

Abstract
β2-Adrenergic receptors (β2-AR) are low abundance, integral membrane proteins that mediate the effects of catecholamines at the cell surface. Whereas the processes governing desensitization of activated β2-ARs and their subsequent removal from the cell surface have been characterized in considerable detail, little is known about the mechanisms controlling trafficking of neo-synthesized receptors to the cell surface. Since the discovery of the signal peptide, the targeting of the integral membrane proteins to plasma membrane has been thought to be determined by structural features of the amino acid sequence alone. Here we report that localization of translationally silenced β2-AR mRNA to the peripheral cytoplasmic regions is critical for receptor localization to the plasma membrane. β2-AR mRNA is recognized by the nucleocytoplasmic shuttling RNA-binding protein HuR, which silences translational initiation while chaperoning the mRNA-protein complex to the cell periphery. When HuR expression is down-regulated, β2-AR mRNA translation is initiated prematurely in perinuclear polyribosomes, leading to overproduction of receptors but defective trafficking to the plasma membrane. Our results underscore the importance of the spatiotemporal relationship between β2-AR mRNA localization, translation, and trafficking to the plasma membrane, and establish a novel mechanism whereby G protein-coupled receptor (GPCR) responsiveness is regulated by RNA-based signals.
Keywords: Cell Surface Receptor, G Protein-coupled Receptors (GPCR), Gene Expression, Receptor Regulation, RNA Transport, β2-Adrenergic Receptor, HuR, RNA-binding Proteins, mRNA Localization, Translational Control
Introduction
G protein-coupled receptors (GPCR)2 play a key role in signal transduction and are important drug targets (1). Although β2-AR is one of the most extensively characterized members of the GPCR family (2), relatively little is known about the nature and kinetics of the specific steps involved in its biosynthesis and trafficking to the cell surface (3). It is generally believed that in mammalian cells, secretory and membrane proteins are translocated across the ER membrane concurrent with their synthesis by membrane-bound ribosomes (4). This co-translational translocation for GPCRs begins with the synthesis of the signal peptide or the first hydrophobic transmembrane sequences of the protein (5, 6) and its recognition by the signal recognition particle that results in mRNA localization to the ER (4). Thus, the localization of integral membrane protein-encoding mRNAs to the ER is achieved by the binding interactions of the ribosome and translation product with the components of the ER (7, 8).
On the contrary, translation-independent mRNA localization usually requires the recognition of the transcript by RNA-binding proteins within the nucleus, which renders the mRNA-protein complex translationally inactive when the complex reaches the cytoplasm (9). HuR, a member of the ELAV (embryonic lethal abnormal vision) family of RNA-binding proteins (10, 11), is ubiquitously expressed and is known to bind a large number of mRNAs that encode many different classes of proteins (12). HuR has two N-terminal RNA recognition motifs (RRM) with a high affinity for U-rich RNA sequences, a nucleocytoplasmic shuttling sequence, and a C-terminal RRM that can recognize the poly(A) tail (13, 14). Although predominantly nuclear, HuR can shuttle between the nucleus and the cytoplasm (10, 14). Binding of HuR to mRNAs in the cytoplasm can stabilize (14) and variably affect their translational processing (15).
Relatively little is known, about how and where mRNAs encoding GPCRs like the β2-AR are localized and translated in intact cells. We recently reported that cellular expression of β2-AR is suppressed at the translational level by 3′-untranslated region (UTR)-binding proteins (16, 17). The current study was designed to address the importance of translational silencing and mRNA localization in β2-AR trafficking to the plasma membrane.
EXPERIMENTAL PROCEDURES
Cell Culture
DDT1-MF2 vas deferens smooth muscle cells, A431 cells, and CHO cells were cultured as described previously (17).
HuR Knockdown in DDT1-MF2 and A431 Cells
We used a stable transfection system to express two different short hairpin interfering (sh) RNAs against HuR. The shRNA expression vector was constructed by inserting synthetic double-stranded oligonucleotides between the BamHI and HindIII sites of the polymerase III gene promoter on vector pSUPER (Ref. 18 Ambion). The shRNA1 sequences were targeted to nucleotides on HuR-coding region “AAC ACG CTG AAC GGC TTG AGG” and shRNA2 sequences were targeted to nucleotides on HuR 3′-UTR “AAC GAC TCA ATT GTC CCG ATA” as described previously (19, 20). DDT1-MF2 and A431 cells were transfected with this vector using Lipofectamine reagent (Invitrogen). Cells were also transfected with pSuper vector harboring a mutation at positions 2 and 9 of the HuR target recognition sequence. G418-resistant clones were collected, and clones with significant reduction in HuR protein (clones 2 and 4, Fig. 1A) were used for studies involving β2-AR mRNA, protein, and immunofluorescence staining and were compared with cells expressing control shRNA. A pcDNA3 (Invitrogen)-based mammalian expression vector was used to clone Myc-tagged HuR that lacked the 3′-UTR of HuR cDNA. Because this chimeric HuR mRNA lacked the 3′-UTR it is insensitive to the shRNA2 that was used for the knockdown of HuR mRNA.
FIGURE 1.

Knockdown of HuR results in reduced steady-state levels of β2-AR mRNA and increased receptor expression. A, immunoblot analyses of proteins extracted from four different clones of DDT1-MF2 cells expressing shRNA specific for HuR mRNA (lanes 2–5) compared with cells expressing nonspecific shRNA (lane 1) using anti-HuR monoclonal antibody and β-tubulin (control) polyclonal antibody. B, RNase protection assays (RPA) for quantitative measurement of β2-AR mRNA (upper panel) and GAPDH mRNAs (lower panel) in the above clones. C, radioligand binding assay using 125I-CYP in the above clones used for Western blot and RPA. Nonspecific binding determined in the presence of 10 μm dl-propranolol (<10%) was subtracted from total binding. The values expressed as percentages of receptor expression, taking the values obtained for receptor expression in control shRNA-transfected cells as 100%. Receptor expression in untransfected cells, and cells transfected with control shRNA were similar. Values represent mean ± S.D. of four separate receptor measurements. D, translational suppression by cycloheximide stabilized β2-AR mRNA in HuR knockdown cells. HuR knockdown and control cells were treated with cycloheximide (50 μg) for the indicated times, and total RNA was extracted and subjected to RPA of β2-AR and GAPDH mRNAs.
Immunoprecipitation (IP), in Vitro Transcription, and UV Cross-linked Label Transfer Assays
IP experiments were carried out as described by Tenenbaum et al., (21). In vitro transcription and UV cross-linked label transfer assays were performed essentially as described previously (17).
