

Abstract
Iron−platinum alloy nanoparticles (FePt NPs) are extremely promising candidates for the next generation of contrast agents for magnetic resonance (MR) diagnostic imaging and MR-guided interventions, including hyperthermic ablation of solid cancers. FePt has high Curie temperature, saturation magnetic moment, magneto-crystalline anisotropy, and chemical stability. We describe the synthesis and characterization of a family of biocompatible FePt NPs suitable for biomedical applications, showing and discussing that FePt NPs can exhibit low cytotoxicity. The importance of engineering the interface of strongly magnetic NPs using a coating allowing free aqueous permeation is demonstrated to be an essential parameter in the design of new generations of diagnostic and therapeutic MRI contrast agents. We report effective cell internalization of FePt NPs and demonstrate that they can be used for cellular imaging and in vivo MRI applications. This opens the way for several future applications of FePt NPs, including regenerative medicine and stem cell therapy in addition to enhanced MR diagnostic imaging.
1. Introduction
Nanoparticles (NPs) have attracted considerable interest as diagnostic and therapeutic tools for biomedicine,1−3 with magnetic NPs offering unique opportunities for magnetic separation, targeted drug delivery, and hyperthermic ablation of cancer and as contrast agents in magnetic resonance imaging (MRI).4−11 MRI is a powerful, noninvasive medical imaging technique where the signal originates predominately from water and lipids. Medical diagnosis requires enhanced contrast between normal and pathological tissues, resulting in the development of exogenous MRI contrast agents.(12) Common T2-agents include superparamagnetic nanoparticles (Fe3O4, γ-Fe2O3, MnFe2O4, FeCo), the clinical MRI applications of which range from imaging lymph-node to liver and spleen, as well as bone marrow and gastrointestinal tract.4,13,14 Despite its widespread usage, superparamagnetic iron oxide (SPIO) has several disadvantages, including rapid clearance by phagocytic cells and hence limited trans-endothelia passage and tissue penetration.4,15 There is therefore a need for the design of biocompatible MRI contrast agents with prolonged intravascular retention, improved tissue delineation, high chemical stability, and improved selectivity when targeting a tissue of interest.
Small NPs avoid fast clearance by the reticulo-endothelial system and therefore should enable long blood circulation half-life, thereby improving their ability to reach specific targets.4,16 Silica is often considered as the most attractive coating for engineered NPs, forming an inert and biocompatible outer shell. Not only does it create a protective shell against chemical degradation, but it also allows surface functionalization with functional alkoxysilanes.(7) An outer coating with functional organic molecules has many advantages, including steric and/or electrostatic repulsion between the NPs, which can increase water solubility and stability in biological environment.(17) Positively charged NPs demonstrate increased cell internalization efficiency and biomedical molecules binding capability for multimodal imaging applications and drug delivery.4,18,19
Characterized by high Curie temperature and magneto-crystalline anisotropy, iron−platinum metallic alloy NPs have emerged as promising candidates for a new generation of magnetic nanomaterials.(20) Over the past decade, several chemical pathways have been developed to synthesize FePt NPs with controlled stoichiometry, size, and shape.(5) Such synthetic routes include co-reduction of Fe and Pt precursors through both low-temperature emulsion protocols and the high-temperature polyol pathway,21−25 along with the thermal decomposition route.26−31 The latter approach often relies on Pt(acac)2, Fe(CO)5, or Collman’s regeant precursors mixed with alkyl amines and acids solubilized in high-boiling-point solvents heated at temperatures of ∼300 °C and above to control the growth, composition, and morphology of the NPs.
