (3.7) | 7 (3.6) | 16 (21.0) | 8 (34.0) | 60 (71.2) | |
| LYS | 3 (13.1) | 14 (13.0) | 7 (10.9) | 11 (10.6) | 19 (62.0) | 38 (100.2) | 92 (209.9) | |
| MET | 2 (1.4) | 11 (1.4) | 0 (1.2) | 3 (1.2) | 20 (6.7) | 6 (10.9) | 42 (22.8) | |
| PHE | 60 (2.3) | 18 (2.2) | 46 (1.9) | 1 (1.8) | 37 (10.7) | 3 (17.3) | 165 (36.2) | |
| PRO | 6 (5.9) | 4 (5.8) | 2 (4.9) | 10 (4.7) | 11 (27.7) | 1 (44.8) | 34 (93.8) | |
| SER | 26 (5.7) | 11 (5.7) | 0 (4.8) | 11 (4.6) | 13 (27.1) | 24 (43.8) | 85 (91.7) | |
| THR | 28 (6.0) | 3 (6.0) | 7 (5.0) | 5 (4.9) | 21 (28.3) | 37 (45.8) | 101 (96.0) | |
| TRP | 0 (1.4) | 52 (1.4) | 0 (1.1) | 0 (1.1) | 0 (6.5) | 0 (10.4) | 52 (21.9) | |
| TYR | 23 (3.5) | 28 (3.5) | 33 (2.9) | 49 (2.9) | 48 (16.7) | 24 (27.0) | 205 (56.5) | |
| VAL | 4 (3.4) | 5 (3.4) | 10 (2.8) | 0 (2.8) | 19 (16.2) | 5 (26.1) | 43 (54.7) | |
| Total | 227 (102.0) | 325 (101.3) | 233 (84.7) | 172 (82.6) | 367 (482.1) | 308 (779.3) | 1632 (1632) |
The numbers in parentheses are the expected values derived by assessing the solvent accessibility of each group.
The pairs shown in bold are those in which the observed value is five times the expected value (individual table entry) or two times the expected value (row or column total entry). These values have been used to create a composite table (Table 7).
In addition to this general survey, the contacts made by the 2′-hydroxyl group in the ribose of RNA molecules were also considered. The contacts made by the oxygen atom in this group (that is not present in the deoxyribose of DNA) were extracted from the non-homologous set of contacts, obtained as described above. The average solvent accessibility of this oxygen was also calculated from the RNA molecules in an uncomplexed state using Naccess (http://wolf.bms.umist.ac.uk/naccess.html).
RESULTS
The classifications used in Table 1 emphasise the diverse nature of RNA recognition by proteins. Each class of recognition site (groove binding and β-sheet binding) is observed with more than one type of RNA structure (single-stranded, single stranded with single loop, single-stranded with multiple loops, double-stranded). The scene is further complicated by the proteins binding tRNAs, as these commonly have a domain exhibiting groove binding and another exhibiting β-sheet binding. The diversity of interactions is also evident when considering the functional groupings of the proteins. Although the proteins involved in protein synthesis all bind RNAs with single strands folded into multiple loops, both the viral proteins and the RNA modification proteins bind a number of different RNA structures, using both the groove and β-sheet modes of binding.
Nucleic acid binding site properties
The interface properties for protein–RNA, protein–ssDNA and protein–dsDNA are summarised in Table 3. Before a detailed comparative analysis is made it should be highlighted that the protein–ssDNA dataset comprises only three structures and hence the results shown may not be representative of such complexes in general.
The RNA binding sites ranged in size from 370 to 2422 Å2, comprised between 3 and 24 sequence segments and included between 5 and 26 intermolecular hydrogen bonds (0.4–2.0 hydrogen bonds per 100 Å2 of interface ASA). The binding sites comprised between 32 and 60% polar atoms. These large variations for many properties further emphasise the diverse nature of the binding sites.
In general the protein sites that bind RNA are slightly smaller than the dsDNA binding sites but have more sequence segments. The RNA binding sites also appear to be less polar than the dsDNA binding sites and less well packed. They show a similar number of intermolecular hydrogen bonds but on average only half the number of bridging water molecules. Although this last result is probably reflective of the lower resolution of the structures in the protein–RNA dataset (mean resolution, excluding two NMR structures, is 2.6 Å) compared to the protein–dsDNA dataset (mean resolution 2.4 Å).