Determination of GST-HuR Binding Affinity to the 21-nt AU-rich Region of β2-AR mRNA
The cDNA for HuR obtained by RT-PCR from DDT1-MF2 cells was cloned into the pET-42a(+) vector (Novagen). The sequences of the insert were verified by DNA sequencing and grown in the presence of kanamycin at 37 °C. Isolation of GST-HUR was done as described previously for GST-TIAR (17). The affinity of purified GST-HuR for 21-nt A+U-rich RNA was determined by gel mobility shift assay as described before for GST-TIAR (17).
Polysome Profile Analysis of β2-AR mRNA from DDT1-MF2 Cells
Cytoplasmic extracts of DDT1-MF2 cells expressing control shRNA and HuR-specific shRNA were made and used for polysome profile analysis of β2-AR mRNA as described previously (17). Twelve fractions were collected, total RNA was extracted from each fraction, and β2-AR and GAPDH mRNAs were quantified by RNase protection assays.
Dual Luciferase Assay
DDT1-MF2 cells expressing control shRNA, HuR-specific shRNA and HuR-specific shRNA along with shHuR insensitive Myc-HuR were cotransfected with the firefly luciferase with or without receptor 3′-UTR and Renilla luciferase. Luciferase activities for firefly or Renilla luciferase were determined according to the manufacturer's instructions (Promega). Firefly luciferase was normalized by Renilla luciferase activities in all experiments and expressed relative to the indicated controls.
Radioligand Binding, Cell Fractionation, and Immunoprecipitation
Binding assays were performed on whole cells using a saturating concentration of 125I-iodocyanopindolol as described previously (17). The total number of β2-AR expressed in DDT1-MF2 cells expressing control shRNA and HuR-specific shRNA was determined by 125I-CYP in a filter binding assay. The number of cell surface β2-AR was determined by binding of the hydrophilic ligand [3H]CGP-12177 as described by Rands et al. (22). For membrane isolation, cells were homogenized by polytron disruption and centrifuged at 450 × g for 5 min at 4 °C. The pellet was saved and the supernatants were centrifuged at 43,000 × g for 30 min. The light membrane fraction (supernatants) saved and the membrane fraction (pellet) was washed and solubilized in binding buffer containing protease inhibitors and used for radioligand binding assay. The light and heavy membrane fractions were incubated with β2-AR antibody (1:25). Controls included non-immune IgG-treated samples of the same dilution and antibody pre-incubated with 10 μm of the corresponding peptide for 2 h at 22 °C. The immune complexes were immobilized by adding protein A-agarose beads to each assay and allowed to rock for 6 h at 4 °C. The antibody-coated complexes were washed three times with buffer containing 0.1% Nonidet P-40. Specific 125I-CYP binding to receptor antibody-protein A-agarose beads was measured by rapid vacuum filtration over glass fiber filters as described for the whole-cell binding assay. In controls, β2-AR antibody was replaced with control IgG. In a second set of controls, β2-AR antibody was preincubated with 10 μm of the corresponding peptide for 2 h at 22 °C. cAMP generation was measured using an enzyme-immunoassay (EIA) system (Amersham Biosciences). Cells were cultured in 96-well plates overnight. Medium was aspirated and treated with isoproterenol (10 μm) dissolved in serum-free medium for 5 min at 37 °C. cAMP generated was calculated using a standard curve per the manufacturer's instructions.
Immunofluorescence Staining, in Situ Hybridization, and Confocal Microscopy
For immunofluorescence staining, cells were grown on 35-mm lysine-coated glass-bottom culture dishes (MatTek Corporation) and fixed in PBS containing 4% paraformaldehyde, 4% sucrose, and 2 mm MgCl2. Fixed cells were washed and permeabilized in 0.3% Triton X-100 in PBS. Nonspecific binding sites were blocked with 3% normal serum (Santa Cruz Biotechnology, Inc.) in PBS for 2 h. The cells were incubated overnight at 4 °C with the specific primary antibodies: rabbit polyclonal β2-adrenergic receptor (1:50) (sc-569; H-20; Santa Cruz Biotechnology, Inc.), monoclonal HUR (1:200) (sc-5261; 3A2; Santa Cruz Biotechnology, Inc.), followed by incubation for 1 h at room temperature with the appropriate Alexa Fluor-conjugated secondary antibodies (1:200) (Molecular Probes). DRAQ5 (Alexis Corporation) (1:500 in PBS for 20 min) was used to visualize the nuclei.
Antisense riboprobes used for in situ hybridization were transcribed in vitro from sense β2-AR cDNA as described previously (17) using digoxigenin-conjugated UTP (Roche Diagnostics). Cells were fixed, washed and permeabilized as described above. Prehybridization was performed in 50% formamide, 5 × SSC, 0.2% SDS, 50 μg/ml heparin, 250 ng/ml salmon sperm DNA, and 250 μg/ml yeast tRNA. Hybridization was performed in the same solution supplemented with 150 μg/ml heparin and 150 ng/ml of digoxigenin-riboprobe. After washing, nonspecific binding sites were blocked with 3% normal serum (Santa Cruz Biotechnology, Inc.) in PBS for 2 h. The probe signal was amplified using biotinylated antidigoxin followed by streptavidin coupled to Cy3. Confocal microscopy was performed using a Zeiss LSM 510 META laser-scanning microscope (Carl Zeiss, Inc.).
RESULTS
ShRNA-mediated Knockdown of HuR Reduces Steady State Levels of β2-AR mRNA While Increasing Receptor Expression
Earlier studies have identified the 3′-UTR of β2-AR mRNA as a target for HuR protein (17, 23). In order to characterize how the interaction of the RNA-binding protein HuR with β2-AR mRNA can influence receptor expression, we performed specific HuR mRNA knockdown by RNA interference on two different cell lines (DDT1-MF2 and A431) that endogenously express β2-AR. Successful attenuation of HuR protein was confirmed by comparing Western blot analyses of extracts from cells expressing a control short hairpin RNA (shRNA) with cells expressing HuR-specific shRNA (Fig. 1A). To study the effect of HuR knockdown on β2-AR mRNA, total RNA was extracted from these cells and β2-AR and GAPDH mRNAs were measured with an RNase protection assay (Fig. 1B). Knockdown of HuR to below 20% of the control levels led to greater than 90% reduction in β2-AR mRNA without any detectable change in GAPDH mRNA (estimated by direct analysis of radioactivity by phoshorimaging). These results suggest that HuR enhances steady state levels of β2-AR mRNA, similar to many other transcripts to which this protein is reported to bind (12, 14).
Receptor expression was quantified in HuR knockdown and control cells using a cell-permeable hydrophobic radioligand, 125I-cyanopindolol (125I-CYP). Paradoxically, attenuation of HuR levels resulted in a 2.0–3.0-fold increase in receptor expression (Fig. 1C; 1.5 to 2 fmol/105 cells in control shRNA-expressing cells compared with 4–4.5 fmol/105 cells in HuR-specific shRNA-expressing cells). A direct comparison of the receptor expression and mRNA levels in different HuR knockdown clones (Fig. 1, B and C) showed that β2-AR expression is negatively correlated with its mRNA levels as reported before (17).