Studies have demonstrated the biomedical potential of FePt NPs,(6) such as in magnetic separation32,33 and hyperthermic ablation34,35 and as T2 MRI contrast agents.36−38 However, toxicity remains a major concern, preventing their use as a bioplatform for diagnosis and therapy.37,39,40
In this article, we describe the fabrication of FePt NPs specifically designed to enhance their magnetic properties for biomedical applications. We demonstrate that, in contrast with previous reports, FePt NPs can be made nontoxic, and we provide the first data on their cellular uptake mechanisms. We report a 6-fold increase in the FePt-based T2 contrast properties compared to those of clinical iron oxide NPs. The relationship between the MRI contrast properties and the NPs’ architecture is explored and rationalized as the basis for the design of NPs as enhanced MRI contrast agents. Finally, we report the first observations of cellular and in vivo MR imaging with FePt NPs. Our results opens the way for several applications of FePt NPs, such as regenerative medicine and stem cell therapy, in addition to enhanced MR diagnostic imaging, thus providing a platform to develop novel diagnostic and therapeutic agents.
2. Results
Figure 1 displays the schematic structure of a clinical iron oxide NP coated with dextran (Feridex, Figure 1A1) and two FePt NPs: the face-centered-cubic (fcc) FePt NP surface functionalized with cysteamine (fcc-FePt-A, Figure 1B1) and the fcc FePt NP coated with silica and surface-functionalized with (3-aminopropyl)triethoxysilane (fcc-FePt-silica-A, Figure 1C1). The preparation of the family of FePt NPs is described in the Supporting Information.27,30,17,41
Figure 1.

Schematic representation (1), TEM images (2), XRD patterns (3), ZFC-FC (4), and hysteresis (5) curves of Feridex (A1−A5), fcc-FePt-A NPs (B1−B5), and fcc-FePt-silica-A NPs (C1−C5).
In agreement with published literature,(42) transmission electron microscopy (TEM) revealed that the commercial iron oxide NPs samples (Figure 1A2) are composed of aggregated crystals approximately 5 nm in diameter, embedded in an organic coating. These aggregates form large particles of a size distribution ranging from 20 to >50 nm. In contrast, the fcc-FePt-A NPs are well dispersed, with diameters of ∼5 nm (Figure 1B2). The fcc-FePt-silica-A NPs are coated with a homogeneous silica shell of ∼17 nm in thickness, leading to a total diameter of ∼40 nm (Figure 1C2).
The powder X-ray diffraction (XRD) pattern of the Feridex iron oxide confirms the presence of magnetite (Fe3O4), the broad peaks at 38°, 45°, and 55° corresponding to the (220), (311), and (400) peaks, respectively (Figure 1A3). Figure 1B3,C3 shows peaks around 51° and 60°, characteristic of fcc-FePt (111) and (200) peaks, respectively. The (111) peak position suggests that the NPs’ composition includes between 40 and 45% of Fe (Table 1). For the silica-coated NPs, the SiO2 XRD characteristic peak is observed at ∼28° (Figure 1C3). The crystal size is ∼3.6 nm for both fcc-FePt-A and fcc-FePt-silica-A NPs (Table 1).
Table 1. XRD, SQUID, and MRI Dataa.
|
Ms (emu/g Fe) |
||||||||
|---|---|---|---|---|---|---|---|---|
| sample | DXRD (nm) | FexPt1−x (%) | Tb (K) | at 2 K | at 300 K | Hc at 2 K (kOe) | r1b (s−1 mM−1) | r2b (s−1 mM−1) |
| fcc-FePt-A | 3.6 ± 0.1 | 42.7 ± 0.7 | 90 | 201 | 112 | 0.7 | 2.5 ± 1.0 | 887 ± 32 |
| fcc-FePt-silica-A | 3.6 ± 0.1 | 43.8 ± 1.4 | 50 | 191 | 83 | 1.3 | 0.3 ± 0.1 | 210 ± 3 |
| Feridex I.V.c (Endorem) | 5.3 ± 0.1 | − | 45 | 101 | 82 | 0.3 | 0.9 ± 0.1 | 148 ± 2 |
DXRD is the crystalline grain size of the NPs; in FexPt1−x, x is the composition based on the FePt lattice constant (SI-Figure 1);(43)Tb is the blocking temperature; Ms is the saturation moment; Hc is the magnetic coercivity of the NPs; and r1 and r2 are the 1H relaxivity values for 1% agarose gel containing Feridex, fcc-FePt-A, and fcc-FePt-silica-A NPs.