The poorer packing of the protein–RNA complexes (as indicated by the larger gap volume index in Table 3) may result from the complex secondary structures that the RNA molecules often form. Many of the interactions with protein occur at features such as bulges or stem–loops (16), where the second unpaired RNA sequence may restrict the very close approach of the protein. In enzymatic proteins that bind DNA, it was observed that they used an enveloping mode of binding, using a large interaction site to surround the DNA double helix (25). Such a mode would not be likely in protein–RNA structures when the RNA forms a complex tertiary structure.
The ssDNA binding sites are the smallest of the three types of complex and show considerably fewer intermolecular hydrogen bonds than either the protein–RNA or the protein–dsDNA complexes (Table 3). This could indicate non-specific binding of the DNA. However, they are better packed than the RNA complexes, as with only one strand of bases the protein can make a close approach without being restricted by the presence of a second strand of bases. In this light it is perhaps surprising that these structures are not better packed than the dsDNA complexes.
The comparison between the three functional subsets of the protein–RNA complexes reveals some interesting differences (Table 4). Those proteins involved in protein synthesis (principally the tRNA amino synthetases) have RNA binding sites more than 1.5 times the size of the RNA modification complexes and twice the size of the viral complexes. These synthetase structures have large binding sites as they comprise at least two structural domains, one that interacts with the acceptor stem and one with the anticodon arm of the RNA (Fig. 1). These effectively form two separate RNA recognition sites. The viral proteins have the most polar and least well packed RNA binding sites. However, it should be considered that these complexes only include a small part of the RNA actually encapsulated in the viral structure. For example, in the case of the coat protein from BMV (PDBcode 1BMV) only 20% of the packaged RNA is ordered and visible in the structure of the complex (32). Hence the full structures of the protein–vRNA complexes may reveal further interaction sites on the coat proteins with many weak interactions combining to form stable multi-site complexes.
Dividing the protein–RNA complexes into three sets (viral proteins, proteins involved in protein synthesis and proteins involved in RNA modification) effectively divides the complexes into those with RNA interactions that are (i) not sequence specific (excluding the MS2 coat protein complex), (ii) partially sequence specific and (iii) highly sequence specific. Hence the sequence-specific complexes appear to achieve their specificity through tight packing and relatively non-polar interfaces. It is surprising that these specific interactions do not feature more hydrogen bonds. RNA modification proteins have the least polar interaction sites but achieve the best packing with the RNA.
The residue interface propensities for the RNA binding sites are compared with those observed for dsDNA binding sites (25) (Fig. 2). For the protein–RNA complexes the highest propensities were observed for lysine, tyrosine, phenylalanine, isoleucine and arginine (in order of decreasing propensity). Hence, aromatic and positively charged amino acids play important roles. It is likely that the aromatics stack adjacent to the unpaired bases in the RNA molecules. The single aromatic amino acids also play key roles in protein–protein interfaces (28,33). In the protein–dsDNA complexes the highest propensities were observed for threonine, arginine, serine, asparagine and glycine (in order of decreasing propensity). The charged and polar residues play important roles in these complexes as they complement the negative charge on the DNA (25). The absence of aromatics reflects the helical dsDNA structure in which the faces of the bases are buried and not accessible for binding interactions.
Atom–atom contacts
In all three types of complex (protein–RNA, protein–ssDNA and protein–dsDNA) van der Waals contacts are significantly more common than hydrogen bond contacts. The van der Waals contacts represent 76.3, 92.6 and 92.2% of the total interactions in the protein–dsDNA, protein–ssDNA and protein–RNA complexes, respectively.
In the protein–RNA and protein–ssDNA complexes ~58% of the contacts made by the protein are to the bases of the nucleic acid, with the remainder made to the backbone. In the protein–dsDNA complexes the opposite trend is observed, with only 24% of contacts made to bases and the remainder to the backbone. This was to be expected, as in the former structures many of the nucleic acids are unpaired and are available to make both hydrogen bond and van der Waals contacts with protein residues. In the dsDNA complexes, the nucleic acids are tightly paired in the regular B-DNA structures and hence the bases are not easily accessible to interacting proteins, and many interactions occur through the backbone.
