

Abstract
Thiamine diphosphate (ThDP)-dependent enzymes are ubiquitously present in all organisms and catalyze essential reactions in various metabolic pathways. ThDP-dependent phosphoketolase plays key roles in the central metabolism of heterofermentative bacteria and in the pentose catabolism of various microbes. In particular, bifidobacteria, representatives of beneficial commensal bacteria, have an effective glycolytic pathway called bifid shunt in which 2.5 mol of ATP are produced per glucose. Phosphoketolase catalyzes two steps in the bifid shunt because of its dual-substrate specificity; they are phosphorolytic cleavage of fructose 6-phosphate or xylulose 5-phosphate to produce aldose phosphate, acetyl phosphate, and H2O. The phosphoketolase reaction is different from other well studied ThDP-dependent enzymes because it involves a dehydration step. Although phosphoketolase was discovered more than 50 years ago, its three-dimensional structure remains unclear. In this study we report the crystal structures of xylulose 5-phosphate/fructose 6-phosphate phosphoketolase from Bifidobacterium breve. The structures of the two intermediates before and after dehydration (α,β-dihydroxyethyl ThDP and 2-acetyl-ThDP) and complex with inorganic phosphate give an insight into the mechanism of each step of the enzymatic reaction.
Keywords: Bacterial Metabolism, Enzyme Mechanisms, Enzyme Structure, Thiamine, X-ray Crystallography, Acetyl Phosphate, Bifidobacteria, Dehydration, Phosphoketolase, Transketolase
Introduction
Bifidobacteria represent a ubiquitous commensal bacterial group in the gastrointestinal tract of humans and animals (1). A unique central hexose fermentation pathway of bifidobacteria is called the “bifid shunt,” which is summarized in the following scheme (2).
SCHEME 1.

Therefore, ATP production by the bifid shunt is 1.25-fold more effective than that by lactic acid fermentation in the well known Embden-Meyerhof glycolytic pathway (2 ATP per glucose). Two thiamine diphosphate (ThDP)3-dependent enzymes, transketolase (TK) and phosphoketolase (PK), catalyze key steps of the bifid shunt (Fig. 1). The structure and mechanism of TK has been extensively studied (3, 4). However, the three-dimensional structure and reaction mechanism of PK has long been enigmatic, although PK was discovered in 1958 (5–7).
FIGURE 1.

The central metabolism of bifidobacteria, the bifid shunt. XFPK is involved in two steps of the bifid shunt.
ThDP is a biologically active form of vitamin B1. ThDP-dependent enzymes are ubiquitously present in all organisms and catalyze various essential reactions in metabolic pathways. These enzymes generally catalyze the conversion of 2-keto acid and require ThDP and divalent cations as cofactors. Formation of ThDP ylide, which is accomplished by deprotonation of the C2 atom on the thiazolium ring, is the first essential activation step (Fig. 2) (8, 9). ThDP-dependent enzymes are divided into four families based on their primary and tertiary structures (10). Three of the four families catalyze oxidative decarboxylation reactions to produce biologically essential metabolites such as acetyl-CoA (9, 11, 12). Acetyl phosphate (acetyl-P)-producing pyruvate oxidase (POX), which belongs to one of the oxidative decarboxylation-catalyzing families, cleaves pyruvate in the presence of inorganic phosphate (Pi) and O2 to produce acetyl-P, CO2, and H2O2 (Fig. 2) (13). TK is a representative member of the fourth family (known as the TK family). The reactions of enzymes belonging to this family are significantly different from those belonging to other families because the TK family enzymes catalyze non-oxidative reactions. TK catalyzes the cleavage of a C-C bond in ketose phosphate (donor substrate) into a two-carbon fragment and aldose phosphate. This cleavage is subsequently followed by condensation of the ThDP-bound two-carbon fragment with aldose (acceptor substrate).
FIGURE 2.

Reaction mechanisms of TK, PK, and acetyl-P-producing POX. The first-half reactions of XFPK and TK are identical, but the second-half- reactions diverge. The reaction catalyzed by POX involves oxidation and radical chemistry. Pyr represents the aminopyrimidine ring of ThDP. Protonated DHEThDP (S-α,β-dihydroxyethyl-ThDP), which is likely the state observed in our crystal structure, is also shown.
PK belongs to the TK family, and there are two types of enzymes with different preferences for sugar phosphate substrates exist. Xylulose-5-phosphate PK (XPK, EC 4.1.2.9) prefers xylulose 5-phosphate (X5P) to fructose 6-phosphate (F6P) (5, 6), whereas X5P/F6P PK (XFPK, EC 4.1.2.22) acts on both X5P and F6P with comparable activities (7, 14–16). These enzymes constitute two homologous (sequence identity > 40%) but phylogenetically distinct groups (supplemental Figs. 1 and 2 and supplemental Table 1) (17). PKs catalyze the cleavage of X5P or F6P (donor) utilizing Pi as the acceptor (phosphorolysis) to produce acetyl-P, water, and glyceraldehyde 3-phosphate or erythrose 4-phosphate.
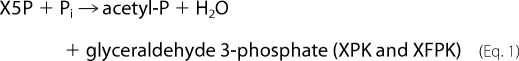 |
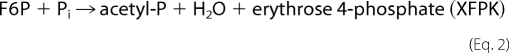 |
The XFPK-type gene has only been found in bifidobacteria, and XFPK catalyzes two steps in the bifid shunt because of its dual-substrate specificity (Fig. 1). Because PK activity against F6P is specific to the bifid shunt, it is employed as the most reliable non-molecular test for identification of bifidobacteria (18, 19).
