

Abstract
The Saccharomyces cerevisiae protein Kes1/Osh4 is a member of the enigmatic family of oxysterol-binding proteins found throughout Eukarya united by a β-barrel structure that binds sterols and oxysterols. In this study, we determined that phosphoinositides are the major determinant in membranes that facilitate Kes1 association both in vitro and in cells. Increased expression of Kes1 in yeast cells decreased the levels of both phosphatidylinositol 4-phosphate (PI4P) and phosphatidylinositol 3-phosphate (PI3P). Phosphoinositide and sterol bindings by Kes1 were necessary for Kes1 to decrease the level of PI4P but not PI3P. Kes1 inhibited vesicular trafficking between the trans-Golgi and plasma membrane as evidenced by accumulation of the vacuolar soluble NSF attachment protein receptors Snc1 in the cytoplasmic vesicles. Sterol and phosphoinositide binding by Kes1 both contributed to its regulation of Snc1 trafficking. This study also describes a previously unknown role for Kes1 in the regulation of the autophagy/cytoplasm to the vacuole trafficking pathway. The Kes1-mediated regulation of the autophagy/cytoplasm to the vacuole trafficking pathway was prevented by increasing expression of the PI3K Vps34, suggesting that it is the Kes1-mediated decrease in PI3P level that contributes to this regulation.
Keywords: Glycerophospholipid, Inositol Phospholipid, Intracellular Trafficking, Lipid-binding Protein, Membrane Trafficking, Phosphatidylinositol, Phospholipid, Phospholipid Metabolism, Yeast Genetics
Introduction
The integrity of cells and organelles is dependent on the proper intracellular trafficking of lipids and proteins. Indeed, a defining characteristic of different organellar membranes is their lipid composition. Information regarding how organelles obtain and maintain lipids and coordinate lipid metabolism with intracellular lipid trafficking is limited. A protein family found throughout Eukarya thought to integrate lipid metabolism with lipid transport is the oxysterol-binding protein family.
The oxysterol-binding protein family of proteins is identified by a common structural domain that binds sterols and oxysterols (1, 2). The yeast Saccharomyces cerevisiae has seven oxysterol-binding protein homologues collectively known as Osh proteins (3). The inactivation of any six OSH genes in combination has no effect or mild effects on growth (3). Inactivation of all seven OSH genes results in substantially decreased growth indicating that the Osh protein family members in yeast share an overlapping essential function (3, 4). Insight into the biochemistry of Osh proteins can be extrapolated from the recently solved crystal structures of Kes1 with and without bound sterol (5). Sterol binding by Kes1 was within the core of the protein through a near complete β-barrel (5). From the crystal structure, it was suggested that the N-terminal 29 amino acids form a flexible lid that protects the sterol ligand within the central tunnel of the β-barrel from solvent. The N-terminal 29 amino acids of Kes1 are also hypothesized to form into an ArfGAP1 lipid-packing sensor motif, an α-helix that preferentially binds to highly curved membranes (6). The presence of an ArfGAP1 lipid-packing sensor-like motif within the N terminus of Kes1 provides a potential mechanism for targeting Kes1 to vesicles.
Beyond sterol binding within the core of the protein, and the potential to bind highly curved membranes via its N-terminal lid, Kes1 has been demonstrated to bind phosphoinositides (PIPs)3 on a discrete region of its surface (7–9). Consistent with a physiological role for PIP binding by Kes1 was the demonstration that inactivation of the KES1 gene relieved the defects in growth, PIP levels, and trans-Golgi to plasma membrane vesicular trafficking associated with decreased function of both the Golgi resident PI4K Pik1 (10) and the phosphatidylinositol (PI)/phosphatidylcholine (PC) transfer protein Sec14 (9–11).
This study furthers our understanding of the role lipid binding has on Kes1 function and the cellular pathways that Kes1 modulates. In this study, we define lipid-binding properties of Kes1 required for membrane association and determine that Kes1 modulates PIP levels, PIP distribution, and vesicular transport routes regulated by PIPs. We identify a bifunctional role for lipid binding by Kes1 in its regulation of PIP metabolism. Kes1-mediated inhibition of phosphatidylinositol 4-phosphate (PI4P) metabolism by Kes1 was dependent on PIP and sterol binding, although inhibition of phosphatidylinositol 3-phosphate (PI3P) metabolism was not. Defects in the Kes1-mediated regulation of PIP metabolism result in inhibition of vesicular trafficking at the Golgi. We also describe a previously unknown role for Kes1 in the regulation of the autophagy/cytoplasm to vacuole targeting (Cvt) pathway.
MATERIALS AND METHODS
Yeast Strains and Media
The S. cerevisiae strains used in this study are wild-type BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0; Euroscarf) and kes1Δ (BY4741, kes1Δ::KanMX4). Rich medium was yeast extract peptone dextrose (YEPD, 1% bacto-yeast extract, 2% bacto-peptone, 2% dextrose) or synthetic complete (SC, 0.67% bacto-yeast nitrogen base without amino acids, 2% dextrose/galactose or 1% raffinose, and nutrients as required for strain and plasmid maintenance). Minimal medium was synthetic defined (SD) with 2% glucose (2% galactose or 1% raffinose was also used as an alternative sugar source), 0.67% bacto-yeast nitrogen base without amino acids, and nutrients as required for strain and plasmid maintenance. For expression of open reading frames under control of the PGAL1 galactose-inducible promoter, raffinose (1%, SRaf) was used as the carbon source to derepress (but not induce) transcription, with the addition of galactose (2%, SGal) to induce transcription.
Plasmid Construction
A plasmid expressing KES1 with a C-terminal His6 tag, KES1-His6, was constructed by using genomic DNA as a template for PCR amplification of the KES1 open reading frame and subcloning into the pET23b (Novagen) Escherichia coli expression vector. Using HindIII and BamHI restriction enzymes, the KES1-His6 DNA fragment was subcloned into the pESC-URA and pESC-LEU S. cerevisiae expression vector placing KES1-His6 under control of a galactose (PGAL1)-inducible promoter.