To test whether translation of β2-AR mRNA is linked to its decay, we treated HuR knockdown and control cells with cycloheximide (50 μg/ml) and compared the change in β2-AR mRNA over time. This concentration of cycloheximide treatment for 3 h resulted in more than 90% inhibition of protein synthesis as measured by trichloroacetic acid precipitation of newly synthesized proteins (data not shown). Cycloheximide treatment and decreased protein synthesis resulted in more pronounced stabilization of β2-AR mRNA in HuR knockdown cells (Fig. 1D, left panel) compared with cells expressing control shRNA (Fig. 1D, right panel).
HuR Binds the 3′-UTR of β2-AR mRNA
A role for the 3′-UTR of β2-AR mRNA in HuR protein association was confirmed by IP of this protein from cytosolic extracts of Chinese hamster ovary (CHO) cells transfected with full-length and 3′-UTR deletion constructs of β2-AR cDNA. RNA isolated from the immunoprecipitate was analyzed by RT-PCR. IP of HuR protein resulted in co-precipitation of β2-AR mRNA when CHO cells were transfected with full-length β2-AR cDNA (Fig. 2A). Using cytosolic extracts from CHO cells transfected with 3′-UTR deletion constructs significantly reduced the amount of β2-AR mRNA that was co-precipitated using HuR antibody. Whereas IP with control IgG1 failed to co-precipitate β2-AR mRNA, the use of T-cell-restricted intracellular antigen-related protein (TIAR) antibody (positive control) resulted in co-precipitation of β2-AR mRNA as demonstrated previously (17).
FIGURE 2.

HuR binds to the 3′-UTR of β2-AR mRNA. A, CHO cells were transfected with full-length (lanes 1–4) or 3′-UTR deletion constructs (lanes 5 and 6) of β2-AR cDNA. 36 h after transfection, cytoplasmic lysates were made in a polysome lysis buffer supplemented with RNase and protease inhibitors. IP reactions were carried out following the method of Tenenbaum et al. (21). Total RNA from the immunoprecipitated complex was isolated and subjected to RT-PCR using β2-AR-specific primers as described previously (17). PCR products were visualized on 1% agarose gels. Lane 1, amplified product from total cytoplasmic extracts without IP. Lane 2, IP using control IgG1. Lanes 3 and 4, IP using TIAR or HuR antibodies respectively. Lanes 5 and 6, IP using TIAR or HuR antibodies, respectively. Transfections, IP, and RT-PCR were performed twice and results were similar. B, identification of multiple binding sites for HuR on β2-AR mRNA. Autoradiogram shows label transfer to HuR protein from in vitro transcribed radiolabeled 3′-UTR transcripts of β2-AR mRNA. Equimolar quantities of radiolabeled β2-AR 3′-UTR transcripts corresponding to the full-length (530-nt) 3′-UTR (lanes 1–3); the proximal 190-nt (lanes 4–6); distal 340 nt (lanes 7–9), and proximal 190-nt region with mutations to disrupt a 21-nucleotide A+U-rich regions (lanes 10–12) were synthesized and incubated with equal quantities of cytosolic lysates from DDT1-MF2 cells (25 μg) and then subjected to UV-cross-linked label transfer. The resulting complexes were treated with RNase A+T1 and subjected to IP using anti-HuR antibody or control IgG1. The immunoprecipitated materials and controls were subjected to SDS-PAGE analysis as described before (17). Lanes 1, 4, 7, and 10, UV cross-linked material that was not subjected to IP; lanes 2, 5, 8, and 11, IP with control IgG1; lanes 3, 6, 9, and 12, IP with anti-HuR antibody. C, non-denaturing gel-shift analysis and affinity determination of GST-HuR to the 21-nt AU-rich region from the proximal 3′-UTR of β2-AR mRNA. A constant amount of RNA was incubated with increasing concentrations (0–25 nm) of purified recombinant GST-HuR (lanes 1–9) or GST (lane 10) protein and analyzed by electrophoresis on native gel.
Additional evidence for HuR association with β2-AR mRNA was obtained by UV-cross-linking of cytosolic lysates from DDT1-MF2 cells with in vitro transcribed 32P-labeled receptor 3′-UTR RNA, followed by IP of the resulting complexes in the presence or absence of specific anti-HuR or control IgG1. Radiolabeled RNA corresponding to the full-length (530 nt, Fig. 2B, lanes 1–3), proximal (190 nt, lanes 4–6), and distal (340 nt, lanes 7–9) 3′-UTRs of β2-AR mRNA were separately assessed to identify the regions that associate with HuR protein. Additionally, specific mutations were introduced into a 21 nt A+U-rich region present in the proximal 190 nt 3′-UTR to identify the binding site for HuR protein (lanes 10–12). Aliquots of the UV cross-linked material, when subjected to SDS-PAGE analysis after RNase digestion, showed significant label transfer to several proteins (Fig. 2B). IP of the cross-linked material without a specific antibody (control IgG1) yielded no radiolabeled bands. In contrast, the use of a specific anti-HuR antibody revealed that all three radiolabeled β2-AR 3′-UTR fragments showed marked label transfer to the HuR protein. Mutations to disrupt the 21 nt A+U-rich region (17) in the 190-nt RNA abolished binding of HuR to this 3′-UTR RNA (Fig. 2B), demonstrating sequence specific label transfer to HuR protein from β2-AR mRNA 3′-UTR sequences.
To address more directly the binding affinity of HuR to the 21 nt A+U-rich region, nondenaturing gel shift experiments were performed using GST-HuR and equimolar concentrations of in vitro transcribed, radiolabeled RNA corresponding to the 21 A+U-rich regions (Fig. 2C). The results of the gel shift analysis demonstrated that GST-HuR and not GST (lane 10) binds with high affinity (∼5 nm) to the 21 nt A+U-rich RNA.