The observed distributions of hydrogen bond and van der Waals contacts made between protein residues and RNA components are shown with expected distributions in Table 5. These data have been used to create a composite table (Table 6). If a row or column total in Table 5 was twice the number of the expected value, the base and residue preferences were included in Table 6 (items c and d). Similarly, if an individual table entry was five times the expected value the contact preference was included in Table 6 (item e). The same criteria were used to extract the atom–atom contact data from the protein–ssDNA and protein–dsDNA included as supplementary material at http://www.biochem.ucl.ac.uk/bsm/RNA. The composite data in Table 6 do show some apparent preferences for specific bases, residues and nucleic residue contacts.
Table 6. Summary of the contact preferences shown by protein–RNA, protein–ssDNA and protein–dsDNA complexes.
| Preferences | Binding sites | ||
| |
RNA |
ssDNA |
dsDNA |
| Hydrogen bonds | |||
| % Contributiona | 8 | 7 | 24 |
| Backbone/basesb | Bases | – | Bases |
| Basesc | G, U | – | G |
| Residuesd | Arg, Tyr | Met, Phe, Trp | Arg |
| NA-residuee | Arg-U, Arg-Phosp, Asn-G, Asn-U, Glu-G, Gly-G, Thr-A, Tyr-Sugar | His-A, Phe-A, Phe-T, Trp-A, Trp-C, Met-T Leu-Sugar, Met-Phosp | Arg-T, Arg-G, Arg-Phosp, His-G |
| van der Waals | |||
| % Contributiona | 92 | 93 | 76 |
| Backbone/basesb | Bases | Bases | – |
| Basesc | G, U | – | – |
| Residuesd | Phe, Tyr | Cys, Ser, Met, Phe, Trp | Arg |
| NA-residuee | Asn-U, Ile-G, Phe-A, Phe-G, Phe-U, Tyr-A,Tyr-U, Tyr-C, Tyr-G, Trp-G, Met-G, Cys-G | Asp-G, Asp-C, Met-T, Phe-A, Phe-T, Pro-T, Trp-A, Cys-T, Cys-Phosp | – |
aPercentage contribution of contacts.
bPreference shown for base contacts or backbone contacts.
cBase preference.
dAmino acid residue preference.
eSpecific amino acid–nucleic acid group contact preference.
In protein–RNA interactions, the van der Waals contacts far outnumber the hydrogen bonding in contacts. The proteins in these complexes show a preference to contact the purine guanine and the pyrimidine uracil, using both van der Waals contacts and hydrogen bonds. The proteins show a preference for the residues arginine, tyrosine and phenylalanine to be present in the RNA binding site.
A preference for hydrogen bonding contacts to guanine was also observed in the protein–dsDNA complexes, as was the preference for the residue arginine to be in the binding site. In the protein–ssDNA complexes, no preference was observed for contacts to any base, but a preference was observed for the residues methionine, phenylalanine and tryptophan, cysteine and serine to be present in the DNA binding sites. In both types of protein–DNA complex, van der Waals contacts were far more prevalent than hydrogen bonding contacts, as observed in the protein–RNA complexes. However, there were far more hydrogen bonding contacts observed in the protein–dsDNA complexes, than in either the protein–RNA or protein–ssDNA complexes (24%, compared to 8 and 7%, respectively).
The ratio of the number of observed contacts made to the nucleic acid bases and backbone are shown in Table 7. This shows that in the RNA complexes, hydrogen bond contacts to the bases and the backbone are present in equal numbers, as observed in the protein–ssDNA complexes. This is in contrast to the protein–dsDNA complexes in which there are half the numbers of hydrogen bonds made to the bases compared to the backbone. This is most likely as a result of the high numbers of unpaired bases in the RNA structures (and in the ssDNA). In both the protein–RNA and the protein–ssDNA complexes there are more than 1.5 times the number of contacts made to the bases compared to the backbone. In contrast, the protein–dsDNA complexes show only a third of the van der Waals contacts are made to the bases.
Table 7. The ratios of intermolecular hydrogen bond and van der Waals contacts made between the protein and the base/backbone component of the nucleic acid for the three datasets of protein–nucleic acid complexes: protein–RNA (complete dataset), protein–dsDNA and protein–ssDNA.
| Contact (base/backbone ratio) | RNA (all) | dsDNA | ssDNA | RNA | ||
| |
|
|
|
Virala |
Synthesisb |
Modificationsc |
| Hydrogen bonds | 1.0 | 0.5 | 0.9 | 1.2 | 0.6 | 2.4 |
| van der Waals contacts | 1.4 | 0.3 | 1.4 | 1.9 | 0.9 | 3.2 |
Ratios are also shown for the three functional subsets of the protein–RNA complexes: viral proteinsa, protein synthesis proteinsb and RNA modification proteinsc.