Kes1 mutant proteins (Kes1K109A) and (Kes13E; R236E/K242E/K243E) were made by site-directed mutagenesis using the QuikChange II XL site-directed mutagenesis kit (Stratagene) with KES1-His6 in pESC-URA as a template. The Kes1K109A-His6 mutant was made using primers 5′-CGT AAT GAA TCC TTA GGT TCT GAG AAA GCC CCT TTG AAC CCA TTT CTA GGT GAG TTG-3′ and 5′-CAA CTC ACC TAG AAA TGG GTT CAA AGG GGC TTT CTC AGA ACC TAA GGA TTC ATT ACG-3′. This mutant possesses decreased sterol binding activity. The Kes13E-His6 mutant was made using primers 5′-CTT TGT GTT ATT GAA TTT TCA GGT GAG GGC TAC TTT TCT GGT GAG GAG AAT TCA TTC AAG GCA AGA ATT TAC-3′ and 5′-GTA AAT TCT TGC CTT GAA TGA ATT CTC CTC ACC AGA AAA GTA GCC CTC ACC TGA AAA TTC AAT AAC ACA AAG-3′.
The lid mutant (Kes12–29Δ-His6) was made by PCR amplification using the KES1-His6-pET23b as a template and the primers 5′-GGA TCC ATG CCT CCA TTC ATT TTA TCT CC-3′ and 5′-AAG CTT CAG CAG CCA ACT CAG CTT CCT TTC-3′. The amplified fragment was subcloned into the pESC-URA plasmid.
Low copy plasmids of KES1 (pRS416) along with the PIP-binding mutant were subcloned from pRS314-KES1 (a gift from Scott Emr). Wild-type KES1 and KES13E were subcloned into pRS416 using SacI and ClaI restriction enzymes. Wild-type KES1 and KES13E were subsequently subcloned into the pRS415 using SacI and XhoI from their appropriate pRS416 counterpart. KES1K109A was constructed from KES1-pRS416 using site-directed mutagenesis. Primers used were the same as mentioned above for the construction of the KES1K109A-His6-pESC-URA. Subsequently, KES1K109A was subcloned into pRS415 using SacI and XhoI restriction enzymes.
KES1
GFP-pRS415 was constructed by PCR amplification from the KES1-GFP yeast strain (Invitrogen) using primers +500 bp of the start codon 5′-AAG CTT ATT CCG TTC GCC TTT TAC TAG-3′ and between the GFP and HIS open reading frame 5′-CTC GAG GCA GGT CTG CAG CGA GGA GCC-3′. The upstream primer contains the HindIII restriction enzyme site, and the downstream primer contains an XhoI site. KES1K109A-GFP-pRS415 and Kes13E-GFP-pRS415 were constructed by site-directed mutagenesis as described above. DNA sequences for all constructs were confirmed by Sanger sequencing at the Analytical Genetics Technology Centre in Toronto, Canada.
Cell Growth Determination
Cells were grown in SRaf medium with the appropriate amino acids to ensure plasmid maintenance to early logarithmic phase and transferred to SGal medium for 16 h. Cell concentration was calculated by measuring A600 nm. Cells were concentrated to 0.1 absorbance units, and a series of serial dilutions were plated on SGal medium and grown for 3 days at 25 °C.
Purification of Kes1-His6
The S. cerevisiae strain BY4741 transformed with KES1-His6-pESC-URA or the kes1Δ strain transformed with the plasmid KES1K109A-His6-pESC-URA, KES13E-His6-pESC-URA, or KES12–29Δ-His6-pESC-URA were grown overnight in SRaf-URA medium and diluted to A600 of 0.25. When cells were in logarithmic phase, galactose was added to a final concentration of 2%, and cells were grown for a further 4 h to induce protein expression. Cells were harvested and lysed in 20 mm Tris-HCl, pH 7.5, and 10% glycerol containing EDTA-free complete protease inhibitor (Roche Applied Science) by French press three times at 8,000 p.s.i. Cell extract was applied to Co2+ resin (Talon, Pierce), and the column was washed with 10 mm imidazole, 50 mm sodium phosphate, pH 7.0, 300 mm NaCl. Proteins were eluted with 150 mm imidazole, 50 mm sodium phosphate, pH 7.0, 300 mm NaCl. The Kes1-His6 protein preparations were dialyzed overnight in 50 mm Hepes, pH 7.2.
Liposome Binding Assays
Lipids were purchased from Avanti Polar Lipids (PC, phosphatidylserine, phosphatidylethanolamine, PI, PA, diacylglycerol, PI4P, and PI-4,5-P2), Echelon (PI3P and PI-3,5-P2), and Sigma (ergosterol). Acyl chain compositions of the lipids used were PC, phosphatidylserine, phosphatidylethanolamine, PA, diacylglycerol (1,2-dioleoyl), PI4P, PI-4,5-P2 (porcine brain), and PI3P, PI-3,5-P2 (1,2-dipalmitoyl).
Liposomes were prepared as described previously (12). A dried film was prepared by evaporation of a mixture of the indicated lipids followed by resuspension in 50 mm Hepes, pH 7.2, containing 120 mm potassium acetate. The liposome suspension was extruded sequentially through polycarbonate filters of 200- and 50-nm pore sizes with the Avanti mini-extruder.
Kes1-His6 binding to liposomes was analyzed using liposome flotation (12). Briefly, Kes1-His6 (0.25 μm) and liposomes (1 mm) were incubated in HKM buffer (50 mm Hepes, pH 7.2, 120 mm potassium acetate, 1 mm MgCl2, 1 mm DTT) for 5 min at room temperature. A 75% (w/v) sucrose solution (in HKM buffer) was added and mixed to the liposome/protein mixture to make a 30% final sucrose solution. HKM buffer containing 25% (w/v) sucrose was gently overlaid on top of the 30% sucrose suspension followed by the addition of HKM buffer containing no sucrose. The samples were centrifuged at 55,000 rpm for 1 h in a Beckman swinging rotor (TLS 55). The two interfacial layers were manually collected and analyzed by SDS-PAGE and Western blot for the presence of Kes1-His6 using monoclonal antibodies versus the His6 epitope.
Western Blotting
Kes1-GFP, Kes1K109A-GFP, and Kes13E-GFP were analyzed by SDS-PAGE and Western blot for the presence of Kes1-GFP using monoclonal antibodies versus GFP and versus Pgk1 to ensure equal loading.