HuR Inhibits de Novo Synthesis of β2-AR When Co-transfected in CHO Cells
To examine the possibility that HuR may inhibit the synthesis of β2-AR, CHO cells were transiently co-transfected with equal quantities (5 μg) of receptor cDNA constructs with and without 3′-UTR sequences along with Myc-tagged HuR cDNAs. Because the deletion of 3′-UTR from β2-AR cDNA results in increased receptor expression (16), CHO cells also were co-transfected separately with 5 μg each of β2-AR cDNA, with and without 3′-UTR sequences to serve as proper controls. The control cells were co-transfected with empty vector in place of HuR cDNA. Receptor expression was measured in each group after 24 and 72 h of transfection (Fig. 3A). Because CHO cells lack endogenous β2-AR, the receptor measured should originate from the transfected β2-AR cDNAs. Cells co-transfected with HuR and full-length receptor cDNAs showed a 75% decrease in receptor expression after 24 h of transfection as compared with a 20% decrease in receptor expression observed in control cells (Fig. 3A, compare lanes 2 and 5). The decreased receptor expression at 24 h of transfection correlated with the increased HuR expression in these cells (Fig. 3B, upper panel, lane 2). Receptor measurement 72 h after transfection showed a significant increase only in the cells that are co-transfected with full-length β2-AR and HuR cDNAs (Fig. 3A, lane 3). Moreover, this increase in β2-AR expression correlated with the loss of HuR overexpression in transiently transfected cells (Fig. 3B, upper panel, lane 3). To confirm that the decreased receptor expression in CHO cells that are co-transfected with β2-AR and HuR cDNAs is due to translational suppression of β2-AR mRNA, we compared steady state levels of receptor mRNA at 0, 24, and 72 h of transfection (Fig. 3C). Receptor mRNA levels were significantly higher in CHO cells that were co-transfected with full-length β2-AR cDNA and Myc-HuR after 24 h of transfection as compared with controls (Fig. 3C, compare lanes 2 and 5). These results suggest that overexpression of HuR suppresses the translation of β2-AR mRNA. Furthermore, after 72 h of transfection, when HuR levels returned to the control values, β2-AR mRNA levels decreased, and receptor expression increased.
FIGURE 3.

HuR inhibits de novo synthesis of β2-AR in a 3′-UTR-dependent manner. A, CHO cells were co-transfected with equal quantities of full-length (lanes 1–3) or 3′-UTR deletion constructs (lanes 4–6) of β2-AR cDNA (5 μg) with HuR cDNA or empty vector. β2-AR expression levels were measured by radioligand binding assay in both groups of cells at 24 h (lanes 2 and 5) and 72 h (lanes 3 and 6) following transfections. The values shown are percentages of receptor expression taking the values obtained for receptor expression at the appropriate time point for cells that were co-transfected with β2-AR cDNA with empty vector as 100% (lanes 1 and 4). Each value for receptor expression represents mean ± S.D. of four separate transfections. B, Western blot analyses using cytoplasmic extracts of CHO cells transfected with full-length β2-AR and HuR cDNA at 0, 24, and 72 h after transfection (upper panel) using HuR antibody. The blots were re-probed using tubulin antibody as a loading control (lower panel). C, autoradiogram shows measurements of steady state levels of β2-AR mRNA in CHO cells transfected with full-length β2-AR (lanes 1–3) or 3′-UTR deletion constructs (lanes 4–6) and HuR cDNAs. Total RNA was extracted from each group at 0, 24, and 72 h after transfections and 50 μg of total RNA was used in RNase protection assay. Lower panel shows RPA of GAPDH as a loading control. D, reversal of translational suppression of reporter transcript in HuR knockdown cells. Reporter luciferase constructs with and without β2-AR 3′-UTR were transfected into DDT1-MF2 cells expressing control shRNA (lanes 1 and 2), HuR-shRNA (lanes 3 and 4), and Myc-tagged HuR that is insensitive to HuR-shRNA expressed (lanes 5 and 6). The values, normalized by co-transfection of Renilla luciferase, represent mean ± S.D. of three separate transfections using luciferase constructs. Error bars indicate S.D.; asterisk, p = 0.05; two asterisks, p = 0.007; three asterisks, p = 0.0004. Statistical analyses were performed using unpaired t-tests (two-tailed p value is given). E, Western blot analyses of cytoplasmic extracts from cells expressing control shRNA (lanes 1 and 2), HuR-specific shRNA (lanes 3 and 4), and cells expressing shRNA insensitive myc-HuR (lanes 5 and 6) with HuR antibody. β-Tubulin is shown as a loading control.
We also used a reporter construct to validate the role of HuR as a β2-AR 3′-UTR-dependent translational regulator in vivo. Introduction of receptor 3′-UTR into the 3′-UTR of a luciferase reporter gene resulted in a 70% decrease in reporter gene expression when transfected into DDT1-MF2 cells expressing control shRNA (Fig. 3, D and E, lanes 1 and 2). Knockdown of endogenous HuR by shRNA enhanced translation from reporter transcripts in a receptor 3′-UTR sequence-dependent manner (Fig. 3, D and E, lanes 3 and 4). The specificity of the role of HuR in translation was further confirmed by overexpressing Myc-tagged HuR that is insensitive to the HuR-shRNA expressed. Expression of HuR restored the translational repression of reporter transcripts containing β2-AR 3′-UTR (Fig. 3, D and E, lanes 5 and 6). β-Tubulin is shown as a loading control (Fig. 3E, lower panel).
Polysome Profile Analysis of Endogenously Expressed β2-AR mRNA in HuR Knockdown and Control DDT1-MF2 Cells
To examine any possible role for HuR protein in β2-AR mRNA association with polyribosomes, we compared the distribution of endogenously expressed β2-AR mRNA in sucrose density gradient fractions. Cytoplasmic extracts of cells expressing control shRNA and HuR-specific shRNA were used. Sucrose density gradient fractionation and RNA extractions were performed as described previously (16, 17). Monosomes and polysomes were well separated as identified by absorbance at 254 nm (Fig. 4A, upper panel). Twelve continuous fractions, from lightest to heaviest, were collected, and total RNA was extracted from each fraction, and the distribution of β2-AR mRNA (Fig. 4A, lower panel) was determined in these fractions by RPA. A substantial portion of endogenously expressed β2-AR mRNA was found to be associated with low molecular weight ribosomes, including 40 S ribosomes, when cells expressing control shRNA were used. On the contrary, when cytoplasmic extracts of HuR knockdown DDT1-MF2 cells were used for polysome analysis, a clear shift in receptor mRNA to heavier polyribosomal fractions was observed (Fig. 4B, upper and lower panels). These results suggest inefficient translation of β2-AR mRNA in control cells and increased polyribosome association and translation in HuR knockdown cells. The polysome profile of glyceraldehyde-3-phosphate (GAPDH) mRNA also was analyzed in control and HuR knockdown cells to serve as a loading control (Fig. 4C). The distribution of GAPDH mRNA was similar in control and HuR knockdown cells, suggesting that HuR knockdown effects were specific to β2-AR mRNA. Densitometric analysis was used to determine the relative distribution of β2-AR mRNA and GAPDH mRNA in different fractions of the gradient (Fig. 4D).
FIGURE 4.