A range of other commercial MRI contrast agent relaxivity values are presented in the Supporting Information (SI-Table 2).
Trademark of AMAG Pharmaceuticals, Lexington, MA.
Zero-field-cooling and field-cooling (ZFC−FC) of Feridex samples highlight a 45 K blocking temperature (Tb, Figure 1A4). At 2 K, i.e., lower than Tb, Feridex hysteresis loop measurements display ferrimagnetic behavior with a small coercivity value of 300 Oe and a saturation moment (Ms) of 101 emu/g of Fe. At 300 K, Feridex is superparamagnetic with no coercivity, as illustrated in Figure 1A5. The Ms value is 82 emu/g of Fe, in agreement with the literature.16,42 In comparison, the fcc-FePt-A NPs have a higher Tb of ∼90 K (Figure 1B4). More importantly, when compared to Feridex, the Ms of fcc-FePt-A NPs is ∼2 times larger at 2 K and 35% stronger at 300 K (Figure 1B5, Table 1). After silica coating, the blocking temperature is reduced to 50 K, and the ZFC slope below Tb is steeper (Figure 1C4). At 2 K and 5 T, the magnetization of fcc-FePt-silica-A NPs is ∼5% lower than that of fcc-FePt-A and 2 times larger than that of Feridex. At 300 K and 5 T, the magnetization of fcc-FePt-silica-A NPs is ∼36% lower than that of fcc-FePt-A NPs and similar to that of Feridex (Figure 1C5, Table 1).
Cytotoxicity of the NPs was evaluated in an A375M cell line using fcc-FePt-A NPs and MTS assay with various concentrations and incubation times.(43) Both dose−response and time course studies were performed (Figure 2A). As expected,44,45 and in contrast with bare iron oxide,46,47 Feridex’s dextran organic coating does not lead to any detectable cytotoxicity. At 120 μg/mL, fcc-FePt-A NPs exhibited cytotoxicity that increases with incubation time. At a concentration of 60 μg/mL, fcc-FePt-A NPs incubated with cells for 7 days showed a ∼15% loss of cell viability. Significantly, there is no loss of cell viability at concentrations of 30 μg/mL and below, even after prolonged incubation periods of up to 7 days. Similar results were obtained with fcc-FePt-A NPs and both MCF7 and U2OS tumor cell lines (SI-Figure 5A,B).(43)
Figure 2.

Viability of A375M cells incubated with fcc-FePt-A NPs (A). Fe released from fcc-FePt-A NPs (orange ◆) in pH 4.8 PBS (B1). Fe released from fcc-FePt-A NPs (orange ◆) and from Feridex (△)(49) in solutions at pH ∼4.8 of RPMI-1640 cell media containing 20 mM sodium citrate. The incubation concentrations for FePt and Feridex(49) were 120 and 25 μg/mL, respectively (B2). Photomicrographs (magnification ×40) of Prussian blue-stained A375M cells cultured on coverslips in cell media (C1), media containing 30 μg/mL of fcc-FePt-A NPs (C2), and media containing 30 μg/mL of Feridex (C3) after overnight incubation. TEM images of A375M cells after 16 h incubation in media containing 30 μg/mL of fcc-FePt-A NPs (D1−D3).
To confirm these results, the chemical stability of fcc-FePt-A NPs in an acidic environment was investigated by incubation of fcc-FePt-A in pH 4.8 buffer solutions. The concentration of FePt NPs incubated in buffer solution is ∼120 μg/mL, to match the highest NPs concentration used in the cytotoxicity study (Figure 2A). In a pH 4.8 phosphate buffered saline (PBS), cysteamine-coated FePt NPs show excellent chemical stability, with only ∼0.6% Fe, i.e. <0.1 μg/mL, of iron released after 7 days of incubation (Figure 2B1). As suggested by the recent literature,48,49 an in vitro lysosomal model, i.e. RPMI-1640 cell media containing 20 mM sodium citrate (pH 4.8), was also used to match the metabolic conditions. In these more extreme conditions (Figure 2B2), more Fe was released—0.3 μg/mL after 6 h and up to ∼3.3 μg/mL after 7 days—but importantly it remained at a level comparable to the release observed with Feridex.