Analysis of Phosphoinositides
Cells were grown overnight in SRaf medium to mid-logarithmic phase and shifted to SGal medium for 4 h to induce Kes1-His6 expression. Five A600 units of cells were harvested, washed, and resuspended in inositol-free medium followed by the addition of 50 μCi of myo-[2-3H]inositol (American Radiolabeled Chemicals) for 1 h. The yeast synthetic medium used contained 11 μm inositol; however, SD/inositol medium was employed during the 1-h [3H]inositol pulse to allow for radiolabel to be incorporated into the rarer PIPs. Cells were lysed using 4.5% perchloric acid containing 0.5-mm glass beads and vortexing. Further processing of extracts was done as described previously to deacylate the PIPs, and the 3H-labeled glycerophosphoinositides were separated using an anion-exchange column coupled to a high performance liquid chromatography system and quantified using an on-line radiometric detector (13).
Live Cell Imaging
Live cells were observed using a Zeiss Axiovert 200 M microscope fitted with a plan-neofluor 100× oil immersion lens. Images were captured using a Zeiss AxioCam HR using Axiovision 4.5 software. Cells were visualized using differential interference contrast, green fluorescent protein (GFP), or rhodamine (for FM4-64, N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl) pyridinium dibromide, Molecular Probes and BODIPY 493/503, Invitrogen). Images were captured using the “auto” setting for the exposure time.
The PI4P reporter contains the pleckstrin homology domain of Osh2 in tandem with GFP (GFP-2xPHOSH2) (14). The PI3P reporter contains the FYVE domain in tandem with GFP (FYVE-GFP) (15). The GFP-Snc1-containing plasmid was from the Hugh Pelham laboratory (MRC Laboratory of Molecular Biology, Cambridge, UK) and was subcloned into the plasmids pRS416 and pRS415 (16). Labeling of cells with FM4-64 was performed as described by Vida and Emr (17). In experiments using the PGAL1 promoter to drive expression of KES1, cells were grown to early logarithmic phase in SRaf and shifted to SGal medium for the indicated time points.
Electron Microscopy
Harvested cells were incubated consecutively in 1.5% KMnO4, 1% sodium periodate, and 1% NH4Cl consecutively. Embedding, image capture, and processing were performed by the Electron Microscopy Facility in the Faculty of Medicine, Dalhousie University (18).
RESULTS
Kes1 Binding to PIPs Is Required for Membrane Association
Kes1 is a soluble protein that binds phospholipids on its surface and sterols in its core. The N-terminal 29 amino acids of Kes1 form a lid that sequesters bound sterols within the core of Kes1 with this same region predicted to contain an ArfGAP1 lipid-packing sensor motif for preferential binding to membranes of high curvature (5–9, 19). To address the role of sterols, phospholipids, and curved membranes in Kes1 membrane association, Kes1 was purified, and its ability to associate with liposomes of various lipid compositions and sizes was determined.
A functional Kes1-His6 protein was expressed in S. cerevisiae cells and purified to apparent homogeneity using Co2+ affinity resin (Fig. 1A). Liposomes of 50 nm representing vesicles (the average diameter of a transport vesicle in S. cerevisiae is 35–50 nm) and 200 nm representing planar membranes were prepared. Liposomes contained PC along with 10% (mol/mol) of another phospholipid and/or 10% (mol/mol) ergosterol. Kes1-His6 bound liposomes containing all the PIPs tested, including PI4P, PI3P, PI-4,5-P2, and PI-3,5-P2 (less so than the other PIPs), as well as phosphatidic acid (PA) (Fig. 1B), but not membranes containing any other phospholipid, including PC, PI, phosphatidylethanolamine, phosphatidylserine, or diacylglycerol (the glycerolipid backbone without a phospho-headgroup). Kes1-His6 was unable to bind membranes containing 10% (mol/mol) ergosterol alone (data not shown). The addition of ergosterol to PIP-containing liposomes resulted in only a small increase in Kes1-His6 membrane association (Fig. 1B). Finally, Kes1-His6 bound 50 nm of liposomes only slightly better than 200 nm liposomes. The results indicate that the major determinant for pure Kes1-His6 association with liposomes is the presence of PIPs, and to a lesser extent PA.
FIGURE 1.

Phospholipid with a free phosphate group is required for Kes1 membrane association. A, Kes1-His6 was purified to apparent homogeneity. Purified protein sample was separated by SDS-PAGE, and proteins were stained using GelCode (Pierce). B, liposomes bound to Kes1-His6 were isolated by flotation. Kes1-His6 (0.25 μm) was assayed for binding to liposomes containing PC and 10% PI4P, PI-4,5-P2, or PA (PI3P, PI-3,5-P2, PI, phosphatidylserine, phosphatidylethanolamine, or diacylglycerol, data not shown) ± 10% ergosterol. Kes1-His6 bound liposomes containing all phosphoinositides and PA but not other lipids tested. Mutant Kes1 (Kes1K109A, Kes13E, and Kes12–29Δ) proteins were assayed for binding to liposomes containing PC + 10% PI4P (C), PC + 10% PI4P + 10% ergosterol (D). Assays contained 1 mm liposomes extruded at 200 and 50 nm. After centrifugation, the top and bottom fractions were collected and analyzed by SDS-PAGE and Western blot. The results were quantified using densitometry. Error bars show mean ± S.E. from a minimum of three different experiments using different sets of extruded liposomes and different protein preparations.
To test further the role of the various lipid binding capabilities of Kes1 in membrane association, mutant versions of Kes1-His6 unable to bind PIPs (Kes13E), with decreased sterol binding capacity (Kes1K109A) and with the N-terminal lid deleted (Kes12–29Δ), were purified to apparent homogeneity, and their ability to bind liposomes containing 10 mol % PIPs and/or ergosterol was determined. Kes13E-His6 was unable to bind PIP-containing liposomes whether ergosterol was present or not, although Kes1K109A-His6 and Kes12–29Δ-His6 bound liposomes in a manner similar to wild-type Kes1 (Fig. 1, C and D). The one exception was the Kes1K109A mutant that bound 50 nm vesicles in the absence of ergosterol much better than wild-type Kes1. The reason for this observation is not currently clear. One explanation may be that Kes1K109A cannot undergo a conformational change within the sterol-binding pocket that allows for release of Kes1-His6 from highly curved liposomes. When ergosterol is present in the membrane, Kes1K109A can bind enough ergosterol to undergo the conformational change needed to be released.
Kes1 Regulates PIP Levels
To determine whether there was a link between the ability of Kes1 to bind membranes containing PIPs in vitro and PIP metabolism in cells, PIP levels were assessed when KES1 was overexpressed. After a 4-h induction from the PGAL1 promoter (by the addition of galactose to the medium) to drive transcription of Kes1, the levels of PI3P and PI4P both decreased by ∼50%. The level of PI-4,5-P2 also decreased, although this could be due to the decrease in its precursor PI4P (Fig. 2).