Polysome profile analysis of β2-AR mRNA in HuR knockdown and control DDT1-MF2 cells. A, upper panel, representative UV absorption profile of the cytoplasmic extracts of DDT1-MF2 cells expressing control shRNA across sucrose density gradients (10–45%) measured at 254 nm. The preparation of the gradients and collection of fractions were performed as described previously (17). A, lower panel, RNase protection assay shows the distribution of β2-AR mRNA in cells expressing control shRNA. B, upper panel, representative UV absorption profile of the cytoplasmic extracts of HuR knockdown DDT1-MF2 cells across sucrose density gradients (10–45%) measured at 254 nm. B, lower panel, RNase protection assay shows the distribution of β2-AR mRNA in cells expressing HuR-specific shRNA. C, shows the distribution of GAPDH mRNA in corresponding fractions. The UV absorption profile and GAPDH mRNA distribution were similar in HuR knockdown and control cells. D, quantification of β2-AR mRNA and GAPDH mRNA distribution in HuR knockdown and control DDT1-MF2 cells.
β2-AR Failed to Traffic to Plasma Membrane and Appeared around the Nucleus in HuR Knockdown Cells
The properties of receptors expressed in HuR knockdown cells were examined by immunofluorescence staining of β2-AR and HuR. The β2-AR staining pattern in HuR knockdown cells showed significant immunoreactivity for receptors around the nucleus (Fig. 5, A and B, arrows). In cells with significant immunoreactivity for HuR, the receptors appeared on the plasma membrane (Fig. 5A, arrowhead), with no detectable β2-AR immunoreactivity around the nucleus. The presence of cells with and without HuR within the same microscopic field provides an internal control for the confocal images presented. These results suggest that HuR-mediated translational suppression of β2-AR mRNA plays a critical role in receptor trafficking to the plasma membrane. Knockdown of HuR showed a similar effect on β2-AR expression in another cell line (A431) that endogenously expresses both receptor and HuR protein (supplemental Fig. S1).
FIGURE 5.

β2-AR in HuR knockdown cells show defective trafficking to the plasma membrane. A, confocal microscopy images of DDT1-MF2 cells shows immunofluorescence staining of HuR (green) and β2-AR (red) in HuR knockdown and control cells. 1) DIC image; 2) nuclear stain with DRAQ5; 3) immunofluorescence staining using monoclonal antibody to HuR; 4) immunofluorescence staining using polyclonal antibody to β2-AR; 5) Merged images. The presence of cells with and without HuR within the same microscopic field provides an internal control for the confocal images presented. B, confocal microscopy images shows a single HuR knockdown cell. C, confocal microscopy images show immunofluorescence staining of β2-AR (red) in DDT1-MF2 cells transfected with full-length β2-AR cDNA. D, confocal microscopy images show immunofluorescence staining of β2-AR (red) in DDT1-MF2 cells transfected with 3′-UTR deletion constructs of β2-AR cDNA. Scale bars 10 μm.
To test the possibility that receptor over-production in HuR knockdown cells may lead to failed trafficking to the plasma membrane, we transfected DDT1-MF2 cells with full-length hamster β2-AR cDNA. Transfection resulted in a 2–3-fold increase in receptor expression, comparable to the levels measured in HuR knockdown cells (Fig. 5C). Immunofluorescence staining and confocal images revealed varying levels of receptor overexpression in different cells. However, all of the overexpressed receptors are present on the plasma membrane (Fig. 5C and supplemental Fig. S2) and do not accumulate around the nucleus. These results demonstrate that receptor over-production is not the reason for the failed trafficking of β2-AR in HuR knockdown cells.
To further test the role of 3′-UTR in receptor trafficking to the plasma membrane, we transfected DDT1-MF2 cells with 3′-UTR deletion constructs of hamster β2-AR cDNA. Transfection resulted in increased receptor expression, comparable to the levels measured in HuR knockdown cells (Fig. 1C). Immunofluorescence staining and confocal images revealed the appearance of most of the overexpressed receptors around the nucleus (Fig. 5D and supplemental Fig. S3). These results suggest a role for 3′-UTR in receptor trafficking to the plasma membrane. Supporting our observation, significant perinuclear appearance of β2-AR-GFP fusion protein was observed when β2-AR-GFP fusion constructs were transfected into HeLa cells (24). These fusion constructs lacked the receptor 3′-UTR.
To further demonstrate the importance of the β2-AR 3′-UTR in mRNA localization and translational control, we used enhanced GFPNLS/myr with and without β2-AR 3′-UTR. This destabilized eGFP, containing an N-terminal myristoylation consensus sequences, limits the diffusion of the fluorescent protein to its site of synthesis (25). DDT1-MF2 cells transfected with GFPNLS/myr with β2-AR 3′-UTR showed localization of GFP to the cell periphery (supplemental Fig. S4). In contrast, when these cells were transfected with GFPNLS/myr without β2-AR 3′-UTR, the GFP distribution was localized to the perinuclear region of the cells suggesting an important role for the receptor 3′-UTR sequences in limiting the distribution of GFP to the cell periphery. In addition, as expected from the inhibitory effect of β2-AR 3′-UTR on translation (16, 17), the fusion of β2-AR 3′-UTR to GFP caused a reduction in GFP expression.
We isolated heavy (plasma membrane) and light membrane fractions from HuR knockdown and control cells and measured the distribution of β2-AR by radioligand binding in these fractions. In cells expressing control shRNA, 80–85% of the receptors were present in the heavy fraction, containing plasma membrane. Similar studies using HuR knockdown cells showed that less than 40% of the total receptors were plasma membrane localized. Thus, more than 60% of the radioligand binding activity was present in the light membrane fraction in HuR knockdown cells (Fig. 6A). Cells expressing control shRNA had less than 10% activity in the light membrane fraction. To further confirm the results of radioligand binding with 125I-CYP, we used a β2-AR antibody to immunoprecipitate the receptor protein from light and heavy membrane fractions separately. Most of the radioligand binding activity was immunoprecipitated from the heavy membrane fraction in cells expressing control shRNA. In contrast, significantly more radioligand binding activity was immunoprecipitated from the light membrane fraction in HuR knockdown cells than from the heavy membrane fraction (Fig. 6B). Control IgG1 did not precipitate any radioactivity above the blank, and preincubation of antibody with the corresponding synthetic peptide for the antibody blocked the IP of the radioligand binding activity (data not shown).
FIGURE 6.
