


Abstract
We describe a fluorescence-based directed termination PCR (fluorescent DT–PCR) that allows accurate determination of actual sequence changes without dideoxy DNA sequencing. This is achieved using near infrared dye-labeled primers and performing two PCR reactions under low and unbalanced dNTP concentrations. Visualization of resulting termination fragments is accomplished with a dual dye Li-cor DNA sequencer. As each DT–PCR reaction generates two sets of terminating fragments, a pair of complementary reactions with limiting dATP and dCTP collectively provide information on the entire sequence of a target DNA, allowing an accurate determination of any base change. Blind analysis of 78 mutants of the supF reporter gene using fluorescent DT–PCR not only correctly determined the nature and position of all types of substitution mutations in the supF gene, but also allowed rapid scanning of the signature sequences among identical mutations. The method provides simplicity in the generation of terminating fragments and 100% accuracy in mutation characterization. Fluorescent DT–PCR was successfully used to generate a UV-induced spectrum of mutations in the supF gene following replication on a single plate of human DNA repair-deficient cells. We anticipate that the automated DT–PCR method will serve as a cost-effective alternative to dideoxy sequencing in studies involving large-scale analysis for nucleotide sequence changes.
INTRODUCTION
High throughput mutation analysis requires two sequential processes, a rapid scanning step to select for molecules with a unique genotype, followed by determination of nucleotide sequence changes in selected molecules. Although a variety of techniques have been advanced to facilitate mutation screening (1), little progress has been made in developing alternative approaches to dideoxy DNA sequencing for mutation characterization. A cost-effective approach for direct mutation identification has become a major challenge in the analysis of mutational spectra using various reporter genes that depend on their phenotypical selection for rare mutations. These marker genes are very important genetic tools in mechanistic studies of chemical mutagenesis and functional analysis of proteins involved in DNA replication and repair. We previously reported a PCR-based method that allows generation of nested termination fragments by integrating both selective DNA amplification and directed chain termination into a single PCR reaction (DT–PCR) (2,3). This was achieved simply by carrying out a PCR reaction with one nucleotide provided at a concentration 5–10 times lower than the other three. This method was successfully applied to rapid mutation screening by coupling it to a SSCP procedure (DT–SSCP) (4,5). DT–PCR also has the potential to be exploited for direct mutation characterization due to its sequencing nature. A limiting dATP reaction provides sequence information about adenine and thymine bases, while a limiting dCTP reaction provides information concerning cytosine and guanine bases. These two complementary reactions provide all the information needed for complete mutation identification. There are two major requirements for retrieval of the sequence information in a practical fashion. Firstly, a labeling system is required to allow separate visualization of two sets of termination fragments so that two complementary reactions will suffice for mutation identification. Secondly, a sensitive system is required to detect both relatively weak signals from the termination fragments generated in the transient linear amplification phase and to generate an evenly separated banding pattern for accurate determination of subtle banding changes.
In this study we describe a fluorescence-based procedure that allows accurate determination of actual base sequence changes without dideoxy DNA sequencing. A comparative study was carried out to test the efficiency of this procedure in characterizing base substitutions in a shuttle vector plasmid that carries the supF gene as a marker and a unique sequence tag (6,7). The method provides simplicity in the generation of terminating fragments and 100% accuracy in mutation identification in the supF gene.
MATERIALS AND METHODS
UV-induced mutations in the supF gene
Plasmid pSP189, which contains the supF gene as a mutational target and an 8 bp signature sequence (7), was irradiated in vitro with 200 J/m2 UVC (254 nm) and transfected into the XP-A cell line XP12BE using lipofectamine. After plasmid rescue by Hirt extraction (8), the DNA was cleaved with DpnI to remove unreplicated plasmid. Replicated plasmid was electroporated into MB7070 bacteria, which carry a lacZ gene with an amber mutation. Plasmids were isolated from white colonies using the Wizard Miniprep kit (Promega, Madison, WI).