Of the 23 hydrogen bonds made between protein residues and the ribose sugar of the RNA, all were made by the oxygen atom of the 2′-hydroxyl group. Of the 308 van der Waals contacts made between the protein and the sugar, 105 (34%) were made by the oxygen atom of the 2′-hydroxyl group. Of the 21 hydrogen bonds between protein and dsDNA all were made by the O4 atom in the pentose sugar ring, whilst 285 (27%) of the van der Waals contacts are made by the C5 carbon in the ribose ring. The oxygen atoms in the 2′-hydroxyl groups in the RNA molecules are highly solvent exposed (mean ASA is 22.2 Å2 in the current dataset) compared with the other oxygens in the sugar (O3*, O4*, O5* have mean ASAs of 7.36, 3.44 and 1.4 Å2, respectively). The 2′-hydroxyl group can be both a hydrogen bond donor and an acceptor and hence can potentially interact with many amino acids of the protein. The protruding nature of the 2′-hydroxyl groups has already been observed in a number of structures including MS2 coat protein and the tRNA synthetases. It has been observed that in such structures there are key ribose groups that, when substituted for deoxyribose, greatly reduce the affinity for the RNA to bind the protein (34).
DISCUSSION
The current analysis presents a similar picture to that observed in DNA binding proteins, in that there is not a single archetypal RNA binding site. In the current dataset, the largest analysed in this way, there are 32 proteins, representing 14 structural families. When the predominant secondary structure element of each binding site was analysed, the sites were equally divided between α-helix and β-strand, with only one example of an αβ interface. The RNAs bound include elongated single-stranded, looped single-stranded, single-stranded with multiple loops and double-helix structures. The size and polarity of the RNA binding sites vary widely, as do the modes of recognition used by the protein and the RNA structures recognised. Thus, the picture presented is far more complicated than that of protein–DNA complexes (25).
Similar modes of secondary structure contacts are observed in proteins binding RNA to those that bind DNA (24). However, when looking more closely at amino acid preferences and base versus backbone contacts, similarities are much harder to find. The unpaired state of many of the bases in RNA structures means that they are more readily available to make contacts with amino acids residues than those in the tightly paired double helices of dsDNA. Hydrogen bond contacts to all parts of the RNA are far less common than in the protein–dsDNA complexes. The ratios of contacts made to the nucleic acid bases and the backbone (Table 7) show the differences between protein–RNA and protein–dsDNA complexes, and the similarities between the contacts made to RNA and ssDNA.
However, some trends do emerge from the contact data. It is evident that van der Waals interactions are more numerous in protein–RNA complexes than hydrogen bonds. A preference for proteins to make contacts with guanine was observed, and arginine, asparagine, phenylalanine, threonine and tyrosine occur in RNA binding sites more often than expected.
One of the features of the current work is the comparison of the observations made for protein–RNA complexes with those for protein–ssDNA and protein–dsDNA complexes. In terms of size, the protein–RNA complexes are intermediate between the two types of protein–DNA complexes, but they are the least well packed of all three types of complex. The poor packing of the protein–RNA complexes is a result of the complex tertiary structure that the RNA chains form. The atom contact analysis showed that the purine base guanine is preferentially contacted by proteins in both RNA and dsDNA structures.
One issue that has not been addressed here is conformational changes on binding. With the recent availability of additional protein–RNA complexes from the ribosome (12–15) it has become evident that almost every complex involves conformational changes in the protein, the RNA or both (35). For the protein it is frequently a case of a transition from an unstructured to a structured state of some part of the binding interface. For example, the structure of the L11 protein has two extended loops that are disordered in the absence of RNA but are defined structures in the complex (36).
Despite the recent addition of ribosomal subunit structures to the PDB and NDB (12–15) there are still a relatively small number of characterised protein–RNA complex structures. Purification and crystallisation difficulties has meant that their presence in the databases lags behind those of the protein–DNA complexes. Many higher resolution structures, like those from the ribosome, are required before firmer conclusions can be drawn about the most common modes of interaction.