FIGURE 2.

Kes1 requires PIP and sterol binding to regulate PI4P level but not PI3P. The kes1Δ strain transformed with the plasmids PGAL1-KES1-His6-pESC-URA, PGAL1-KES1-K109A-His6-pESC-URA, or PGAL1-KES13E-His6-pESC-URA were grown to mid-logarithmic phase at 25 °C. For expression of the KES1 gene from the PGAL1 promoter, 2% galactose was added to the medium for 4 h. Cells were washed in inositol-free medium and labeled with myo-[3H]inositol for 1 h. Phosphoinositides were extracted, deacylated, separated by high performance liquid chromatography and quantified using an on-line radiometric detector. Data are presented as percentage of the total number of counts/min in inositol-containing phospholipids and are expressed as mean ± S.E. of a minimum of three separate experiments.
To assess whether the ability of Kes1 to bind to sterols or PIPs contributes to its ability to regulate PIP levels, KES1K109A and KES13E were expressed from the PGAL1 promoter in cells where the endogenous KES1 gene was inactivated, and PIP levels were determined. Expression of either KES1K109A or KES13E resulted in a PI4P level between that of cells expressing an empty vector and cells expressing wild-type Kes1 (Fig. 2). In contrast, expression of either Kes1 mutant resulted in a decrease in PI3P level similar to that observed for wild-type Kes1 (Fig. 2). This suggests a bifurcation in the role of Kes1 with respect to its regulation of PIP levels. PIP and sterol binding appear to contribute to its regulation of PI4P but not PI3P level.
PIP Binding Is Required for Kes1 Golgi Association
To assess the contribution of PIP and sterol binding to Kes1 membrane association in cells, a functional GFP-tagged version of Kes1 (10) and the Kes1K109A and Kes13E mutants were placed under control of the KES1 promoter on a low copy (1–3 per cell) plasmid and transformed into kes1Δ cells. A Western blot versus the GFP tag determined all proteins were expressed at a similar level in vivo indicating the mutations did not affect Kes1 abundance (Fig. 3A). Kes1-GFP was diffused throughout the cytoplasm as well as in punctate spots (Fig. 3B) that have previously been demonstrated to be the Golgi apparatus (10). The Kes1K109A-GFP mutant was also primarily present in punctate regions in the cell; however, this was clearly not the case for Kes13E-GFP, which was much more diffuse. The intracellular localization of Kes1 mutants supports our in vitro data that PIP binding by Kes1 is the primary determinant for its localization to membranes.
FIGURE 3.

Kes1 binding to PIPs is required for membrane association in cells. KES1-GFP, KES1K109A-GFP, and KES13E-GFP are expressed under their own promoter in a low copy plasmid (1–2 copies) in a cell with an inactivated KES1 gene. A, Western blot of Kes1-GFP, Kes1K109A-GFP, and Kes13E-GFP shows equal expression of wild-type and mutant Kes1 proteins. B, cells were visualized by using fluorescence (GFP) and differential interference contrast filters.
Kes1 Regulates PIP Distribution
As we observed that increased expression of KES1 decreased PI4P and PI3P levels, we went on to assess the distribution of these PIPs using well characterized fluorescent reporters that bind PI4P (GFP-2×PHOSH2) (14) and PI3P (FYVE-GFP) (15). Re-distribution of these PIP reporters can be due to a change in the ratio of the PIP at different organelles or due to a decrease in the total PIP available for binding (14, 15). Consistent with previous observations, in wild-type cells the distribution of the PI4P reporter is partitioned between the plasma membrane and punctate regions in the cytoplasm (previously identified as Golgi) (Fig. 4A), whereas the PI3P reporter primarily decorated the vacuolar membrane and was present in small punctate spots (previously identified as endosomes) (Fig. 4B) (14, 15). Increased expression of KES1 resulted in a more diffuse localization of the PI4P binding probe (Fig. 4A). Increased expression of KES1 resulted in a decrease in PI4P level and, coupled with the fact that Kes1 is a PI4P-binding protein, both could be contributing to the decrease in the PI4P available for binding by the PI4P-binding probe. Localization of the PI3P reporter remained similar to that observed in wild-type cells (Fig. 4B).
FIGURE 4.

Sterol and PIP binding by Kes1 is required for proper PI4P distribution. Wild type BY4741 cells containing the empty vector (pESC-LEU) or the plasmid PGAL1-KES1-pESC-LEU were also transformed with the PI4P reporter (GFP-2xPHOSH2) (A) or the PI3P reporter (FYVE-GFP) (B). Cells were grown to mid-logarithmic phase in raffinose medium and transferred to galactose medium for 16 h. C, kes1Δ strain containing the plasmids KES1-pRS415, KES1K109A-pRS415, or KES13E-pRS415 was transformed with the PI4P reporter (GFP-2×PHOSH2). Cells were visualized by using fluorescence (GFP) and differential interference contrast filters.
To determine whether there is a role for PIP or sterol binding by Kes1 in altering PI4P availability, we expressed Kes1K109A and Kes13E in cells with an inactivated KES1 gene and determined the localization of the PI4P reporter (Fig. 4C). The reporter still localized to intracellular punctate spots suggesting that neither mutant can bind to Golgi PI4P at a level that significantly affects its availability. This is consistent with the increased expression of both of these mutants resulting in only a modest decrease in PI4P levels when compared with increased expression of wild-type Kes1.
Increased Levels of Kes1 Cause Defects in Distinct Vesicular Transport Pathways
The effect on PIP levels and PI4P distribution suggests a role for Kes1 in the regulation of PIP-dependent processes. PI4P is primarily synthesized by two PI4Ks, the Golgi resident Pik1 and Stt4 at the plasma membrane, with Pik1 being required for vesicular transport at the trans-Golgi (20–24). PI3P is synthesized on endosomal and pre-autophagosomal structure membranes by the PI3K Vps34 and is required for endosome trafficking to the vacuole and for the autophagy/Cvt pathway from the cytoplasm to the vacuole (25–28).
The lipophilic dye FM4-64 is incorporated into the plasma membrane and travels via endosomes to the vacuole (17). FM4-64 trafficking was examined in cells expressing KES1 from the PGAL1 promoter (Fig. 5A). FM4-64 was incorporated into the plasma membrane and reached the vacuole with similar kinetics regardless of the level of KES1. Not surprisingly, expression of the Kes1 PIP- and sterol-binding mutants also did not alter the trafficking of the lipophilic dye to the vacuole (Fig. 5A). Interestingly, although the transit of FM4-64 to the vacuole was not altered, the images did reveal that the vacuole was fragmented upon increased expression of KES1.