Fluorescent DT–PCR
To determine mutations in the supF gene a 203 bp fragment of pSP189 was amplified with primers PS189NT800 (5′-GGCGACACGGAAATGTTGAA) and PS189TR700 (5′-GCTAAGGATCCGGGTACCG). To verify the independence of identical mutations, a 317 bp fragment containing both the supF gene and the signature sequence was analyzed with primers PS189NT800 and PS189JR700 (5′-TCAGGGGGGCGGAGCCTAT). Fluorescent primers were synthesized by Li-cor (Lincoln, NE). Primer PS189NT800 was 5′-end-labeled with IRD-800, primers PS189TR700 and PS189JR700 with IRD-700. For directed termination at dATP sites the 203 bp fragment was amplified using 1.5 pmol of each end-labeled primer in the presence of 10 µM each dCTP, dTTP and dGTP and 1 µM dATP (10:1 ratio) (2 µM dATP was used for the 317 bp fragment). The remaining cocktail consisted of 1× PCR buffer (50 mM KCl, 10 mM Tris–HCl pH 8.3 and 0.001% w/v gelatin), 1.5 mM MgCl2, 2 U AmpliTaq DNA polymerase (Perkin Elmer, Branchburg, NJ) and 10 ng plasmid DNA in a 20 µl reaction. DT–PCR was performed in a PTC-100 Programmable Thermal Controller (MJ Research, Watertown, MA) under the following amplification conditions: 95°C for 1 min for initial denaturation, followed by 35 cycles of 1 min at 94°C for denaturation, 1 min at 55°C for annealing and 1 min at 72°C for extension. The extension time for the final cycle was lengthened to 5 min.
For directed termination at dCTP sites the DT–PCR reactions were performed under the same conditions as above except that 1–2 µM dCTP was provided in the presence of 10 µM each dATP, dTTP and dGTP. Fluorescent DT–PCR products could be stored for several months at –20°C without signal reduction. Yellow light illumination was used to prevent photodegradation of the 800 nm fluorochrome.
Mutation analysis using a Li-cor DNA sequencer
Fluorescent DT–PCR products were directly analyzed for mutations using a dual dye automated DNA sequencer (Li-cor Model 4200 IR2 system). Prior to electrophoresis, 8 µl from each 20 µl of DT–PCR reaction was mixed with 2 µl of 5× IR2 Stop/Loading Buffer (Li-cor) plus a drop of mineral oil. After 2 min denaturation at 95°C, samples were chilled on ice and 2 µl of each was loaded onto a 41 cm length gel with 36 wells (6% Sequagel; DiaMed, Atlanta, GA). The gel was mounted on the Model 4200 IR2 sequencing system. Electrophoresis was performed according to the manufacturer’s recommendations. Fluorescence-labeled termination fragments were detected in real time and the image files generated were visually processed in Adobe Photoshop 4.0.
Dideoxy DNA sequencing
Both DNA strands of each supF mutant plasmid were sequenced using primers PS189NT and PS189F (5′-GAGCGTCGATTTTTGTGAT) and the ABI Taq FS dye terminator kit. Sequencing was carried out with an ABI 377 automated DNA sequencer (Applied Biosystems, Foster City, CA). Dideoxy sequence ladders used in DT–PCR were generated using IRD-labeled primers and a Thermal Sequenase Cycle Sequencing Kit (Amersham Pharmacia Biotech, Cleveland, OH) according to the manufacturer’s instructions.
RESULTS AND DISCUSSION
Fluorescent visualization of termination fragments
DT–PCR allows generation of two sets of termination fragments that define all the sites requiring incorporation of the limiting nucleotide (2,3). Detection of DT–PCR products using two IRD-labeled primers and a Li-cor DNA sequencing system provide a sensitive, non-radioactive approach for independent visualization of the two sets of termination fragments generated in the same reaction. Four sets of characteristic termination fragments were generated in DT–PCR reactions with a different nucleotide limited in each reaction (Fig. 1). The band patterns of termination fragments corresponded well to their dideoxy sequence ladders, but the dideoxy and DT–PCR patterns were not identical. A doublet was usually observed in the DT–PCR reaction at each limiting nucleotide site and a triplet was observed at two consecutive limiting sites, and so on. Full profiles of termination fragments were generated at mononucleotide runs with more than two consecutive limiting sites in all DT–PCR reactions with the notable exception of the limiting dGTP reaction. The partial profiles of termination fragments generated at mononucleotide runs of guanine (Fig. 1, limiting dGTP) were not restricted to specific sites, reflecting an intrinsic property of Taq DNA polymerase, but their kinetic mechanism was not clear. Although the relative intensities of terminating bands varied from site to site in a sequence context-dependent manner, the band patterns of termination fragments were highly reproducible. The doublet phenotype also assisted the recognition of point mutations by concomitant gain and loss of two related termination fragments. Even though base substitutions were readily identifiable using four separate DT–PCR reactions (Fig. 1), this approach was redundant because only half of the sequence information generated in these reactions was used. In addition, difficulties were also expected in identifying mutations involving mononucleotide runs of dGTP.