By looking for physical and structural features that characterise RNA binding sites on proteins, it may be possible to predict the location of such sites on proteins for which complexes have not yet been solved. This has successfully been achieved for protein–protein binding sites (37,38) using combinations of interface properties, including interface propensities. Knowing the characteristics of RNA binding sites may also be helpful in designing novel RNA binding proteins.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to acknowledge the support of all those involved in the Nucleic Acid Databank (NDB), and thank Gabriele Varani for helpful discussions. This work was carried out with funding from the Department of Energy (USA) (grant number DE-FG02096ER62166.A000). This is a publication from the BBSRC Bloomsbury Centre for Structural Biology.
References
- 1.Moore P.B. (1998) The three-dimensional structure of the ribosome and its components. Annu. Rev. Biophys. Biomol. Struct., 27, 35–58. [DOI] [PubMed] [Google Scholar]
- 2.Ramakrishnan V. and White,S.W. (1998) Ribosomal protein structures: insights into the architecture, machinery and evolution of the ribosome. Trends Biochem. Sci., 23, 208–212. [DOI] [PubMed] [Google Scholar]
- 3.Luhrmann R., Kastner,B. and Bach,M. (1990) Structure of spliceosomal snRNP’s and their role in pre-mRNA splicing. Biochim. Biophys. Acta, 1087, 265–292. [DOI] [PubMed] [Google Scholar]
- 4.Tarasow T.M. and Eaton,B.E. (1998) Dressed for success: realising the catalytic potential of RNA. Biopolymers ,48, 29–37. [Google Scholar]
- 5.Scott W.G. and Klug,A. (1996) Ribozymes: structures and mechanism in RNA catalysis. Trends Biochem. Sci., 21, 220–224. [PubMed] [Google Scholar]
- 6.Scott W.G. (1998) RNA catalysis. Curr. Opin. Struct. Biol., 8, 720–726. [DOI] [PubMed] [Google Scholar]
- 7.Moras D. (1992) Aminoacyl-tRNA synthetases. Curr. Opin. Struct. Biol., 2, 138–142. [Google Scholar]
- 8.Varani G. and Nagai,K. (1998) RNA recognition by RNP proteins during RNA processing. Annu. Rev. Biophys. Biomol. Struct., 27, 407–445. [DOI] [PubMed] [Google Scholar]
- 9.Steitz T.A. (1990) Structural studies of protein nucleic-acid interaction: the sources of sequence specific binding. Q. Rev. Biophys., 23, 205–210. [DOI] [PubMed] [Google Scholar]
- 10.Harrison S.C. (1991) A structural taxonomy of DNA-binding domains. Nature, 353, 715–719. [DOI] [PubMed] [Google Scholar]
- 11.Luscombe N.M., Austin,S.E., Berman,H.M. and Thornton,J.M. (2000) An overview of the structures of protein–DNA complexes. Genome Biol., 1, 1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wimberly B.T., Brodersen,D.E., Clemons,W.M., Morgan-Warren,R.J., Carter,A.P., Vonrhein,C., Hartsch,T. and Ramakrishnan,V. (2000) Structure of the 30S ribosomal subunit. Nature, 407, 327–339. [DOI] [PubMed] [Google Scholar]
- 13.Agalarov S.C., Prasad,G.S., Funke,P.M., Stout,C.D. and Williamson,J.R. (2000) Structure of the S15,S18-rRNA complex: assembly of the 30S ribosome central domain. Science, 288, 107–112. [DOI] [PubMed] [Google Scholar]
- 14.Schluenzen F., Tocilj,A., Zarivach,R., Harms,J., Gluehmann,M., Janell,D., Bashan,A., Bartels,H., Agmon,I., Franceschi,F. and Yonath,A. (2000) Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell, 102, 615–623. [DOI] [PubMed] [Google Scholar]
- 15.Berman H.M., Olson,W.K., Beveridge,D.L., Westbrook,J., Gelbin,A., Demeny,T., Hsieh,S.H., Srinivasan,A.R. and Schneider,B. (1992) The nucleic-acid database: a comprehensive relational database of 3-dimensional structures of nucleic acids. Biophys. J., 63, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai K. (1996) RNA–protein complexes. Curr. Opin. Struct. Biol., 6, 53–61. [DOI] [PubMed] [Google Scholar]
- 17.Steitz T.A. (1999) RNA recognition by proteins. In Gesteland,R.F., Cech,T.R. and Atkins,J.F. (eds), The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 427–450.