FIGURE 5.

Kes1 expression inhibits Snc1-GFP transport. A, kes1Δ strain transformed with the plasmids PGAL1-KES1-His6-pESC-URA, PGAL1-KES1K109A-His6-pESC-URA, or PGAL1-KES13E-His6-pESC-URA were grown to mid-logarithmic phase at 25 °C in raffinose medium and then shifted to galactose-containing medium for 16 h. Cells were labeled for 5 min with 40 μm FM4-64 and incubated post-label in fresh medium containing galactose for the indicated time points. kes1Δ strains containing plasmids PGAL1-KES1-His6-pESC-URA(B) and PGAL1-KES1K109A-His6-pESC-URA or PGAL1-KES13E-His6-pESC-URA (C) were transformed with a plasmid expressing Snc1-GFP. Cells were grown to mid-logarithmic phase at 25 °C in raffinose medium and shifted to galactose medium for 16 h and visualized.
Snc1 is a vacuolar soluble NSF attachment protein receptor routinely monitored using a functional GFP-Snc1, which cycles between the trans-Golgi and plasma membrane to facilitate continuous rounds of membrane transport/fusion. Transport of Snc1 from the Golgi to the plasma membrane is dependent on PI4P production in the Golgi. The bulk of Snc1 is normally found on the plasma membrane, at the leading edge of the bud, where vesicular transport is directed to allow for delivery of components required for membrane expansion and cell growth (29). Consistent with the overexpression of KES1 decreasing PI4P levels, we observed a disruption of GFP-Snc1 trafficking as observed through the accumulation of GFP-Snc1 intracellularly (Fig. 5B). Overexpression of the KES1K109A (decreased sterol binding) and KES13E (no PIP binding) mutants showed improved but not complete restoration of Snc1 localization to the plasma membrane of the emerging bud (Fig. 5C). The ability of Kes1 to bind to both sterols and PIPs appears to contribute to its inhibition of Snc1 trafficking.
Accumulation of Lipid Droplets and Intracellular Membranes
EM images after induction of KES1 expression were obtained, and it was evident that not only was the vacuole fragmenting (as was evident from FM4-64 treatment) but also membranous structures were accumulating in the cytoplasm (Fig. 6A). After 4 h of KES1 induction from the PGAL1 promoter, the membrane accumulation defect was prominent. Temporally, these coincide with the decreased levels of PI4P and PI3P. The EM also showed that there appeared to be lipid droplet accumulation when KES1 was overexpressed, with their identity as lipid droplets confirmed using the neutral lipid staining dye BODIPY 493/503 (Fig. 6B) (30, 31).
FIGURE 6.

Increased Kes1 causes vacuole fragmentation, lipid droplet formation, and membrane accumulation. A, wild type strain BY4741 was transformed with the empty vector pESC-URA, and the kes1Δ strain was transformed with the plasmid PGAL1-KES1-His6-pESC-URA. Cells were grown to mid-logarithmic phase at 25 °C in raffinose medium and shifted to galactose medium for 4 or 16 h and then processed and visualized by electron microscopy. B, kes1Δ strain transformed with PGAL1-KES1-His6-pESC-URA was grown to mid-logarithmic phase at 25 °C in raffinose medium and shifted to galactose-containing medium for 16 h. Cells were stained with 50 μm BODIPY 493/503 for 30 min, washed with PBS, and visualized using differential contrast and fluorescence microscopy (RFP). C, kes1Δ strain transformed with PGAL1-KES1-His6-pESC-URA, PGAL1-KES1K109A-His6-pESC-URA, or PGAL1-KES13E-His6-pESC-URA were grown to mid-logarithmic phase at 25 °C in raffinose medium and shifted to galactose medium for 16 h. Cells were concentrated to 0.1 absorbance units, and a series of serial dilutions were plated on SGal medium and grown for 3 days at 25 °C. D, kes1Δ strain transformed with PGAL1-KES1K109A-His6-pESC-URA or PGAL1-KES13E-His6-pESC-URA was grown to mid-logarithmic phase at 25 °C in raffinose medium and shifted to galactose medium for 16 h before being prepared for electron microscopy. Abbreviations used are as follows: V, vacuole; LD, lipid droplet.
A growth curve in liquid medium after induction of KES1 expression from the PGAL1 promoter, by shifting cells to galactose-containing medium, resulted in a slowing of growth at ∼8 h of induction (data not shown). Growth arrest was apparent after 16 h, easily visualized through a growth assay on solid medium (Fig. 6C). Although cells ceased to grow, viability was not affected.
To correlate the known lipid-binding characteristics of Kes1 with the growth arrest and membrane lipid droplet anomalies, we assessed if defects in the ability of Kes1 to bind either of these lipids would cause fewer defects. Increased expression of Kes1 with mutations in PIP or sterol binding reduced cell growth arrest (Fig. 6C). However, vacuole fragmentation, lipid droplet, and membrane accumulation were still present (Fig. 6D).
Inactivation of the Autophagy/Cvt Pathway Prevents Kes1-dependent Intracellular Membrane Accumulation
To attempt to determine the pathway(s) resulting in membrane accumulation in cells overexpressing Kes1, we performed a systematic genetic array screen (29, 32–34) versus all (∼5000) nonessential S. cerevisiae genes. The goal was to identify genes whose inactivation allowed for a reprieve from the growth inhibition associated with expression of KES1 from the PGAL1 promoter. Of genes known to affect membrane trafficking, only those required for the autophagy/Cvt trafficking pathway restored growth and included the following: ATG24/SNX4, a sorting nexin that has a PX domain that binds to PIPs (35); ATG9, involved in membrane delivery to the PAS (36–39); and ATG22, a vacuolar integral membrane protein involved in amino acid efflux from the vacuole (36, 40, 41). The bulk of the remaining genes identified were transcription factors and proteins involved in protein stability that most likely were affecting Kes1 abundance (supplemental Table 1).