XPK is a key enzyme in the pentose catabolism in various microbes, including filamentous fungi and yeasts (17). This pentose catabolism is called the “PK pathway” (20). Furthermore, the PK pathway is the central pathway in the metabolism of heterofermentative lactic acid bacteria including the genera Lactobacillus and Leuconostoc. Heterofermentative lactic acid bacteria, representatives of beneficial gut microbes (probiotics), produce lactic and acetic acids as the main end products. These short-chain fatty acids are important for hosts, not only because they prevent the growth of harmful bacteria by lowering the intestinal pH but also because they serve as an energy source for intestinal epithelial cells. In addition, short-chain fatty acids can modulate intestinal immune and inflammatory responses via G-protein-coupled receptors (21).
Recently, kinetic analysis of PK-2 from Lactobacillus plantarum, belonging to the XPK group, revealed that the reaction follows a ping-pong bi-bi mechanism, leading to the proposal that the first-half of the reaction by PK proceeds through the same mechanism as TK and forms an α,β-dihydroxyethyl ThDP (DHEThDP) intermediate (Fig. 2) (22). However, the subsequent reaction catalyzed by PK is distinct from TK at the following two points; 1) the dehydration reaction occurs, and 2) the acceptor substrate Pi is believed to attack the possible 2-acetyl-ThDP (AcThDP) intermediate, similar to the case of acetyl-P-producing POX.
Recently, preliminary x-ray crystallographic studies of XPK from Lactococcus lactis (23) and XFPK from Bifidobacterium breve 203 (BbXFPK) (24) are reported. In this study we report the crystal structure of BbXFPK as the first three-dimensional structure of PK. The structures of the resting form, the two intermediates before and after dehydration (DHEThDP and AcThDP), the complex with the acceptor substrate Pi, and four mutant enzymes have been determined. This study revealed a structural basis for the reaction mechanism of a ThDP-dependent enzyme involving dehydration and nucleophilic attack of Pi to the AcThDP intermediate.
EXPERIMENTAL PROCEDURES
Enzyme Preparation and Assay
Construction of the expression vector, protein expression, purification, and kinetic analysis employing F6P as the donor substrate were performed as described previously (24). Mutants of BbXFPK were constructed using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) using the primers summarized in supplemental Table 2.
Crystallography
Native (resting) BbXFPK and its mutants were crystallized according to the method described previously (24). Structures of AcThDP intermediate (BbXFPK/AcThDP), DHEThDP intermediate (BbXFPK/DHEThDP), and BbXFPK in complex with the acceptor substrate Pi (BbXFPK/Pi) were trapped using cryocrystallography technique. Crystals of BbXFPK/AcThDP were prepared by soaking native BbXFPK crystals in mother liquor (24% (v/v) PEG 6000, 0.1 m Bicine buffer, pH 9.0) containing 27 mm F6P for various time periods at room temperature. BbXFPK/DHEThDP crystals were obtained by soaking in 54 mm F6P solution for 15 s. Crystals of BbXFPK/Pi were obtained by cocrystallization with 5 mm potassium phosphate. The mother liquor containing 20% ethylene glycol was used as a cryoprotectant, except for the BbXFPK/Pi complex for which 20% glycerol was used. Selenomethionine-labeled enzyme was expressed in Escherichia coli B834 (DE3) (Novagen, Madison, WI). Diffraction data were collected using beamlines at SPring-8 (Harima, Japan) and Photon Factory (Tsukuba, Japan). Diffraction data were processed using HKL2000 (25). Initial phases were calculated using SnB (26) and Solve/Resolve (27). Initial model building was performed using ARP/wARP (28). Manual model rebuilding and refinement was achieved using Coot (29) and REFMAC5 (30). Parameters for refinement were generated by the PRODRG server (31). Details of data collection and refinement statistics are given in Table 1 and supplemental Table 3. Figures were prepared using PyMol (DeLano Scientific, Palo Alto, CA).
TABLE 1.
Data collection and refinement statistics
SAD, single anomalous diffraction; r.m.s.d., root mean square deviation.