- 18.Mattaj I.W. (1993) RNA recognition: a family matter? Cell, 73, 837–840. [DOI] [PubMed] [Google Scholar]
- 19.Nagai K. (1992) RNA–protein interactions. Curr. Opin. Struct. Biol., 2, 131–137. [Google Scholar]
- 20.Guzman R.N., Turner,R.B. and Summers,M.F. (1998) Protein–RNA recognition. Biopolymers, 48, 181–195. [DOI] [PubMed] [Google Scholar]
- 21.Draper D.E. (1995) Protein–RNA recognition. Annu. Rev. Biochem., 64, 593–620. [DOI] [PubMed] [Google Scholar]
- 22.Cusack S. (1999) RNA–protein complexes. Curr. Opin. Struct. Biol., 9, 66–73. [DOI] [PubMed] [Google Scholar]
- 23.Arnez J.G. and Moras,D. (1997) Structural and functional considerations of the aminoacylation reaction. Trends Biochem. Sci., 22, 211–216. [DOI] [PubMed] [Google Scholar]
- 24.Draper D.E. (1999) Themes in RNA–protein recognition. J. Mol. Biol., 293, 255–270. [DOI] [PubMed] [Google Scholar]
- 25.Jones S., van Heyningen,P., Berman,H.M. and Thornton,J.M. (1999) Protein–DNA interactions: a structural analysis. J. Mol. Biol., 287, 877–896. [DOI] [PubMed] [Google Scholar]
- 26.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor W.R. and Orengo,C.A. (1989) Protein structure alignment. J. Mol. Biol., 208, 1–22. [DOI] [PubMed] [Google Scholar]
- 28.Jones S. and Thornton,J.M. (1996) Principles of protein–protein interactions. Proc. Natl Acad. Sci. USA, 93, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald I.K. and Thornton,J.M. (1994) Satisfying hydrogen-bonding potential in proteins. J. Mol. Biol., 238, 777–793. [DOI] [PubMed] [Google Scholar]
- 30.Milburn D., Laskowski,R.A. and Thornton,J.M. (1998) Sequences annotated by structure: a tool to facilitate the use of structural information in sequence analysis. Protein Eng., 11, 855–859. [DOI] [PubMed] [Google Scholar]
- 31.Orengo C.A. (1999) CORA—Topological fingerprints for protein structural families. Protein Sci., 8, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z.G., Stauffacher,C., Li,Y.G., Schmidt,T., Bomu,W., Kamer,G., Shanks,M., Lomonossoff,G. and Johnson,J.E. (1989) Protein–RNA interactions in an icosohedral virus at 3.0 angstroms resolution. Science, 245, 154–159. [DOI] [PubMed] [Google Scholar]
- 33.Argos P. (1988) An investigation of protein subunit and domain interfaces. Protein Eng., 2, 101–113. [DOI] [PubMed] [Google Scholar]
- 34.Talbot S.J., Goodman,S., Bates,S.R.E., Fishwick,C.W.G. and Stockley,P.G. (1990) Use of synthetic oligonucleotides to probe RNA–protein interactions in the MS2 translational operator complex. Nucleic Acids Res., 18, 3521–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williamson J.R. (2000) Induced fit in RNA–protein recognition. Nature Struct. Biol., 7, 834–837. [DOI] [PubMed] [Google Scholar]
- 36.Wimberly B.T., Guymon,R., McCutcheon,J.P., White,S.W. and Ramakrishnan,V. (1999) A detailed view of a ribosomal active site: the structure of the L11–RNA complex. Cell, 97, 491–502. [DOI] [PubMed] [Google Scholar]
- 37.Jones S. and Thornton,J.M. (1997) Analysis of protein–protein interaction sites using surface patches. J. Mol. Biol., 272, 121–132. [DOI] [PubMed] [Google Scholar]
- 38.Jones,S. and Thornton,J.M. (1997) Prediction of protein–protein interaction sites using patch analysis. J. Mol. Biol., 272, 133–143. [DOI] [PubMed] [Google Scholar]
- 39.Fauchere,J. and Pliska,V. (1983) Hydrophobic parameters of amino acid side chains from the partitioning of N-acetyl amino acid amides. Eur. J. Med. Chem., 18, 369–375. [Google Scholar]