Most components of the autophagy and Cvt pathways are common to both, including the PI3K Vps34. However, some are specific for each pathway (42, 43). ATG24, identified in the SGA screen, is thought to be specific for the Cvt pathway. To elucidate if inactivation of autophagy, the Cvt pathway, or both affects membrane accumulation upon expression of KES1, genes known to be specific to each pathway (ATG17, ATG31, and ATG29 for autophagy and ATG11, ATG19, and ATG24 for the Cvt pathway) were inactivated, and EM images were examined. Inactivation of either ATG17 or ATG24 ameliorated the membrane accumulation defect associated with increased expression of Kes1 (Fig. 7). Inactivation of ATG11 also appears to ameliorate the membrane accumulation defects, although it may not be to the same extent as ATG17 or ATG24 inactivation.
FIGURE 7.
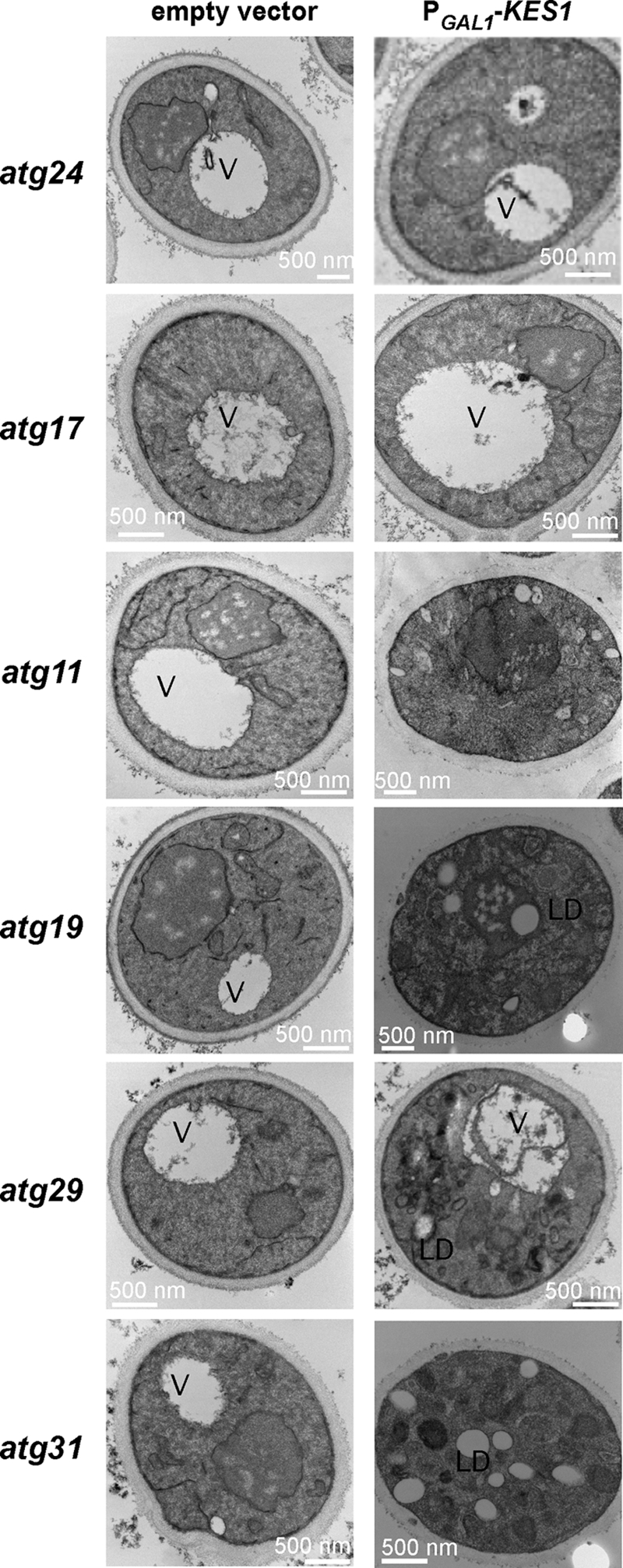
Kes1 causes membrane and lipid droplet accumulation. The empty vector pESC-URA or the plasmid PGAL1-KES1-His6-pESC-URA was transformed into yeast strains with inactivated genes specific for autophagy (atg17Δ, atg29Δ, and atg31Δ) or the Cvt pathway (atg11Δ, atg19Δ, and atg24Δ). Cells were grown to mid-logarithmic phase at 25 °C in raffinose medium and shifted to galactose medium for 16 h prior to visualization by electron microscopy. Abbreviations used are as follows: V, vacuole; LD, lipid droplet.
Increased Expression of the PI3K Vps34 Prevents Kes1-dependent Membrane Accumulation
The autophagy/Cvt pathways require the assembly of the PI3K Vps34 into a specific macromolecular protein complex. To assess if the decrease in PI3P level, observed upon increased expression of Kes1, could be mediating the membrane and lipid droplet accumulation, the VPS34 gene was introduced on a high copy plasmid (∼20–40 copies per cell) into cells where Kes1 expression was increased. Increased expression of VPS34 ameliorated the membrane and lipid droplet accumulation (Fig. 8), although increased expression of the Golgi resident PI4K Pik1 did not. Lipid droplets are required for the progression of autophagy with inhibition of autophagy resulting in lipid droplet build up (44). The increase in lipid droplet formation appears to be due to the decrease in PI3P leading to inhibition of the autophagy/Cvt pathway. The data are consistent with a Kes1-mediated decrease in PI3P levels contributing to the regulation of autophagy/Cvt.
FIGURE 8.

Increased expression of the PI3K, Vps34, prevents membrane and accumulation defects associated with expression of KES1. The plasmid PGAL1-KES1-pESC-LEU was transformed into a kes1Δ yeast strain expressing either the PIK1 or VPS34 gene on a high copy plasmid. The yeast strains were grown to mid-logarithmic phase at 25 °C in raffinose medium and shifted to galactose-containing medium for 16 h prior to visualization by electron microscopy. Abbreviations used are as follows: V, vacuole; LD, lipid droplet.
DISCUSSION
This study determined that PIPs are a major determinant in membranes that facilitate Kes1 association. Kes1 affected PIP metabolism as increased expression of Kes1 decreased the level of both PI4P and PI3P. Kes1 expression inhibited cycling of the vacuolar soluble NSF attachment protein receptors Snc1 between the trans-Golgi and plasma membrane. This is in line with previous work that implied Kes1 inhibited vesicular trafficking pathways mediated by the Golgi PI4K Pik1 and the PI/PC transfer protein Sec14 (9, 10). Increased expression of KES1 also resulted in a decreased ability of a PI4P-binding probe to associate with Golgi membranes. As increased expression of Kes1 resulted in both a decrease in PI4P level, and as Kes1 itself is a PI4P-binding protein, both properties could be contributing to the inability of the PI4P-binding probe to associate with membranes. A role for Kes1 in affecting binding of yeast proteins that use PI4P to target to specific membranes is a real possibility that should be explored.