Figure 1.

Visualization of termination fragments of the supF gene using IRD-labeled primers. Four fluorescent DT–PCR reactions each with a different nucleotide limited were carried out for six samples and electrophoresed side by side on a denaturing 6% polyacrylamide gel mounted on an automated Li-cor DNA sequencer. The partial fluoroimage was visualized using IRD-labeled primer PS189NT800. The termination fragments visualized by primer PS189RT700 are not shown. The first lane in each group was a dideoxy sequence ladder generated as described in Materials and Methods. Substitution mutations were identified using correlated gain and loss of terminating bands in related DT–PCR reactions. For example, samples 3 and 6 had a transition from C:G to T:A and a transversion from C:G to A:T, respectively, at the sites indicated by arrows.
Characterization of base substitutions
The procedure of dual dye labeling coupled with automated fluorescent detection allowed inspection of any sequence change in a DNA fragment using two complementary DT–PCR reactions with limiting dATP and dCTP. A base substitution in a template DNA results in simultaneous changes in band gain and loss at the site of mutation, but no band shift at other sites. The partial fluoroimages in Figure 2 demonstrate the characterization of base substitutions in a 203 bp fragment of pSP189 containing the supF gene. Two examples of the data are explained as follows. (i) Lane 4 in Figure 2A had a two band addition as indicated by an arrow in the limiting dATP reaction. Based on the doublet nature, these two bands represented a single base substitution to adenine at this position. This change was accompanied by a band missing in a string of four bands in Figure 2B of the limiting dCTP reaction. Because these two changes corresponded to the same mutation, sample 4 had a transversion from C:G to A:T at the position shown in the sequence below the image. (ii) Lane 3 in Figure 2B had two bands missing in the middle of a five band tract in the limiting dCTP reaction, indicating a tandem substitution from CC to other bases at the missing site. Correspondingly, there was a three band addition as indicated by an open arrow in Figure 2C of the limiting dATP reaction, indicating that the tandem substitution was from CC:GG to TT:AA. Base substitutions in the three other samples were characterized in the same fashion (Fig. 2).
Figure 2.

Partial fluoroimages showing determination of substitution mutations in a 203 bp fragment of the pSP189 plasmid. Two fluorescent DT–PCR reactions with limiting dATP and dCTP were carried out for six samples, including a wild-type plasmid (wt), and electrophoresed on a denaturing 6% polyacrylamide gel. DNA fragments visualized in the left panel (A and B) are extensions of primer PS189NT700 while those visualized in the right panel (C and D) are extensions of primer PS189TR800. The first lane in each group is a dideoxy sequence ladder. Arrows on the gel indicate positions of band gains and losses and letters following each are the actual base changes associated with these gains and losses. The wild-type sequence with its fluorescent labeling in each strand is shown below the fluoroimages (E). Substitution mutations in mutant samples are indicated by uppercase bases in the sequence. Samples 1, 2, 4 and 5 had a single base pair substitution as indicated by bases above the wild-type sequence. Sample 3 had a tandem substitution CC→TT in the upper strand. See text for interpretations on how substitutions were actually determined for two representative samples, 3 and 4.
Factors influencing mutation identification
The accurate identification of point mutations was achieved through the specificity of terminating profiles at all limiting nucleotide sites, especially those with mononucleotide runs. Despite the existence of several possible combinations, complementary reactions with limiting dATP and dCTP provided the best terminating band patterns for mutation characterization when coupled with Taq DNA polymerase. Other non-proofreading DNA polymerases such as Exo– Pfu and Exo– Vent DNA polymerases were also used for DT–PCR analysis and slightly different terminating band patterns were generated. It is worth noting that Exo– Pfu DNA polymerase produced more specific and intensified termination fragments in limiting dCTP reactions. This feature was useful in analyzing ambiguous base substitutions in G:C-rich regions, especially in mononucleotide runs involving four or more cytosines or guanines. Proofreading DNA polymerases such as Pfu turbo and Vent polymerases were not suitable for DT–PCR because the proofreading activity prevented formation of termination fragments (data not shown).