The high chemical stability of fcc-FePt-A was investigated further. Prussian blue staining experiments were carried out and showed that FePt NPs have greater stability compared to Feridex (Figure 2C1−C3).(43) Indeed, photomicrographs obtained after overnight incubation show no stain for both blank control sample (Figure 2C1) and cells labeled with fcc-FePt-A NPs (Figure 2C2). In contrast, Feridex-labeled cells show significantly staining (Figure 2C3) characteristic of the release of Fe in the cells. These results confirm that the cysteamine coating provides fcc-FePt-A NPs with high chemical stability.
The cysteamine coating is positively charged, resulting in higher cellular uptake by human non-phagocytic A375M tumor cells (Figure 2D1−D3) compared to Feridex. FePt NPs appear to be internalized by an endocytotic pathway. Indeed, TEM studies suggest a macropinocytosis cellular uptake mechanism by the large protrusions of the plasma membrane around NP clusters (Figure 2D2).50,51 The intracellular localization route is endosome, early lysosome, and late lysosome, which are basic cell biology phenomena. Both endosomes and lysosomes are confined subcellular organelles, and, as both expected and commonly observed in other NPs systems, the FePt NPs appear typically “entrapped” inside these subcellular vesicles (Figure 2D3).
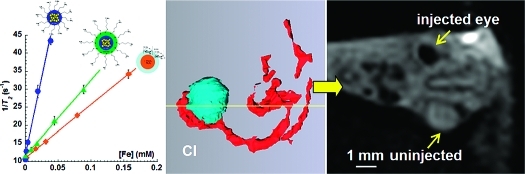
The effectiveness of fcc-FePt-A and fcc-FePt-silica-A as MRI contrast agents was investigated by measuring the dependence of the longitudinal relaxation rate (T1−1) and transverse relaxation rate (T2−1) of 1% agarose gels.(43) Relaxivity (ri) quantifies the enhancement of either the longitudinal or the transverse relaxation rates of the protons in water. As expected at high magnetic field, the FePt NPs and Feridex have a weak influence on the water 1H longitudinal relaxation rate (Table 1 and SI-Figure 7A).16,43 In contrast, FePt NPs have a significant effect upon the aqueous transverse relaxation rate (Figure 3A1). The relaxivity r2 of the fcc-FePt-silica-A NPs is 40% larger than that of Feridex, which is attributed to the stronger magnetic moment of the fcc-FePt-silica-A NPs (SI-Figure 7B) at the MRI magnetic field (7 T).(43) Importantly, the relaxivity r2 of the fcc-FePt-A NPs is more than 6 times larger than that of Feridex. Figure 3A2 displays T2-weighted MRI images of various concentrations of fcc-FePt-A NPs in agarose gels. The signal intensity reduces and the image appears darker as the NPs concentration increases. The FePt NPs produce an observable change in image contrast at concentrations as low as 6 μg/mL, which is well within the safety range indicated by the cytotoxicity data (Figure 2A). Similarly, signal intensity loss was observed in cells labeled with fcc-FePt-A NPs as the cell density increases from 10 × 103 to 1000 × 103 cells/mL (Figure 3B).
Figure 3.

T2−1 (s−1) vs [Fe] (mM) of the water in 1% w/v agarose gel containing (blue ●) fcc-FePt-A, (green ▲) fcc-FePt-silica-A, and (orange ◆) Feridex (A1). 1H longitudinal relaxation time T2-weighted MRI images of 1% agarose gel/water solution containing fcc-FePt-A NPs with [Fe] = 0.00 mM (a), 0.02 mM (CFePt = 6 μg/mL) (b), and 0.04 mM (CFePt = 12 μg/mL) (c) (A2). T2-weighted cellular imaging by MRI (3D spin echo, TE = 30 ms, TR = 1 s) of A375M cells loaded with fcc-FePt-A NPs with a cell density of 10 × 103 cells/mL (a), 100 × 103 cells/mL (b), and 1000 × 103 cells/mL (c) (B). Embryo injected in ovo with cell culture media (1 μL) containing no NPs (C) and 20 μg/mL fcc-FePt-A (D). 3D surface reconstruction of embryo eyes (blue) and blood vessels (red) showing the position in yellow of the transverse and coronal images (C1,D1); dorsal view of transverse image of the embryo’s head aligned through the eyes (C2,D2); and anterior view of coronal image of the embryo’s head aligned through the eyes (C3,D3). 2D slices from 128 × 128 × 128 3D Rare-8 MRI data set of day 4 quail embryo egg (TR/TE = 250/25 ms), field of view of 30 mm and pixel dimensions of 0.234 mm/pixel. Labels: y, yolk; a, albumen; b, brain; in-e, injected eye; un-e, un-injected eye; h, heart. Scale bar indicates 1 mm. All the MRI measurements were completed at 7.1 T.