| Data set | Native resting | Native/AcThDP | Native/DHEThDP | Native/Pi | SAD peak |
|---|---|---|---|---|---|
| Data collection statistics | |||||
| Space group | I422 | ||||
| Unit cell (Å) | a = 174.8 | a = 174.4 | a = 173.5 | a = 173.9 | a = 174.0 |
| b = 174.8 | b = 174.4 | b = 173.5 | b = 173.9 | b = 174.0 | |
| c = 163.8 | c = 163.8 | c = 163.5 | c = 163.5 | c = 163.8 | |
| Beam line | PFAR-NE3A | PF-BL5A | PFAR-NW12A | PF-BL6A | Spring-8 BL38B1 |
| Wavelength (Å) | 1.00000 | 1.00000 | 1.00000 | 0.97800 | 0.97875 |
| Resolution (Å)a | 50-1.70 (1.73-1.70) | 50-1.90 (1.93-1.90) | 50-2.10 (2.14-2.10) | 50-2.30 (2.38-2.30) | 50-2.60 (2.69-2.60) |
| Total reflections | 2,049,689 | 1,442,105 | 1,079,159 | 633,806 | 575,648 |
| Unique reflections | 137,590 | 98,940 | 72,417 | 55,669 | 38,884 |
| Completeness (%)a | 100 (100) | 99.9 (100) | 100 (100) | 100 (100) | 99.9 (100) |
| Rmerge (%)a | 6.5 (32.2) | 7.0 (34.9) | 7.1 (29.8) | 10.8 (35.5) | 6.7 (22.1) |
| I/σIa | 49.6 (7.4) | 52.6 (9.0) | 42.8 (11.0) | 24.9 (4.7) | 32.9 (9.4) |
| Redundancya | 14.9 (14.8) | 14.6 (14.7) | 14.9 (15.0) | 11.4 (10.4) | 7.8 (7.7) |
| Refinement statistics | |||||
| Resolution range (Å) | 34.18-1.70 | 34.21-1.90 | 49.07-2.10 | 40.88-2.30 | |
| No. of reflections | 130,627 | 93,681 | 68,524 | 52,685 | |
| R-factor/Rfree (%)b | 15.0/18.1 | 16.2/19.7 | 16.1/20.6 | 17.7/22.6 | |
| r.m.s.d. from ideal | |||||
| Bond lengths (Å) | 0.037 | 0.030 | 0.028 | 0.023 | |
| Bond angles (°) | 2.722 | 2.212 | 2.066 | 1.904 | |
| Average B-factor (Å2) | |||||
| Protein | 18.1 | 22.7 | 20.9 | 28.1 | |
| Water | 30.2 | 33.4 | 32.1 | 32.6 | |
| ThDP | 16.5 | 18.9 | 19.1 | 20.9 | |
| Mg2+ | 13.2 | 16.1 | 15.4 | 21.2 | |
| Ramachandran plot (%)c | |||||
| Favored | 96.6 | 96.6 | 96.1 | 95.5 | |
| Allowed | 3.4 | 3.4 | 3.9 | 4.4 | |
| Outlier | 0.0 | 0.0 | 0.0 | 0.1 | |
a Values for highest resolution shell are given in parentheses.
bRfree actor was calculated using 5% of the unique reflections.
c Calculated by RAMPAGE (45).
RESULTS
Overall Structure
The crystal structures (residues 5–806) of the resting form, the AcThDP intermediate, the DHEThDP intermediate, and the complex with Pi were determined at 1.7, 1.9, 2.1, and 2.3 Å, respectively (Table 1). The dimeric and monomeric structures of BbXFPK are shown in Fig. 3, A and B. BbXFPK consists of three α/β-fold domains; N-terminal (PP domain, residues 5–378), middle (Pyr domain, residues 379–611), and C-terminal (residues 612–806) domains. The crystals contain one molecule per asymmetric unit, and a crystallographic 2-fold axis is consistent with the molecular axis of the dimer. The active site is positioned at the interface between the PP and Pyr domains from different subunits to form a deep and narrow substrate channel, and the reactive C2 atom of ThDP is only accessible from the solvent. These observations suggest that the minimum functional unit of the enzyme is a homodimer. The overall architectures of the monomer and the tightly packed homodimer of BbXFPK are basically similar to those of TK (Fig. 3C) (3), with a root mean square deviation of 2.6 Å for 545 α-carbon atoms. However, they share very low sequence homology, with a sequence identity of 15% based on structural alignment with TK from Saccharomyces cerevisiae (ScTK). Three active peaks of BbXFPK appeared on gel filtration chromatography with different estimated sizes (24). A sample from the major homohexameric peak successfully crystallized, whereas those from the two minor peaks (homodimer and homotetramer) failed to crystallize. We examined the crystal packing, but there was no strong interaction that possibly interconnects the dimer units (data not shown). The biological assembly might have collapsed under the crystallization condition.
FIGURE 3.

Overall structures of homodimer (A) and monomer (B) of BbXFPK and the monomer structure of ScTK (C). The N-terminal PP (blue), middle Pyr (yellow), and C-terminal (purple) domains are colored differently. ThDP molecules are shown as spheres. A, the subunit located at the distant side is shown in gray.
Structures of Resting Form, AcThDP and DHEThDP Intermediates, and the Pi Complex
In all structures determined here, ThDP binds in a typical V-conformation (Fig. 4). The pyrimidine and thiazolium rings are held mainly through the hydrophobic residues of the PP and Pyr domains (supplemental Fig. 3A). The diphosphate group of ThDP and the hexacoordinated Mg2+ are anchored to the PP domain through many interactions (supplemental Fig. 3B). Several histidine residues in the active site as well as other residues important for catalysis are completely conserved between BbXFPK and TK (discussed later).
FIGURE 4.

|Fo| − |Fc| omit electron density maps of resting ThDP (A), AcThDP (B), DHEThDP (C), Pi complex (D), and tricyclic ring form of ThDP in H64A mutant (E). The maps were contoured at 4σ (A, B, D, and E) or 3σ (C). A, a catalytically important Glu-479 residue is also shown. Interactions between the N4′ group of pyrimidine and the reactive C2 atom of the thiazolium ring and between the N1′ atom of the pyrimidine and Glu479 are shown as dotted lines. B, the inset shows planarity of the acetyl group. C, this panel is stereographic. The inset shows nonplanarity of the DHE moiety. E, the mutated Ala-64 residue is also shown.
In the initial reaction step of all ThDP-dependent enzymes, formation of reactive ThDP ylide is achieved by cofactor-assisted deprotonation of the C2 atom of the thiazolium ring by the N4′-imino group of the pyrimidine ring, and the imino tautomer is stabilized by a completely conserved Glu residue (9, 32). In the resting structure, interactions between the N4′ atom and the reactive C2 atom (2.9 Å) and between the N1′ atom of pyrimidine and the conserved Glu479 (2.7 Å) residue are present (Fig. 4A). Replacement of this residue (E479A) completely abolished the activity (Table 2). These results indicate that the initial activation mechanism of BbXFPK to form ThDP ylide is identical to that of other ThDP-dependent enzymes.