Rapid analysis of a UV-induced mutational spectrum
Fluorescent DT–PCR was used to generate a UV-induced spectrum of mutations in the supF gene following replication in human DNA repair-deficient cells. Blind analysis of 78 supF samples revealed 35 unique substitution events including eight tandem substitutions, a single 42 bp deletion and a plasmid with no detectable mutation in the 203 bp fragment analyzed. Although each mutant plasmid contained only one mutation in the coding sequence of the supF gene, secondary substitutions occurred outside the coding region and were also detected in three plasmids. Consistent with the nature of UV-induced mutations, 93.4% of the single and tandem base substitutions were G:C→A:T transitions. The distribution of these mutations within the sequence of the supF gene is shown in Figure 3. Because some mutations occurred in multiple samples, mutational hotspots containing at least 5% of total mutations were observed at bp 108, 124, 139, 156, 168 and 172. Classification of mutational hotspots can be complicated by possible jackpot mutations when multiple identical mutations from the same plate were analyzed. This problem was resolved by introducing an 8 bp random sequence into the pSP189 vector system so that each individual plasmid had a unique identification tag (7). Fluorescent DT–PCR was also useful in determining the independence of identical mutations by detecting distinct terminating band patterns in the signature sequence-containing region located 56 bp 5′ to the supF gene (Fig. 4). Rapid screening of all identical mutations using a single DT–PCR reaction revealed no identical signature sequence except for two plasmids containing the same mutation at bp 156.
Figure 3.

The distribution of 76 UV-induced substitution mutations in the supF suppressor tRNA gene. The top line shows the coding sequence of the marker gene. Each letter beneath the lower line indicates a single mutation and adjacent underlined letters represent tandem mutations. Except for two substitutions at bp 156, as indicated by asterisks, which had identical signature sequences, all other mutations were independent in origin. A 42 bp deletion (bp 142–184) is not shown in the figure.
Figure 4.
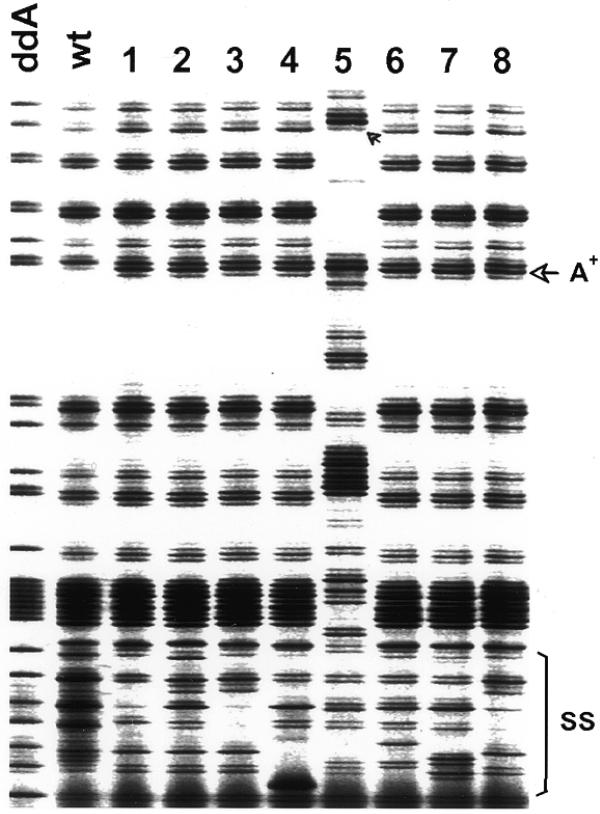
Screening of the signature sequence to demonstrate independence of identical mutations. The partial fluoroimage shows both the variation of terminating band patterns in the signature sequence element (SS) and eight identical mutations that occurred at bp 172 as indicated by arrows. Although most banding changes in the signature sequence element were caused by nucleotide substitutions, sample 5 also involved a multi-nucleotide insertion that led to concomitant upward banding shifts. Terminating fragments shown in this image were generated from one of the DNA strands by primer PS189JR700 in a limiting dATP reaction. The results from the complementary strand are not shown.