The in vivo potential of FePt NPs as MRI contrast agents was demonstrated by injecting NPs in cell culture media into the eyes of day 4 quail embryos in ovo. Figure 3C,D displays 3D MRI image data sets after injection. The transverse and coronal images through the embryo’s head are presented. The uninjected eye (un-e) acts as an internal control. There is no difference in the contrast between the uninjected and the uppermost eye (in-e) which had been injected with a 1 μL of culture medium only (Figure 3C2,C3). However, when the injection medium contained 20 μg/mL of fcc-FePt-A NPs, a very distinct hypointensity appears in the injected eye (Figure 3D2−D3). This was consistently observed when varying the NPs concentration.
This thus demonstrates the capability of nontoxic FePt NPs as potent MRI probes for cellular imaging, even at very low NP concentrations. Labeling the cells and these magnetic NPs will allow, for instance, cell migration to be studied, with their biodistribution and behavior monitored in vivo, especially as we have demonstrated that the effect of contrast agents can be observed at non-cytotoxic concentrations lower than 30 μg/mL.
3. Discussion
The major hurdle preventing the wider use of FePt NPs in biomedicine is their potential toxicity. In the literature, there are many inconsistencies in FePt cytotoxicity, leaving the subject rather confused. A summary of the cytotoxicity data of FePt NPs is presented in Table 2. While it was reported that 24 h incubation of FePt-cysteine NPs had no significant toxicity at concentrations below 5 μg/mL, these compounds resulted in 50% cell damage at 15.5 μg/mL;(39) FePt-Fe3O4 NPs showed no significant cell viability damage up to 3 days of incubation at 10 μg/mL.(37) In contrast, FePt-Fe2O3 and FePt-CoS2 were reported to induce significant cell viability damage of over 50% even at concentrations of NPs as low as 1.5 and ∼8.3 μg/mL, respectively.37,39 A recent toxicity study with phospholipids-coated fcc-FePt NPs showed 50% cell viability damage above 1.25 μg/mL Fe concentration (∼7.8 μg/mL Fe40Pt60 NPs).(52) In this latter study, the high toxicity was attributed to fast Fe release from phospholipids-coated fcc-FePt NPs. It was reported that ∼6% Fe was already released after 6 h of incubation, and the percentage of Fe released increased to ∼20% after 24 h of incubation in pH 4.8 PBS.(52) While iron is essential for almost all living organisms, it is toxic. It can act as a catalyst in the Fenton reaction, which generates a wide range of free radical species, including hydroxyl radicals. These are among the most reactive free radical species known and have the ability to react with a wide range of cellular constituents, including pyrimidine bases of DNA, and also to trigger lipid peroxidation in cellular membranes, leading to cell death.(53)
Table 2. Summary of FePt Nanoparticles Cytotoxicity Studies Reported in the Literaturea.