TABLE 2.
Kinetic parameters for wild-type and mutant BbXFPK
| Enzyme | Kcat | Apparent Km |
Cofactor in mutant structuresa | |
|---|---|---|---|---|
| F6P | Pi | |||
| min−1 | mm | |||
| WTb | 1,540 ± 60 | 9.7 ± 0.3 | 1.2 ± 0.2 | |
| H64A | NAc | NA | NA | Tricyclic ring form of ThDP |
| H64N | NA | NA | NA | |
| H97A | NA | NA | NA | (CDNGd) |
| H97N | NA | NA | NA | (CDNGd) |
| H142A | 12.2 ± 0.9 | 7.4 ± 0.6 | 1.7 ± 0.2 | AcThDP |
| H142N | 39.2 ± 1.2 | 7.2 ± 0.8 | 2.6 ± 0.5 | |
| H320A | NA | NA | NA | AcThDP |
| H320N | NA | NA | NA | |
| Q321A | 59.5 ± 2.5 | 4.4 ± 0.3 | 1.6 ± 0.2 | |
| S440A | 350 ± 11 | 53.5 ± 1.8 | 0.57 ± 0.04 | |
| E479A | NA | NA | NA | |
| Y501F | 197 ± 4 | 1.4 ± 0.2 | 25.5 ± 1.7 | |
| H548A | 100 ± 3 | 2.3 ± 0.7 | 0.52 ± 0.16 | |
| N549A | NA | NA | NA | |
| H553A | NA | NA | NA | ThDP |
| H553N | NA | NA | NA | |
| K605A | NA | NA | NA | |
a Crystallographic data collected after soaking in 27 mm F6P for 5 min. See supplemental Table 3 for details.
b The data are taken from (24).
c NA, no activity was detected.
d CDNG, crystals did not grow.
When we collected diffraction data from crystals soaked in 27 mm F6P for 5 min in the absence of Pi, extra electron density was observed (Fig. 4B). The density corresponds to three branched carbon or oxygen atoms and is covalently attached to the C2 atom of the thiazolium ring. The appearance of the electron density did not change when the soaking time was varied in a range from 1 to 60 min (data not shown). The central Cα atom appears to be sp2-hybridized because the map of the covalent adduct is trigonal planar (Fig. 4B, inset). Based on these observations, we concluded that the covalent adduct is an acetyl group. It has been proposed that AcThDP formed after dehydration is a stable intermediate of PK in the absence of the acceptor, Pi (22, 33). One of the two terminal atoms is located close to the NE2 atom of His-553 (2.5 Å) and the N4′ atom of the pyrimidine ring (3.2 Å) (Fig. 5A, gray). Superimposition with the ScTK-DHEThDP structure illustrates that this atom overlaps with the Cα hydroxyl group of DHEThDP (Fig. 5B), suggesting that it is an oxygen atom. Careful inspection of the heights of electron density peaks and temperature factors supported this assignment of terminal oxygen and carbon atoms. We refined the structure using the parameters of keto-AcThDP. The resolution was not sufficiently high to determine whether it is a keto or an enol form. The planar character of the Cα atom is consistent with the similar AcThDP adduct of L. plantarum POX (LpPOX) (34). A water molecule is located 3.2 Å from the terminal Cβ atom of the acetyl group (discussed later).
FIGURE 5.

Stereo view of superimposed structures of covalent ThDP adducts in BbXFPK and ScTK. Superimposition of AcThDP (gray) and DHEThDP (yellow) in BbXFPK (A), AcThDP in BbXFPK (gray) and DHEThDP in ScTK (green) (B), and DHEThDP in BbXFPK (yellow) and ScTK (green) (C). Labels for residues from the Pyr domain of the other subunit are underlined. The water molecules near the covalent adducts are shown as spheres.
Then we prepared a crystal by soaking in 54 mm F6P for a short time period (15 s), and a clear electron density peak corresponding to the Cβ hydroxyl group of DHEThDP was observed (Fig. 4C). The electron density map for the DHE moiety is relatively weak but is sufficiently clear in shape, and is interpreted as DHEThDP. An electron density peak that corresponds to the water molecule in the AcThDP structure was also observed, but the distance from the Cβ hydroxyl atom (Oβ) was abnormally close (2.0 Å). We repeatedly refined the crystal structure by changing the occupancies of these elements (the DHE moiety and the water molecule) and then examined the resultant |Fo| − |Fc| difference map. We finished the refinement by setting the occupancies to 0.7 for the water molecule (B factor = 9.8 Å2), 0.3 for the Oβ atom (B factor = 16.3 Å2), and 0.5 for the other three atoms of the DHE moiety (Cα, Oα, and Cβ; average B factor = 24.9 Å2). The average B factor of the remaining moiety of the ThDP cofactor was 18.8 Å2. Thus, this crystal was assumed to be a mixture of free ThDP with the water (0.5 fraction), DHEThDP without the water (0.3), and AcThDP with the water (0.2). This result indicates that the DHEThDP intermediate is transient under this condition. The map around the central Cα atom is not planar, suggesting that this atom has an sp3-hybridized character (Fig. 4C, inset). Because of limited resolution (2.1 Å), low occupancy (0.3), and the electron density overlaps with those of other states, the hybridization state of the Cα atom cannot be determined unambiguously. However, crystallographic refinement with parameters of the sp2 hybridized (flat) Cα atom resulted in the appearance of significant difference peaks near the Oα and Oβ atoms in the |Fo| − |Fc| map (data not shown). Observation of the sp3-hybridized Cα atom suggests that the resonance hybrid between the neutral enamine and zwitterionic α-carbanion in our DHEThDP structure is unlikely present. Instead, the structure is likely a protonated state at the Cα atom with the S-configuration (S-α,β-dihydroxyethyl-ThDP; Fig. 2, boxed with a broken line). Although the crystal structure was obtained at pH 9.0, the local environment may stabilize the protonated state. In contrast, the DHEThDP intermediate of ScTK was observed as an sp2-hybridized enamine character (Fig. 5C) (4). The terminal Cβ hydroxyl group interacts with the NE2 atom of His-142 (2.9 Å), the main chain oxygen atom of Gly-155 (2.9 Å), and the N4′ group of the pyrimidine moiety (3.2 Å). The branched Cα hydroxyl group interacts with the NE2 atom of His-553 (2.6 Å), but the N4′ atom of pyrimidine moiety is distant (3.6 Å).