The PIP and sterol-binding properties of Kes1 contributed to the capacity of Kes1 to decrease PI4P levels but not PI3P. As sterol and PIP binding by Kes1 both contributed to its regulation of PI4P levels, this implies that both properties could be required to affect the activity of an enzyme that metabolizes PI4P. Possibilities include inhibition of the PI4K Pik1 or activation of a PIP phosphatase such as Sac1.
This study also described a role for Kes1 in regulation of the autophagy/Cvt pathway that could be prevented by increasing expression of the PI3K Vps34 but not the PI4K Pik1. The data suggest that the Kes1-mediated decrease in PI3P levels contributes to its regulation of autophagy. The PI3K-dependent step, as well as four of the five genes whose inactivation prevented Kes1-mediated membrane and lipid droplet accumulation, ATG9, ATG11, ATG17, and ATG24, are involved in formation of the PAS membrane (or cargo assembly at the PAS) pointing to this step as a possible site of action for Kes1-mediated regulation of this pathway. The sole PI3K in yeast, Vps34, is associated with two protein complexes, one of which synthesizes PI3P for endosome function, with the other being required for Cvt/autophagy function. Kes1 could inhibit the ability of Vps34 to either assemble with the Cvt-autophagy complex or catalyze the synthesis of PI3P required for complex assembly.
Recent work has linked aspects of vesicular trafficking with autophagy. A role for the Golgi in autophagy was proposed when it was observed that the activation of the small G proteins Arf1 and Arf2 by the GTPases Sec7, Gea1, and Gea2 is essential for autophagy to occur (45). Cells lacking Sec7 function can undergo autophagy under starvation conditions, indicating that the autophagy machinery can be assembled at the PAS. Nevertheless, the expansion of the precursor membrane cisterna from the PAS to autophagosomes is impaired. This suggests that the Golgi complex provides lipids for the biogenesis of autophagy vesicles (45). Another study determined that a third form of the TRAPP complex, containing Trs85 that is a guanine nucleotide exchange factor for Ypt1, localizes to the PAS and plays a direct role in the Cvt and autophagy pathways (46, 47). The TRAPPIII complex targets its guanine nucleotide exchange factor activity for Ypt1 at the PAS indicating that certain TRAPP subunits can direct core guanine nucleotide exchange factor components to different parts of the cells (47). Previous studies determined that PI4P at the trans-Golgi is crucial for the downstream TRAPPII complex, which Kes1 regulates (10). Kes1 inhibition of trans-Golgi-endosome trafficking could in turn regulate the autophagy/Cvt pathway. However, the role of Kes1 in regulation of autophagy (and PI3P level) was independent of sterol and PIP binding, both of which were required for its regulation of trans-Golgi trafficking. How Kes1 regulates the autophagy/Cvt pathway and whether it is dependent on trans-Golgi trafficking needs to be determined.
Kes1 is a soluble protein that binds PIP-enriched membranes and extracts sterols from membranes into the core of the protein. The role of sterol binding by Kes1 is still unclear, although this study implies that sterol binding by Kes1 influences PI4P metabolism and vesicular trafficking. What is clear is that sterol binding by Kes1 does not appear to be a major influence on its membrane association. Possibilities for roles for sterol binding by Kes1 that deserve further investigation include the effect it may have on Kes1 association with other proteins (and in turn affecting function of specific proteins such as the PI4K Pik1 or the PI4P phosphatase Sac1). As PIPs and sterols are transported from the trans-Golgi to other sites in the cell, Kes1, through its PIP- and sterol-binding properties, could act to coordinate membrane transport according to a specific lipid environment.
Supplementary Material
Acknowledgment
We thank Christopher Stefan of Cornell University for assistance with PIP determinations.
This work was supported in part by a discovery award from the National Sciences and Engineering Research Council (to C. R. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Refs. 48–91.
- PIP
- phosphoinositide
- Cvt
- cytoplasm to vacuole trafficking
- PA
- phosphatidic acid
- PAS
- pre-autophagosomal structure
- PC
- phosphatidylcholine
- PI
- phosphatidylinositol
- PI3P
- phosphatidylinositol 3-phosphate
- PI4P
- phosphatidylinositol 4-phosphate
- PI-3,5-P2
- phosphatidylinositol 3,5-bisphosphate
- PI-4,5-P2
- phosphatidylinositol 4,5-bisphosphate
- Raf
- raffinose
- FM4-64
- N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl) pyridinium dibromide
- Gal
- galactose.