| cytotoxicity |
||||||||
|---|---|---|---|---|---|---|---|---|
| material | Fe precursorb | coating | cells | [NPs] (μg/mL) | [Fe] (μg/mL) | tinc (h) | cell damage (%) | ref |
| FePt-CoS2 | Fe(CO)5 | CoS2 shell | HeLa | 1.5 | ∼0.007 | 24 | 50 | (39) |
| FePt-Cys | Fe(CO)5 | cysteine | HeLa | 5 | ∼0.7 | 24 | ∼10 | (39) |
| 15.5 | ∼2.5 | 24 | 50 | |||||
| FePt-Fe2O3 | Fe(CO)5 | Fe2O3 shell, l-DOPA coating | HeLa | ∼8.3 | ∼0.045c | 24 | 50 | (37) |
| ∼3.8d | ||||||||
| FePt-Fe3O4 | Fe(CO)5 | Fe3O4 shell | HeLa | 40 | e | 24 | ∼10 | (37) |
| 10 | e | 72 | ∼10 | |||||
| 20 | e | 72 | ∼30 | |||||
| FePt-COOH | Fe(CO)5 | bilayer of phospholipid (DSPE-PEG(2000) carboxylic acid lipid) and oleic acid/oleylamine | HeLa | ∼10.7 | 1.72 | 24 | ∼40 | (52) |
| A431 | ∼10.7 | 1.72 | 24 | ∼40 | ||||
| SK-BR3 | ∼10.7 | 1.72 | 24 | ∼20 | ||||
| HEK-293 | ∼10.7 | 1.72 | 24 | ∼30 | ||||
| A2780 | ∼7.8 | 1.25 | 24 | 50 | ||||
| FePt-SiO2-A | Na2Fe(CO)4 | SiO2 shell | A375M, MCF7, U2OS | 200 | ∼2 | 168 | 0 | f |
| FePt-A | Na2Fe(CO)4 | cysteamine | A375M, MCF7, U2OS | 30 | ∼5.3 | 168 | 0 | f |
| 60 | ∼10.5 | 72 | ∼10 | |||||
Listed are NPs’ structure, precursors, and coating; “cells” stands for the cell line; [NPs] is the nanoparticle concentration; tinc is the incubation time; cell damage is that after incubation.
Pt precursor always Pt(acac)2.
Fe contribution from Fe2O3 shell not included.
Fe contribution from Fe2O3 shell included.
[Fe] cannot be calculated.
Data presented in this work.
In this study, the fcc-FePt-silica-A NPs have good biocompatibility and, as expected, the silica coating forms an inert shell (SI-Figure 5C).(43) In contrast to previously published results, our study demonstrates that cysteamine-coated fcc-FePt NPs also have very high chemical stability in pH 4.8 PBS. Fe release after 24 h of incubation is only ∼0.3%, with only a slight increased to 0.6% after 7 days of incubation (Figure 2B1). In an in vitro model (i.e., cell medium containing sodium citrate),48,49 fcc-FePt-A NPs were found to release as little Fe as Feridex (Figure 2B2), with ∼5% Fe found to be released after 24 h and 20% after 7 days. This confirms that the low pH in the endosome/lysosome environment is not sufficient to dissolve the NPs and that the presence of endogenous citrate or similar metallic chelates is instrumental to the NPs degradation by binding to solubilized Fe atoms.48,49 The results presented herein are significant as they suggest that, when considering the Fe release, the safety of fcc-FePt-A NPs is comparable to that of Feridex under similar experimental conditions. The chemical stability of the fcc-FePt-A NPs is also supported by the Prussian blue staining experiments (Figure 2C1−C3).