The fourth structure, a complex with the acceptor substrate, Pi, was prepared by cocrystallization in the presence of Pi. A clear tetrahedral electron density peak was observed near the C2 atom of the thiazolium ring (Fig. 4D). The four structures of the wild-type BbXFPK determined in this work did not show any significant conformational changes around the active site. This observation indicates that the formation of reaction intermediates and binding of the acceptor substrate do not induce remarkable conformational change, which is consistent with previous findings in ScTK (4) and LpPOX (34).
Substrate Binding Channel of BbXFPK
The substrate binding channel of ScTK has been identified by the complex structure with erythrose 4-phosphate (35). Moreover, the crystal structures of TK from E. coli (EcTK) in complex with uncleaved donor substrates, tetrahedral covalent X5P-ThDP and F6P-ThDP adducts, have been reported (36). In the tetrahedral substrate-cofactor adducts, the C2-Cα bond between the cofactor and substrate is distorted from the planarity, and the leaving group is perpendicularly orientated to the thiazolium ring of ThDP (Fig. 6). The out-of-plane distortions of the C2-Cα bond and the perpendicular orientation of the leaving groups are considered to facilitate the elimination of the first product (9, 37). Therefore, the covalent F6P-ThDP adduct of BbXFPK is also expected to adopt a similar conformation. Superimposition of the resting BbXFPK and F6P-ThDP adduct of EcTK revealed that F6P can bind in the active site channel of BbXFPK without any steric hindrance, leading to the estimation of residues involved in substrate recognition (Fig. 6). To verify the importance of these residues in substrate recognition and catalysis, we constructed mutants of residues located in this site (His-64, His-142, His-320, Gln-321, Ser-440, His-548, Asn-549, and His-553) (Table 2). The His residues near the sugar-derived hydroxyl groups (His-64, His-142, His-320, and His-553) are completely conserved, and mutation of these residues significantly impaired the activity (discussed later). In EcTK, Arg-358, Arg-520, His-461, and Ser-385 recognize the distal phosphate group of F6P. In the X5P-ThDP adduct, the phosphate is slightly displaced, and the hydrogen bonds with Arg-358 and Arg-520 disappear. In the BbXFPK structure, only Ser-440 is conserved, but additional residues (His-548, Gln-321, and Asn-549) appear to strengthen the interactions. Mutation of these residues also significantly impaired the activity (Table 2). We expected the presence of a distinct residue conservation pattern between the XPK and XFPK groups around this region. However, the directly interacting residues are completely conserved (supplemental Fig. 1A), and we could not find any significant pattern that can predict the substrate preference to X5P and F6P. Surrounding residues that do not directly interact with the substrate may be responsible for the substrate specificity. As shown in supplemental Table 1, the substrate preferences of XFPK and XPK groups are not very significant. The XPK-type enzyme from L. plantarum prefers X5P to F6P, but the Vmax/Km ratio is only about 15.
FIGURE 6.

Stereo view of superimposition between BbXFPK and tetrahedral donor substrate adducts of EcTK. Resting BbXFPK (yellow for protein and green for cofactor), X5P-ThDP (2R8O, black), and F6P-ThDP adduct of EcTK (2R8P, gray) are shown. Residues expected to participate in F6P binding are shown as stick models. Labels in parentheses are those of EcTK.
Dehydration Mechanism
Observation of the DHEThDP and AcThDP adducts after soaking the crystals in F6P for very short and relatively long periods of time, respectively, supports the previous notion that the dehydration occurs in the absence of Pi (22, 33). The water molecule observed near AcThDP is held by four hydrogen bonds with the side chain N atoms of His-97 and His-142 and the main chain O atoms of Gly-154 and Gly-155 (Fig. 5A). The water molecule is slightly displaced in the DHEThDP structure, and the hydrogen bonds with His-142 and Gly-155 are disengaged. This site appears to be suitable for occupation by the water molecule derived from the dehydration reaction. A similar water binding site is present in ScTK, although TK does not catalyze dehydration (Fig. 5, B and C). The water molecule in ScTK is located at a significantly distant position from the Cβ hydroxyl group of DHE (3.0 Å) compared with that in BbXFPK. The Thr-115 residue in ScTK corresponding to Gly-154 in BbXFPK is significantly displaced because of the difference in the main chain trace, and hence, there is no hydrogen bond to the water molecule.