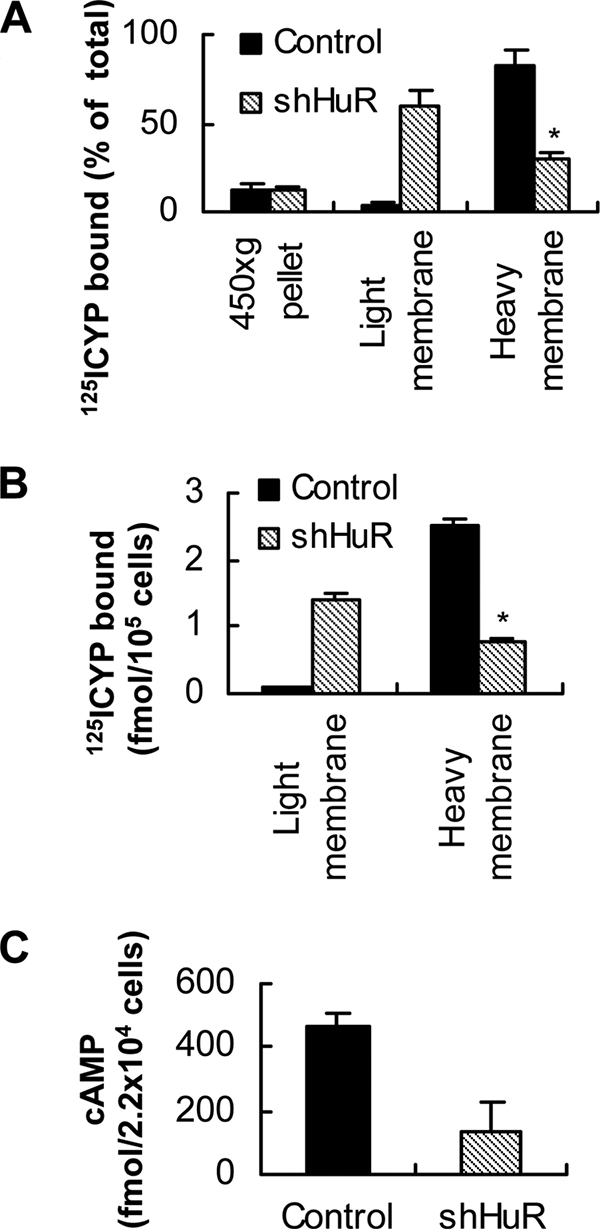
Subcellular fractionation of 125I-CYP binding activity and cAMP induction in HuR knockdown and control cells. A, distribution of 125I-CYP binding activity in light and heavy membrane fractions in HuR knockdown and control cells. The values represent mean ± S.D. of four separate assays. Error bars indicate ± S.D.; asterisk, p = 0.002. B, immunoprecipitation (IP) of 125I-CYP binding activity from light and heavy membrane fractions using β2-AR antibody used for immunofluorescence staining. The values represent mean ± S.D. of three separate IP. Error bars indicate S.D. and asterisk, p = 0.001. C, agonist-induced cAMP generation in HuR knockdown and control cells. Statistical analysis was performed using unpaired t test (two-tailed p value is given).
We extended these studies by measuring the cell surface-associated receptors using a hydrophilic β-antagonist, [3H]CGP-12177. The number of receptors present at the cell surface as defined by the binding of [3H]CGP-12177 was ∼45% lower in cells expressing HuR-specific shRNA as compared with cells expressing nonspecific shRNA (data not shown). Thus, the results of immunofluorescence staining, cell fractionation, and IP of β2-AR using light and heavy membrane fractions, as well as measurement of cell surface receptors using a hydrophilic ligand, all suggest that HuR knockdown results in defective receptor trafficking to the plasma membrane. To test the functional coupling of the receptors expressed in HuR knockdown cells to G proteins, whole-cell adenylyl cyclase activity was measured. HuR knockdown and the appearance of receptors around the nucleus significantly reduced the amount of cAMP induced in response to β2-AR agonist isoproterenol treatment (Fig. 6C).
β2-AR mRNA and Cytoplasmic HuR Are Localized to the Cell Periphery in DDT1-MF2 Cells, and HuR Knockdown Resulted in Failed mRNA Trafficking
Knockdown of the nucleocytoplasmic shuttling RNA-binding protein, HuR, resulted in increased translation of β2-AR mRNA and defective trafficking of β2-AR to the plasma membrane. Based on these results, we hypothesize that HuR is involved in β2-AR mRNA translational silencing and localization in the cytoplasm. A well-accepted paradigm of translation-independent mRNA localization is that specific RNA-binding proteins associate with mRNA in the nucleus, rendering the mRNA translationally inactive when the mRNA-protein complex reaches the cytoplasm (9). To validate our hypothesis, we performed fluorescent in situ hybridization (FISH) analysis to localize endogenous β2-AR mRNA in DDT1-MF2 cells (Fig. 7A). β2-AR mRNA was localized to the peripheral cytoplasmic region of these cells. Immunofluorescence staining of endogenous HuR combined with FISH analysis showed nonrandom distribution and co-localization of β2-AR mRNA and cytoplasmic HuR to the peripheral cytoplasmic regions of DDT1-MF2 cells (Fig. 7A, arrows). To test the co-localization of β2-AR mRNA and HuR in polarized cells, we used actively growing DDT1-MF2 cells and performed FISH analyses, combined with immunofluorescence staining of HuR protein. β2-AR mRNA and HuR are co-localized to the leading edge of the growth cones (Fig. 7B) suggesting active transport of β2-AR mRNA. These results also support the role of HuR in β2-AR mRNA trafficking.
FIGURE 7.

β2-AR mRNA is co-localized with cytoplasmic HuR at the cell periphery and HuR knockdown results in defective trafficking. A, confocal images of DDT1-MF2 cells show FISH analysis using digoxigenin-labeled riboprobes directed against β2-AR mRNA and immunofluorescence staining of HuR protein in cells expressing control shRNA. 1) DIC image; 2) nuclear stain with DRAQ5; 3) immunofluorescence staining of HuR (green); 4) β2-AR mRNA (red); 5) merged images. B, FISH analysis of β2-AR mRNA combined with immunofluorescence staining of HuR protein in DDT1-MF 2 cells with well defined growth process. C, knockdown of HuR resulted in decreased levels of β2-AR mRNA that failed to traffic to the cell periphery. The presence of HuR knockdown (inset) and control cells within the same microscopic field provides an internal control for the confocal images presented. D, enlarged view of HuR knockdown DDT1-MF2 cell shows accumulation β2-AR mRNA around nucleus. The staining pattern shown is similar in Fig. 7, A–C and D. Scale bars, 10 μm.
To directly test the role of HuR in β2-AR mRNA localization, we co-cultured HuR knockdown and control cells and performed FISH analysis of β2-AR mRNA combined with immunofluorescence staining of HuR. The knockdown of HuR (Fig. 7B, inset) decreased β2-AR mRNA levels significantly, confirming our RPA data (Fig. 1B), and the mRNA appeared around the nucleus (Fig. 7, C and D, arrows). To provide an internal control for the confocal images, a cell with a significant quantity of HuR also is shown within the same microscopic field (Fig. 7C, arrowhead). These results suggest that there is a tight coupling between β2-AR mRNA translational repression and transport, and that HuR plays a critical role in this process.
DISCUSSION
Our earlier studies demonstrated that the 3′-UTR sequence and their binding proteins regulate the translational control of β2-AR mRNA (16, 17). In the present study, we provide new information related to the functional significance of the 3′-UTR binding protein, HuR, in regulating receptor expression and function. Our results suggest that HuR regulates receptor trafficking to the plasma membrane by localizing translationally silenced β2-AR mRNA to the cell periphery.