The high chemical stability of cysteamine-coated FePt NPs certainly contributes to the NPs’ noteworthy low cytotoxicity. Of course, iron release is only one parameter that contributes to toxicity. It is quite possible that differences in the NPs preparation, surface coating, ligand exchange, and extraction protocols could account for the spread of the cytotoxicity data reported in the literature. The toxicity may not necessarily be due to the material itself through the release of its constituent. In the present study, we used Collman’s reagent, Na2Fe(CO)4, to control the FePt NPs stoichiometry and to ensure the simultaneous presence of Fe and Pt species during the formation of FePt alloy NPs; the use of Fe(CO)5 has been shown to form NPs with a core and a shell rich in platinum and iron, respectively.(31) After ligand exchange, the extraction process was repeated up to six times to ensure removal of hazardous chemicals such as chloroform, oleic acid, and oleylamine, while the thiol group provides a strong bond between the cysteamine and the NPs’ surface, preventing the ligands from becoming lethal. During the review of this publication, cysteamine-coated FePt NPs were reported to induce, after 24 h, very low cell damage on Veron cells;(38) however, it is noticeable that the strong bonding to the surface of the NPs, protecting them from the cells’ environment, can be contrasted with, for instance, a coating made with a bilayer structure of phospholipids and oleic acid/oleylamine.(52) Indeed, even though this phospholipid approach provides aqueous solubility, it is also characterized by a weaker bonding strength and a more dynamic behavior, allowing the coating to slowly disperse in the cell. By doing so, the “free” ligands can alter the cell viability as well as the release of the NP constituents as the surface of the particles is exposed to the cell, and the NPs’ chemical integrity can be compromised further. Such outcomes have been reported by Xu et al., although they elegantly focused on the opportunity this provided to use the NPs as metal reservoirs to kill cancer cells.(52)
While the very high chemical stability of fcc-FePt-A NPs was demonstrated in this report, cytotoxicity is observed at high concentrations above 30 μg/mL FePt and long incubation times. This could be explained by contributions from (i) the very small release of Fe, which has been found to be <1% after 168 h and (ii) the high concentration of positive surface charges, which could also interact with the more negatively charged cell membranes and induce membrane disruption.(54) In addition, ligands with strong anchoring groups can reduce the toxicity, as illustrated in this publication, and it is noticeable that the present results were obtained with commercially available ligands (i.e. cysteamine), so one should expect enhanced viability from, for instance, custom-made dithiolate ligands, such as those developed recently for quantum dots.55,56 These could provide cell viability at NP concentrations higher than 30 μg/mL.
In this context, the little consistency observed when considering FePt NPs’ cytotoxicity data available in the literature stresses that the measured toxicity should always be associated with the nanocolloids, i.e. inorganic NPs + organic stabilizer. The interplay between the components will indeed contribute to the cytotoxicity results, with, for instance, a weak bonding to the NPs’ surface increasing the probability of the nanocolloids being toxic. It then appears that, to be able to compare cytotoxicity data in a meaningful way, one must to develop and use standardized approaches including the correlation between (i) the evolution of the nanocolloids’ integrity, relying on both the inorganic NPs and the organic stabilizers, and (ii) the cell viability. It should be noticed that, in the context of apparently toxic NPs, the nanocolloids’ solution stability could easily be measured by, for instance, time-resolved transmission monitored sedimentation experiments, as while the ligands come off the NPs’ surface, the NPs tend to aggregate and fall out of solution.
The cellular internalization of Feridex is poor due to the weak binding of the neutral dextran coating to the plasma membrane, which limits the capability of cell internalization by the fluid-phase endocytosis pathway.57,58 Enhancing cellular uptake of dextran-coated NPs is possible with transfection agents and further surface functionalization but was not relevant to the present study.(44) In contrast, the amine-coated FePt NPs, fcc-FePt-A and fcc-FePt-Silica-A, are efficiently internalized directly without requiring the use of any external mediation such as transfection agents or electroporation, illustrated with non-phagocytic human cells in Figure 2D1. The uptake appears to be mediated via macropinocytosis, a nonspecific endocytic process, which involves the internalization of large areas of plasma membrane together with significant amounts of fluid (Figure 2D2). Irregular macropinosome vesicles larger than 1 μm are usually generated when membrane protrusions fuse back to the plasma membrane (SI-Figure 6A).43,50,51 Other pathways could be involved, e.g., lipid raft-dependent mechanisms or the clathrin-dependent pathway;50,51,59,60 however, we did not observe any evidence supporting their involvement. Further studies are needed to clarify the exact cellular internalization mechanism. However, and as expected once inside the cells, the FePt NPs that are stabilized by Coulombic interactions appear to be confined in specific subcellular organelles, most likely endosomes and lysosomes (Figure 2D3). This is of significant importance when developing FePt NPs-based biomedical platforms and can be attributed to the interplay between the cationic surface of the NPs and the negatively charged cell membrane.