In a possible reaction mechanism of PK, a proton acceptor (B1) is required to deprotonate the C3 hydroxyl group of the tetrahedral donor substrate-ThDP adduct for the elimination of the first product (Fig. 2). The candidates for the B1 catalyst, His-64 and His-320 of BbXFPK, are positioned close to the C3 hydroxyl group of F6P-ThDP (Fig. 6). The subsequent dehydration reaction requires a proton donor (B2) to protonate the C1 hydroxyl group leaving from DHEThDP (Fig. 2). The candidates for the B2 catalyst, His-142 (2.9 Å), His-553 (3.4 Å), and the N4′ group of the pyrimidine moiety (3.2 Å), are positioned close to the Cβ hydroxyl group of DHEThDP (Fig. 5A). These His residues (His-64, His-142, His-320, and His-553) are completely conserved in all PKs (supplemental Fig. 2). Mutations of these residues completely abolished the activity except in H142A and H142N (Table 2). We also performed crystallographic analysis of H64A, H142A, H320A, and H553A mutants because the tetrahedral F6P-ThDP or DHEThDP intermediates were expected to be trapped in the B1 and B2 catalyst mutants, respectively (Fig. 2). The electron density maps of the data collected from crystals of these mutants soaked in 27 mm F6P for 5 min are shown in Fig. 4E and supplemental Fig. 4. As summarized in Table 2, we could not obtain any of these expected intermediates; only adduct-free ThDP (H64A and H553A) or AcThDP (H142A and H320A) were observed. The ThDP cofactor in H64A was in a unique tricyclic ring form (discussed below). Based on these results, His-142 and His-320 are excluded from the candidates of B1 and B2 catalysts. Although the DHEThDP intermediate was not observed in the H64A structure, His-64 is a better candidate for the B1 catalyst than His-320. In the case of ScTK, the His-64 counterpart (His-30) and the His-320 counterpart (His-263) are thought to act in concert during the catalytic step, but the latter has been assigned to be the B1 catalyst (3, 38). His-553 and the N4′ group of the pyrimidine moiety are possible B2 candidate, but the latter cannot be investigated by a mutational method. If His-553 is the B2 catalyst, the reason why an adduct-free ThDP is trapped by the H553A mutation is unclear. This residue may also be important for the preceding steps of dehydration. For example, His-553 seems to stabilize the tetrahedral donor substrate-ThDP adduct by forming an interaction with the C2 hydroxyl group (Fig. 6). In the case of ScTK, the His-553 counterpart (His-481) is thought to stabilize the transition state of the step catalyzed by B1 (38). Another candidate for the B2 catalyst is His-97 (Fig. 5A). His-97 is located at a relatively distant position from the Cβ hydroxyl group of DHEThDP (4.2 Å) but sufficiently close to the water molecule near AcThDP. As shown in Table 2, H97A and H97N mutants showed no catalytic activity. We tried to determine their structures in the presence of F6P but failed due to poor reproducibility of crystallization. This may be because this residue is also involved in fixation of the diphosphate moiety of ThDP (Fig. 5).
Structure of a Tricyclic Ring Form of ThDP
To our surprise, in the electron density map of the H64A mutant, the N4′ atom of the pyrimidine ring and the C2 atom of the thiazolium ring were close to each other and within covalent bonding distance, exhibiting a tricyclic character (Fig. 4E). Furthermore, this tricyclic ring thiamine cofactor was observed from crystals of H64A mutants without soaking in F6P (data not shown). The central ring is six-membered and is clearly different from the shape of a carbinolamine form, which has a seven-membered central ring formed by a covalent bond between the carbonyl carbon of a substituted acetyl group on the C2 atom of the thiazolium ring and the N4′ atom of the pyrimidine ring (34, 39). The cofactor derivative was, instead, interpreted as a tricyclic form observed in the active site of acetolactate synthase (40). The electron density map was reasonably fit with a pyramidal N3 atom because the thiazolium and central rings form an acute angle. The effect of deleting the side chain of His-64 appears to propagate to another subunit, and a region holding the pyrimidine and thiazolium rings is significantly displaced toward the outside of subunit interface (supplemental Fig. 5). As a result, the pocket for the cofactor rings is relatively open in the H64A mutant and provides flexibility. The flexibility might have permitted migration of the positive charge from N3 to S1 to facilitate the tricyclic ring formation as in the case of acetolactate synthase (40). It is also possible that x-ray radiation damage caused the cyclization. The drastic reduction in the catalytic activity of H64A is attributed to the inactivation of the cofactor by the significant change of its form. Therefore, the abovementioned possibility that His-64 works as the B1 catalyst is not ruled out.
Nucleophilic Attack on AcThDP by Pi
The Pi binding site of BbXFPK is formed by His-64, His-320, Gln-321, Tyr-501, and Asn-549 (Fig. 7A). Mutations of these residues dramatically decreased the activity (Table 2). In particular, replacement of Tyr-501 by phenylalanine significantly elevated the Km value for Pi. The residue corresponding to Tyr-501 is generally Phe in TKs (Phe-442 in ScTK). The distance between an oxygen atom of Pi and the Cα carbon of AcThDP is 3.1 Å, and the O(Pi)···Cα—Oα angle is 115° (Fig. 7A). This spatial configuration is within the limits of the Bürgi-Dunitz trajectory (41), suggesting that the nucleophilic attack by the acceptor substrate (Pi) occurs on the keto form and not on the enol form (Fig. 2). In contrast, the nucleophilic attack in TK is reversely performed by the α-carbanion DHEThDP on the acceptor substrate (aldose in this case).