Comparative analysis
To test the accuracy of fluorescent DT–PCR analysis, the same set of plasmids was analyzed independently from both DNA strands using ABI 377 assisted dideoxy DNA sequencing. Comparison of both methods revealed identical results in both characterizing mutations in the supF gene and in ascertaining the independence of hotspot mutations. It is worth noting that double-stranded sequencing was essential for the dideoxy method to obtain an accurate mutational spectrum because multiple errors of miscalling were made by the ABI 377 sequencer when only a single strand of DNA was analyzed. The high accuracy of fluorescent DT–PCR was enhanced by a double confirmation mechanism of verifying a result from two complementary reactions. This is because a point mutation always leads to concomitant gain and loss of related terminating bands in two complementary DNA strands. Furthermore, the requirement for template DNA was different in the two methods. About 10 ng plasmid DNA was routinely used for each DT–PCR reaction and no significant reduction in signal intensities was observed with an up to 10 000-fold reduction in template concentration. In contrast, 400–500 ng purified template DNA was typically required for a 5 kb plasmid like pSP189 to be sequenced properly by the ABI 377 sequencer. The comparative study demonstrated that the fluorescent DT–PCR analysis has 100% accuracy in determining all types of base substitutions, requires no special reagents and uses much less template DNA than automated dideoxy sequencing.
Conclusions
The procedure of dual dye labeling coupled with automated fluorescent detection described in this study provides a sensitive system to retrieve the sequence information generated in two complementary DT–PCR reactions. We demonstrated its 100% efficiency in determining substitution mutations in a reporter gene with high G/C content and its application to rapid analysis of a UV-induced mutation spectrum in mammalian cells. In a separate study we also showed its usefulness in accurately determining different types of deletions and insertions in the lys2–Bgl frameshift assay from yeast genomic DNA (9). Because of its simplicity in the generation of termination fragments, high accuracy in mutation determination and feasibility for computerized data processing, fluorescent DT–PCR will serve as a cost-effective alternative to dideoxy DNA sequencing for large-scale analysis of nucleotide sequence changes.
Acknowledgments
ACKNOWLEDGEMENTS
Funding for this work was provided by grants CA69449 to G.P.H. and ES06070/CA84469 to G.P.P.
References
- 1.Cotton R.G.H. (1997) Slowly but surely towards better scanning for mutations. Trends Genet., 13, 43–46. [DOI] [PubMed] [Google Scholar]
- 2.Chen J. and Hebert,P.D.N. (1998) Directed termination PCR: a one-step approach for mutation detection. Nucleic Acids Res., 26, 1546–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J.Z. and Hebert,P.D.N. (1999) Directed termination of the polymerase chain reaction: kinetics and application in mutation detection. Genome, 42, 72–79. [PubMed] [Google Scholar]
- 4.Chen J.Z. and Hebert,P.D.N. (1999) Intraindividual sequence diversity and a hierarchical approach to the study of mitochondrial DNA mutations. Mutat. Res., 434, 205–217. [DOI] [PubMed] [Google Scholar]
- 5.Chen J.Z. and Hebert,P.D.N. (1999) Terminal branch haplotype analysis: a novel approach to investigate newly risen variants of mitochondrial DNA in natural populations. Mutat. Res., 434, 219–231. [DOI] [PubMed] [Google Scholar]
- 6.Kraemer K.H. and Seidman,M.M. (1989) Use of supF, an Escherichia coli tyrosine suppressor tRNA gene, as a mutagenic target in shuttle-vector plasmids. Mutat. Res., 220, 61–72. [DOI] [PubMed] [Google Scholar]
- 7.Parris C.N. and Seidman,M.M. (1992) A signature element distinguishes sibling and independent mutations in a shuttle vector plasmid. Gene, 117, 1–5. [DOI] [PubMed] [Google Scholar]
- 8.Hirt B. (1967) Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol., 26, 365–369. [DOI] [PubMed] [Google Scholar]
- 9.Chen J.Z., Qiu,J., Shen,B. and Holmquist,G.P. (2000) Mutational spectrum analysis of RNase H(35) deficient Saccharomyces cerevisiae using fluorescence-based directed termination PCR. Nucleic Acids Res., 28, 3649–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]