The process of mRNA localization typically utilizes cis-targeting elements and trans-recognition factors to direct the translationally silenced ribonucleoprotein particles to their cellular destinations (9). Whereas the list of known localized mRNAs has grown significantly over the past several years, the prevalence, variety, and importance of mRNA localization events are still not known. Over the years, mRNA localization has primarily been considered important in specialized biological processes such as morphogen gradient formation and asymmetric cell division as occurs in development (26). A recent study to comprehensively evaluate mRNA localization during early Drosophila embryogenesis showed tight correlation between mRNA distribution and subsequent protein localization (27). The results presented in this report describe the first example, to our knowledge, of the importance of mRNA targeting to the peripheral cytoplasmic region in GPCR trafficking to the plasma membrane.
We propose that the assembly of the β2-AR mRNA-HuR protein complex occurs in the nucleus, rendering it translationally inactive when the RNA-protein complex reaches the cytoplasm. Continued binding of HuR protein to β2-AR mRNA prevents translation until the mRNA is localized to the peripheral cytoplasmic regions. In that regard, HuR knockdown resulted in increased polyribosome association of β2-AR mRNA, suggesting a role for this protein in sequestering β2-AR mRNA away from polyribosomes. Although our results suggest increased translation of β2-AR mRNA in HuR knockdown DDT1-MF2 cells, it is also possible that an increased protein half-life for the receptor that accumulated around the nucleus in these cells could also contribute to the increased receptor expression.
β2-AR mRNA and cytoplasmic HuR co-localize to the cytoplasmic periphery, suggesting an important role for this protein in localizing translationally silenced β2-AR mRNA to the cell periphery. Thus, the appearance of β2-AR around the nucleus in HuR knockdown cells may have resulted from failed receptor mRNA trafficking in the cytoplasm and translational initiation immediately upon nuclear exit. The results of the immunofluorescence staining and FISH analysis emphasize the spatiotemporal relationship between β2-AR mRNA localization and translation and its importance in receptor trafficking to the plasma membrane. These results support our hypothesis that translocation of receptor mRNA to the cell periphery before translation is an important step in efficient plasma membrane trafficking of β2-AR. In addition to the above, increased cytoplasmic shuttling and accumulation of HuR protein in the cytoplasmic compartments observed in full-length receptor over-expressing cells, as compared with controls (Fig. 5C and supplemental Fig. S2), and cells overexpressing 3′-UTR deletion constructs (Fig. 5D and supplemental Fig. S3) also suggest an important role for HuR in β2-AR mRNA distribution and localization in the cytoplasm.
The mechanisms involved in translational de-repression of β2-AR mRNA after localization in the peripheral cytoplasmic compartments are not understood. It is possible that membrane-associated kinases may control the anchoring, complex disassembly, and translational activation of β2-AR mRNA, as has been suggested recently (28) for β-actin mRNA. Because such membrane-associated kinases are usually at the intersection of signal transduction pathways (29), these kinases can provide an important regulatory step in GPCR synthesis.
The roles for HuR in stabilizing a number of short-lived mRNAs containing an A+U-rich sequence are well documented (12, 14) and are consistent with our results of decreased β2-AR mRNA in response to HuR knockdown. However, in published studies, HuR interaction with the mRNA resulted in an increased target protein production due to increased mRNA stability (12, 19). Although our results appear contradictory to the reports above, HuR also is known to repress translation of Wnt-5a (30) and the GPCR κ opioid receptor mRNAs (31). In addition, HuR inhibits the internal ribosomal entry site-mediated translation of p27 mRNA (32). A recent study indicated that HuR is necessary for translational de-repression of a cationic amino acid transporter mRNA that is under translational repression by microRNA (33). In addition to these roles, HuR also functions as a pre-mRNA splice regulator (34) and as a negative posttranscriptional modulator of inflammation by inhibiting TNF mRNA translation (35). The translational suppression of TNF mRNA by HuR was abolished in the absence of TIA-1 suggesting a synergy between HuR and TIA-1 (35). We previously demonstrated that TIAR can bind and suppress the translation of β2-AR mRNA (17). Whether or not TIAR and HuR act in synergy to suppress the translation of β2-AR mRNA remains to be determined.
Our data further suggest that HuR mediated post-transcriptional regulation of gene expression is probably governed by the cis-elements in each independent mRNA species. We speculate that, in addition to translational silencing, HuR also functions as a coordinator in regulating the association of other RNA-binding proteins necessary for trafficking of β2-AR mRNA to the cell periphery. Thus, HuR is emerging as a multifunctional protein that regulates several post-transcriptional events. Our study identifies a new role for HuR in localization of translationally suppressed β2-AR mRNA to the cell periphery.
Little is known about the mechanisms that govern the localization of mRNAs encoding G protein-coupled receptors within intact cells, nor their importance in protein distribution. According to the current model, the partitioning of integral membrane protein-encoding mRNA to the ER requires both translation and an encoded signal sequence (4). Based on this model, the recognition of this signal sequence by the signal recognition particle (SRP) directs mRNAs encoding membrane proteins to the ER, enabling co-translational protein translocation (7). This model also suggests that the amino acid sequence of the encoded protein contains information necessary and sufficient for protein localization to the plasma membrane (8). On the basis of the results presented in this report, we propose a new model (Fig. 8) that suggests the localization of translationally silenced β2-AR mRNA to the cell periphery is an important step in receptor synthesis and function. Of particular interest in our proposed model is the separation of the two processes involving mRNA localization and protein translocation.
FIGURE 8.

Proposed model shows the importance of β2-AR mRNA localization to the cell periphery in receptor trafficking to the plasma membrane. β2-AR mRNA is recognized by the RNA-binding protein, HuR. Upon export from the nucleus, β2-AR mRNA-HuR protein complex associates with motor proteins and cytoskeletal elements and is transported to the cell periphery. Continued association of HuR protein silences translational initiation while chaperoning the mRNP complex to the cell periphery (upper half). When HuR expression is down-regulated (lower half), β2-AR mRNA translation is initiated prematurely in perinuclear polyribosomes, leading to the overproduction of receptors but defective trafficking to the plasma membrane.
Our findings, although based on a single member of the GPCR family, suggest a change in perspective, implicating that mRNA localization is a basic regulatory step in GPCR trafficking and function. Inappropriate targeting of GPCR mRNAs can lead to aberrant protein distributions within the cell that may interfere with normal regulatory pathways with potential relevance to human disease (36). This study also identifies a new unanticipated function for HuR protein as an RNA chaperone. Whether these findings are broadly applicable to other related GPCRs is currently unknown and is an interesting area of investigation.