FIGURE 7.

Stereo view of superimposition of the Pi binding site of BbXFPK with ScTK (A) and LpPOX (B). A, shown is superimposition of the AcThDP and Pi complex structures of BbXFPK (yellow for protein and green for cofactor) and the resting structure of ScTK (PDB code 1TRK, gray). Labels in parentheses are those of ScTK. B, shown is superimposition of the AcThDP intermediate and Pi complex structures of BbXFPK (C and P atoms are in green and orange, respectively), and Pi complexed with α-hydroxyethyl-ThDP adduct of LpLOX (2EZT, C and P atoms are in gray).
This part of the reaction of BbXFPK is analogous to that of acetyl-P-producing POX, which also produces acetyl-P after nucleophilic attack by Pi (9, 34, 42). However, the reaction of POX is distinct from that of PK because the nucleophilic attack occurs on the radical state of AcThDP (or 2-hydroxyethyl-ThDP) and involves electron transfer to a flavin adenine dinucleotide cofactor. The Pi molecule bound to the α-hydroxyethyl-ThDP adduct of LpPOX is also located in the vicinity of the Cα atom of the covalent adduct, but its spatial configuration is different from that of BbXFPK (Fig. 7B).
DISCUSSION
Our study expanded the structural insight into the reaction mechanism of PK. The reaction of PK is distinct from that of other ThDP-dependent enzymes at the point of dehydration. In this study we trapped the structures of two key intermediates before and after dehydration and showed that AcThDP accumulates in the absence of Pi as a stable intermediate. However, the identity of the critical factor for dehydration that discriminates PK from TK remains to be determined. An 1H NMR study has shown that AcThDP does not form in the active site of TK (43), indicating that the TK reaction is completely devoid of dehydration. Mutagenesis and crystallographic analysis of BbXFPK indicated that the most probable candidate for the B1 catalyst is His-64. Identity of the B2 catalyst remains elusive, but possible factors responsible for the dehydration step are assigned to be His-553, the N4′ group of the pyrimidine, or His-97. It is confusing that there is no clear structural difference from TK in the catalytic center region around the Cβ hydroxyl group of DHEThDP. However, the environment around the water molecule observed near the AcThDP adduct seems to be critical for the dehydration reaction. This reaction is elimination of a hydroxyl group from an enamine tautomer catalyzed by a catalytic acid. The dehydration reaction of PK is analogous to the second β-elimination step of the dehydration reaction catalyzed by enolase, which is involved in major glycolytic pathways (44). Observation of the tricyclic ring form of ThDP in the active site of BbXFPK is noteworthy, as this form is not usually observed in ThDP-dependent enzymes. Unlike the case of ALS, a point mutation resulted in the formation of this unique but catalytically inactive form of the cofactor.
Supplementary Material
Acknowledgment
We thank the staff of the Photon Factory and SPring-8 for the X-ray data collection.
Note Added in Proof
The crystal structure of XFPK from B. longum is reported by Takahashi et al. (Takahashi, K., Tagami, U., Shimba, N., Kashiwagi, T., Ishikawa, K., and Suzuki, E. (2010) FEBS Lett. 584, 3855–3861).
This work was supported by the Program for the Promotion of Basic Research Activities for Innovative Bioscience (PROBRAIN) in Japan.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1–3 and Figs. 1–5.
- ThDP
- thiamine diphosphate
- TK
- transketolase
- PK
- phosphoketolase
- POX
- pyruvate oxidase
- acetyl-P
- acetyl phosphate
- XPK
- xylulose 5-phosphate phosphoketolase
- X5P
- xylulose 5-phosphate
- F6P
- fructose 6-phosphate
- XFPK
- xylulose 5-phosphate/fructose 6-phosphate phosphoketolase
- DHEThDP
- α,β-dihydroxyethyl ThDP
- AcThDP
- 2-acetyl-ThDP
- BbXFPK
- XFPK from B. breve
- ScTK
- TK from S. cerevisiae
- LpPOX
- POX from L. plantarum
- EcTK
- TK from E. coli
- Bicine
- N,N-bis(2-hydroxyethyl)glycine.