REFERENCES
- 1.Fairn G. D., McMaster C. R. (2008) Cell. Mol. Life Sci. 65, 228–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raychauduri J., Prinz W. (2010) Annu. Rev. Cell Dev. Biol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beh C. T., Cool L., Phillips J., Rine J. (2001) Genetics 157, 1117–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beh C. T., Rine J. (2004) J. Cell Sci. 117, 2983–2996 [DOI] [PubMed] [Google Scholar]
- 5.Im Y. J., Raychaudhuri S., Prinz W. A., Hurley J. H. (2005) Nature 437, 154–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drin G., Casella J. F., Gautier R., Boehmer T., Schwartz T. U., Antonny B. (2007) Nat. Struct. Mol. Biol. 14, 138–146 [DOI] [PubMed] [Google Scholar]
- 7.Fairn G. D., McMaster C. R. (2005) Biochem. J. 387, 889–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knodler A., Mayinger P. (2005) BioTechniques 38, 858, 860,, 862 [DOI] [PubMed] [Google Scholar]
- 9.Li X., Rivas M. P., Fang M., Marchena J., Mehrotra B., Chaudhary A., Feng L., Prestwich G. D., Bankaitis V. A. (2002) J. Cell Biol. 157, 63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fairn G. D., Curwin A. J., Stefan C. J., McMaster C. R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15352–15357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang M., Kearns B. G., Gedvilaite A., Kagiwada S., Kearns M., Fung M. K., Bankaitis V. A. (1996) EMBO J. 15, 6447–6459 [PMC free article] [PubMed] [Google Scholar]
- 12.Bigay J., Casella J. F., Drin G., Mesmin B., Antonny B. (2005) EMBO J. 24, 2244–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stefan C. J., Audhya A., Emr S. D. (2002) Mol. Biol. Cell 13, 542–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roy A., Levine T. P. (2004) J. Biol. chem. 279, 44683–44689 [DOI] [PubMed] [Google Scholar]
- 15.Burd C. G., Emr S. D. (1998) Mol. Cell 2, 157–162 [DOI] [PubMed] [Google Scholar]
- 16.Lewis M. J., Nichols B. J., Prescianotto-Baschong C., Riezman H., Pelham H. R. (2000) Mol. Biol. Cell 11, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vida T. A., Emr S. D. (1995) J. Cell Biol. 128, 779–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright R. (2000) Microsc. Res. Tech. 51, 496–510 [DOI] [PubMed] [Google Scholar]
- 19.Xu Y., Liu Y., Ridgway N. D., McMaster C. R. (2001) J. Biol. Chem. 276, 18407–18414 [DOI] [PubMed] [Google Scholar]
- 20.Demmel L., Beck M., Klose C., Schlaitz A. L., Gloor Y., Hsu P. P., Havlis J., Shevchenko A., Krause E., Kalaidzidis Y., Walch-Solimena C. (2008) Mol. Biol. Cell 19, 1046–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faulhammer F., Kanjilal-Kolar S., Knödler A., Lo J., Lee Y., Konrad G., Mayinger P. (2007) Traffic 8, 1554–1567 [DOI] [PubMed] [Google Scholar]
- 22.Strahl T., Hama H., DeWald D. B., Thorner J. (2005) J. Cell Biol. 171, 967–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walch-Solimena C., Novick P. (1999) Nat. Cell Biol. 1, 523–525 [DOI] [PubMed] [Google Scholar]
- 24.Audhya A., Foti M., Emr S. D. (2000) Mol. Biol. Cell 11, 2673–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slessareva J. E., Routt S. M., Temple B., Bankaitis V. A., Dohlman H. G. (2006) Cell 126, 191–203 [DOI] [PubMed] [Google Scholar]
- 26.Stack J. H., DeWald D. B., Takegawa K., Emr S. D. (1995) J. Cell Biol. 129, 321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stack J. H., Herman P. K., Schu P. V., Emr S. D. (1993) EMBO J. 12, 2195–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schu P. V., Takegawa K., Fry M. J., Stack J. H., Waterfield M. D., Emr S. D. (1993) Science 260, 88–91 [DOI] [PubMed] [Google Scholar]
- 29.Curwin A. J., Fairn G. D., McMaster C. R. (2009) J. Biol. Chem. 284, 7364–7375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petschnigg J., Wolinski H., Kolb D., Zellnig G., Kurat C. F., Natter K., Kohlwein S. D. (2009) J. Biol. Chem. 284, 30981–30993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurat C. F., Wolinski H., Petschnigg J., Kaluarachchi S., Andrews B., Natter K., Kohlwein S. D. (2009) Mol. Cell 33, 53–63 [DOI] [PubMed] [Google Scholar]
- 32.Tong A. H., Evangelista M., Parsons A. B., Xu H., Bader G. D., Pagé N., Robinson M., Raghibizadeh S., Hogue C. W., Bussey H., Andrews B., Tyers M., Boone C. (2001) Science 294, 2364–2368 [DOI] [PubMed] [Google Scholar]
- 33.Tong A. H., Lesage G., Bader G. D., Ding H., Xu H., Xin X., Young J., Berriz G. F., Brost R. L., Chang M., Chen Y., Cheng X., Chua G., Friesen H., Goldberg D. S., Haynes J., Humphries C., He G., Hussein S., Ke L., Krogan N., Li Z., Levinson J. N., Lu H., Ménard P., Munyana C., Parsons A. B., Ryan O., Tonikian R., Roberts T., Sdicu A. M., Shapiro J., Sheikh B., Suter B., Wong S. L., Zhang L. V., Zhu H., Burd C. G., Munro S., Sander C., Rine J., Greenblatt J., Peter M., Bretscher A., Bell G., Roth F. P., Brown G. W., Andrews B., Bussey H., Boone C. (2004) Science 303, 808–813 [DOI] [PubMed] [Google Scholar]
- 34.Sopko R., Huang D., Preston N., Chua G., Papp B., Kafadar K., Snyder M., Oliver S. G., Cyert M., Hughes T. R., Boone C., Andrews B. (2006) Mol. Cell 21, 319–330 [DOI] [PubMed] [Google Scholar]
- 35.Teasdale R. D., Loci D., Houghton F., Karlsson L., Gleeson P. A. (2001) Biochem. J. 358, 7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukada M., Ohsumi Y. (1993) FEBS Lett. 333, 169–174 [DOI] [PubMed] [Google Scholar]
- 37.Reggiori F., Tucker K. A., Stromhaug P. E., Klionsky D. J. (2004) Dev. Cell 6, 79–90 [DOI] [PubMed] [Google Scholar]
- 38.Noda T., Kim J., Huang W. P., Baba M., Tokunaga C., Ohsumi Y., Klionsky D. J. (2000) J. Cell Biol. 148, 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Legakis J. E., Yen W. L., Klionsky D. J. (2007) Autophagy 3, 422–432 [DOI] [PubMed] [Google Scholar]
- 40.Yang Z., Huang J., Geng J., Nair U., Klionsky D. J. (2006) Mol. Biol. Cell 17, 5094–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suriapranata I., Epple U. D., Bernreuther D., Bredschneider M., Sovarasteanu K., Thumm M. (2000) J. Cell Sci. 113, 4025–4033 [DOI] [PubMed] [Google Scholar]
- 42.Nair U., Klionsky D. J. (2005) J. Biol. Chem. 280, 41785–41788 [DOI] [PubMed] [Google Scholar]
- 43.Xie Z., Klionsky D. J. (2007) Nat. Cell Biol. 9, 1102–1109 [DOI] [PubMed] [Google Scholar]
- 44.Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A. M., Czaja M. J. (2009) Nature 458, 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Vaart A., Griffith J., Reggiori F. (2010) Mol. Biol. Cell 21, 2270–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nazarko T. Y., Huang J., Nicaud J. M., Klionsky D. J., Sibirny A. A. (2005) Autophagy 1, 37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lynch-Day M. A., Bhandari D., Menon S., Huang J., Cai H., Bartholomew C. R., Brumell J. H., Ferro-Novick S., Klionsky D. J. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 7811–7816 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.