Supplementary Material
Acknowledgments
We thank Drs. Jeffery L. Twiss of DuPont Hospital for Children Research, Wilmington, DE and Carme Gallego of Universitat de Lleida, Spain for the gift of eGFP construct. We also thank Dr. Yusuf A. Hannun (MUSC) for suggestions towards improving this manuscript.
This work was supported, in whole or in part, by grants from the National Institutes of Health (to B. G. T.) and the American Heart Association (0555470U, to B. G. T. and 0765356U, to K. J.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- GPCR
- G protein-coupled receptors
- β2-AR
- β2-adrenergic receptor
- ARE
- A+U-rich element(s)
- TIA-1
- T-cell-restricted intracellular antigen-1
- TIAR
- T-cell-restricted intracellular antigen-related protein
- IP
- immunoprecipitation
- FISH
- fluorescent in situ hybridization
- CYP
- cyanopindolol.
REFERENCES
- 1.Drake M. T., Shenoy S. K., Lefkowitz R. J. (2006) Circ. Res. 99, 570–582 [DOI] [PubMed] [Google Scholar]
- 2.Dohlman H. G., Thorner J., Caron M. G., Lefkowitz R. J. (1991) Annu. Rev. Biochem. 60, 653–688 [DOI] [PubMed] [Google Scholar]
- 3.Guan X. M., Kobilka T. S., Kobilka B. K. (1992) J. Biol. Chem. 267, 21995–21998 [PubMed] [Google Scholar]
- 4.Lingappa V. R., Blobel G. (1980) Recent Prog. Horm. Res. 36, 451–475 [DOI] [PubMed] [Google Scholar]
- 5.Wallin E., von Heijne G. (1995) Protein Eng. 8, 693–698 [DOI] [PubMed] [Google Scholar]
- 6.Köchl R., Alken M., Rutz C., Krause G., Oksche A., Rosenthal W., Schülein R. (2002) J. Biol. Chem. 277, 16131–16138 [DOI] [PubMed] [Google Scholar]
- 7.Walter P., Johnson A. E. (1994) Annu. Rev. Cell Biol. 10, 87–119 [DOI] [PubMed] [Google Scholar]
- 8.Blobel G. (2000) Chem. Bio. Chem. 1, 86–102 [DOI] [PubMed] [Google Scholar]
- 9.Czaplinski K., Singer R. H. (2006) Trends Biochem. Sci. 31, 687–693 [DOI] [PubMed] [Google Scholar]
- 10.Keene J. D. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 5–79874760 [Google Scholar]
- 11.Ma W. J., Cheng S., Campbell C., Wright A., Furneaux H. (1996) J. Biol. Chem. 271, 8144–8151 [DOI] [PubMed] [Google Scholar]
- 12.López de, Zhan M., Lal A., Yang X., Gorospe M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2987–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan X. C., Steitz J. A. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 15293–15298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan X. C., Steitz J. A. (1998) EMBO J. 17, 3448–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antic D., Keene J. D. (1998) J. Cell Sci. 111, 183–197 [DOI] [PubMed] [Google Scholar]
- 16.Subramaniam K., Chen K., Joseph K., Raymond J. R., Tholanikunnel B. G. (2004) J. Biol. Chem. 279, 27108–27115 [DOI] [PubMed] [Google Scholar]
- 17.Kandasamy K., Joseph K., Subramaniam K., Raymond J. R., Tholanikunnel B. G. (2005) J. Biol. Chem. 280, 1931–1943 [DOI] [PubMed] [Google Scholar]
- 18.Brummelkamp T. R., Bernards R., Agami R. (2002) Science 296, 550–553 [DOI] [PubMed] [Google Scholar]
- 19.Mazan-Mamczarz K., Galbán S., López de Silanes I., Martindale J. L., Atasoy U., Keene J. D., Gorospe M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8354–8359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdelmohsen K., Pullmann R., Lal A., Kim H. H., Galban S., Yang X., Blethrow J. D., Walker M., Shubert J., Gillespie D. A., Furneaux H., Gorospe M. (2007) Mol. Cell 25, 543–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tenenbaum S. A., Lager P. J., Carson C. C., Keene J. D. (2002) Methods 26, 191–198 [DOI] [PubMed] [Google Scholar]
- 22.Rands E., Candelore M. R., Cheung A. H., Hill W. S., Strader C. D., Dixon R. A. (1990) J. Biol. Chem. 265, 10759–10764 [PubMed] [Google Scholar]
- 23.Blaxall B. C., Pellett A. C., Wu S. C., Pende A., Port J. D. (2000) J. Biol. Chem. 275, 4290–4297 [DOI] [PubMed] [Google Scholar]
- 24.Kallal L., Gagnon A. W., Penn R. B., Benovic J. L. (1998) J. Biol. Chem. 273, 322–328 [DOI] [PubMed] [Google Scholar]
- 25.Aakalu G., Smith W. B., Nguyen N., Jiang C., Schuman E. M. (2001) Neuron 30, 489–502 [DOI] [PubMed] [Google Scholar]
- 26.St. Johnston D. (2005) Nat. Rev. Mol. Cell Biol. 6, 363–375 [DOI] [PubMed] [Google Scholar]
- 27.Lécuyer E., Yoshida H., Parthasarathy N., Alm C., Babak T., Cerovina T., Hughes T. R., Tomancak P., Krause H. M. (2007) Cell 131, 174–187 [DOI] [PubMed] [Google Scholar]
- 28.Hüttelmaier S., Zenklusen D., Lederer M., Dictenberg J., Lorenz M., Meng X., Bassell G. J., Condeelis J., Singer R. H. (2005) Nature 438, 512–515 [DOI] [PubMed] [Google Scholar]
- 29.Paquin N., Chartrand P. (2005) Nat. Struct. Mol. Biol. 12, 1026–1027 [DOI] [PubMed] [Google Scholar]
- 30.Leandersson K., Riesbeck K., Andersson T. (2006) Nucleic Acids Res. 34, 3988–3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bi J., Tsai N. P., Lu H. Y., Loh H. H., Wei L. N. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13810–13815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kullmann M., Göpfert U., Siewe B., Hengst L. (2002) Genes Dev. 16, 3087–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattacharyya S. N., Habermacher R., Martine U., Closs E. I., Filipowicz W. (2006) Cell 125, 1111–1124 [DOI] [PubMed] [Google Scholar]
- 34.Izquierdo J. M. (2008) J. Biol. Chem. 283, 19077–19084 [DOI] [PubMed] [Google Scholar]
- 35.Katsanou V., Papadaki O., Milatos S., Blackshear P. J., Anderson P., Kollias G., Kontoyiannis D. L. (2005) Mol. Cell. 19, 777–789 [DOI] [PubMed] [Google Scholar]
- 36.Conn P. M., Ulloa-Aguirre A., Ito J., Janovick J. A. (2007) Pharmacol. Rev. 59, 225–250 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.