REFERENCES
- 1.Guarner F., Malagelada J. R. (2003) Lancet 361, 512–519 [DOI] [PubMed] [Google Scholar]
- 2.Scardovi V., Trovatelli L. D. (1965) Ann. Microbiol. 15, 19–29 [Google Scholar]
- 3.Schneider G., Lindqvist Y. (1998) Biochim. Biophys. Acta 1385, 387–398 [DOI] [PubMed] [Google Scholar]
- 4.Fiedler E., Thorell S., Sandalova T., Golbik R., König S., Schneider G. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 591–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heath E. C., Hurwitz J., Horecker B. L., Ginsburg A. (1958) J. Biol. Chem. 231, 1009–1029 [PubMed] [Google Scholar]
- 6.Hurwitz J. (1958) Biochim. Biophys. Acta. 28, 599–602 [DOI] [PubMed] [Google Scholar]
- 7.Schramm M., Klybas V., Racker E. (1958) J. Biol. Chem. 233, 1283–1288 [PubMed] [Google Scholar]
- 8.Breslow R. (1958) J. Am. Chem. Soc. 80, 3719–3726 [Google Scholar]
- 9.Kluger R., Tittmann K. (2008) Chem. Rev. 108, 1797–1833 [DOI] [PubMed] [Google Scholar]
- 10.Duggleby R. G. (2006) Acc. Chem. Res. 39, 550–557 [DOI] [PubMed] [Google Scholar]
- 11.Jordan F. (2003) Nat. Prod. Rep. 20, 184–201 [DOI] [PubMed] [Google Scholar]
- 12.Tittmann K. (2009) FEBS J. 276, 2454–2468 [DOI] [PubMed] [Google Scholar]
- 13.Muller Y. A., Schulz G. E. (1993) Science 259, 965–967 [DOI] [PubMed] [Google Scholar]
- 14.Goldberg M. L., Racker E. (1962) J. Biol. Chem. 237, 3841–3842 [PubMed] [Google Scholar]
- 15.Sgorbati B., Lenaz G., Casalicchio F. (1976) Antonie Van Leeuwenhoek 42, 49–57 [DOI] [PubMed] [Google Scholar]
- 16.Meile L., Rohr L. M., Geissmann T. A., Herensperger M., Teuber M. (2001) J. Bacteriol. 183, 2929–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sánchez B., Zúñiga M., González-Candelas F., de los Reyes-Gavilán C. G., Margolles A. (2010) J. Mol. Microbiol. Biotechnol. 18, 37–51 [DOI] [PubMed] [Google Scholar]
- 18.Scardovi V. (1986) in Bergey's Manual of Systematic Bacteriology (Sneath P. H. A., Mair N. S., Sharpe M. E., Holt J. G. eds) pp 1418–1434, Williams and Wilkins, Baltimore [Google Scholar]
- 19.Vlková E., Nevoral J., Jencikova B., Kopecný J., Godefrooij J., Trojanová I., Rada V. (2005) J. Microbiol. Methods. 60, 365–373 [DOI] [PubMed] [Google Scholar]
- 20.Panagiotou G., Andersen M. R., Grotkjaer T., Regueira T. B., Hofmann G., Nielsen J., Olsson L. (2008) PloS one. 3, e3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maslowski K. M., Vieira A. T., Ng A., Kranich J., Sierro F., Yu D., Schilter H. C., Rolph M. S., Mackay F., Artis D., Xavier R. J., Teixeira M. M., Mackay C. R. (2009) Nature 461, 1282–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yevenes A., Frey P. A. (2008) Bioorg Chem. 36, 121–127 [DOI] [PubMed] [Google Scholar]
- 23.Petrareanu G., Balasu M. C., Zander U., Scheidig A. J., Szedlacsek S. E. (2010) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 805–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki R., Kim B.-J., Iwamoto Y., Katayama T., Ashida H., Wakagi T., Shoun H., Fushinobu S., Yamamoto K. (2010) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 941–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 26.Rappleye J., Innus M., Weeks C. M., Miller R. (2002) J. Appl. Crystallogr 35, 374–376 [Google Scholar]
- 27.Terwilliger T. C., Berendzen J. (1999) Acta. Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perrakis A., Morris R., Lamzin V. S. (1999) Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 29.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 30.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta. Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 31.Schüttelkopf A. W., van Aalten D. M. (2004) Acta. Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 32.Kern D., Kern G., Neef H., Tittmann K., Killenberg-Jabs M., Wikner C., Schneider G., Hübner G. (1997) Science 275, 67–70 [DOI] [PubMed] [Google Scholar]
- 33.Frey P. A. (1989) Biofactors 2, 1–9 [PubMed] [Google Scholar]
- 34.Wille G., Meyer D., Steinmetz A., Hinze E., Golbik R., Tittmann K. (2006) Nat. Chem. Biol. 2, 324–328 [DOI] [PubMed] [Google Scholar]
- 35.Nilsson U., Meshalkina L., Lindqvist Y., Schneider G. (1997) J. Biol. Chem. 272, 1864–1869 [DOI] [PubMed] [Google Scholar]
- 36.Asztalos P., Parthier C., Golbik R., Kleinschmidt M., Hübner G., Weiss M. S., Friedemann R., Wille G., Tittmann K. (2007) Biochemistry 46, 12037–12052 [DOI] [PubMed] [Google Scholar]
- 37.Tittmann K., Wille G. (2009) J. Mol. Catal. B Enzym. 61, 93–99 [Google Scholar]
- 38.Wikner C., Nilsson U., Meshalkina L., Udekwu C., Lindqvist Y., Schneider G. (1997) Biochemistry 36, 15643–15649 [DOI] [PubMed] [Google Scholar]
- 39.Gruys K. J., Halkides C. J., Frey P. A. (1987) Biochemistry 26, 7575–7585 [DOI] [PubMed] [Google Scholar]
- 40.Pang S. S., Duggleby R. G., Schowen R. L., Guddat L. W. (2004) J. Biol. Chem. 279, 2242–2253 [DOI] [PubMed] [Google Scholar]
- 41.Bürgi H. B., Dunitz J. D. (1983) Acc. Chem. Res. 16, 153–161 [Google Scholar]
- 42.Tittmann K., Wille G., Golbik R., Weidner A., Ghisla S., Hübner G. (2005) Biochemistry 44, 13291–13303 [DOI] [PubMed] [Google Scholar]
- 43.Tittmann K., Golbik R., Uhlemann K., Khailova L., Schneider G., Patel M., Jordan F., Chipman D. M., Duggleby R. G., Hübner G. (2003) Biochemistry 42, 7885–7891 [DOI] [PubMed] [Google Scholar]
- 44.Reed G. H., Poyner R. R., Larsen T. M., Wedekind J. E., Rayment I. (1996) Curr. Opin. Struct. Biol. 6, 736–743 [DOI] [PubMed] [Google Scholar]
- 45.Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.