By considering a family of FePt NPs, we established that, despite the attractiveness of the silica coating, it weakens the NPs’ magnetic moment and their applicability in clinical MRI. This may be the result of a reduction of the magnetic effective volume and/or an alteration of the material. In support of this interpretation, the strong base environment associated with the silica coating can partially oxidize the FePt surface, resulting in a thin layer of softer magnetic material like iron oxide or iron silicide, which because of its thinness is not visible by XRD or TEM.61,62
The fcc-FePt-A NPs demonstrated a very high MRI relaxivity r2, which can be attributed to the combination of (i) an intrinsically high magnetic moment, (ii) a thin organic coating, and (iii) their small size, resulting in effective dispersion across a sample. While this combination created very effective T2 MRI contrast agents even at μg/mL concentrations, these results also underline the importance of engineering the interface of NPs and demonstrate that a strong magnetic moment such as that of FePt, combined with a thin coating layer having an open molecular structure allowing very close aqueous proximity, are essential parameters for the design of new generations of diagnostic and therapeutic MRI contrast agents.
Finally, we demonstrate that FePt NPs can be successfully used as a potent cellular and in vivo MRI probes. This is illustrated by both cell MRI (Figure 3B) and distinct hypointensity of ∼1 mm in size observed in the in vivo environment into which the FePt NPs were injected (Figure 3D and SI-Figure 8).(43) Their more than 6 times higher relaxivity r2 allows using less NPs than with iron oxide contrast agents such as Feridex, which makes FePt NPs very promising contrast agents to identify small lesions such as lymph node metastases.
4. Conclusion
In conclusion, we have presented the synthesis and characterization of a family of FePt NPs: fcc-FePt-A NPs and fcc-FePt-silica-A NPs. The reduction of magnetic properties observed with both SQUID magnetometry and MRI relaxivity of silica-coated NPs is attributed to the formation of very thin layers of oxides and silicides. In sharp contrast with some previous reports, our biocompatibility studies clearly demonstrate and explain the absence of cytotoxicity at concentrations below 30 μg/mL, even after 7 days of incubation. The biocompatibility is mainly attributed to the strong surface coating and the extraction protocols. Another striking feature for cellular imaging is the ability of the FePt NPs to efficiently enter tumor cells without requiring any external mediation. The cellular uptake by non-phagocytic tumor cells occurs via an apparent macropinocytosis mechanism. The 7.1 T MRI studies, in vitro and in vivo, have confirmed that both fcc-FePt-A and fcc-FePt-silica-A NPs are stronger T2 contrast agents than commercial Feridex, with fcc-FePt-A T2 relaxivity (r2) being more than 6 times larger. The superior capability of MRI contrast enhancement associated with the FePt NPs makes them an ideal platform for the design of diagnostic and therapeutic agents for image-guided magnetic drug delivery and hyperthermic tumor ablation. The findings of this study underline the importance of engineering magnetic NP interfaces combining intrinsically strong magnetic moment with coating layers. Finally, our studies have also demonstrated that FePt NPs can be used successfully for cellular and in vivo imaging, thus confirming the potential of FePt NPs in regenerative medicine and stem cell therapy.
Acknowledgments
The authors gratefully acknowledge the Scottish Universities Physics Alliance for funding of a SUPA Advanced Research Fellowship (P.A.) and the James and Enid Nicol Trust for funding a studentship (S.C.). The authors thank Dr. Lorna Eades (EaSt CHEM, University of Edinburg) for the ICP-OES measurements, Prof. Kate Storey (University of Dundee) for use of the microinjection equipment, Dr. Steve Francis (University of St Andrews) for the FTIR measurements, and Profs. David Cole-Hamilton and Peter Bruce (University of St Andrews) for discussion and access to instruments. S.L.D. acknowledges the Wellcome Trust (WT081039) for funding.
Supporting Information Available
Experimental section including the NPs synthesis and characterization, protocols, cytotoxicity, cellular uptake, and MRI application data. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Shubayev V. I.; Pisanic T. R.; Jin S. H. Adv. Drug Deliv. Rev. 2009, 61, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel M. C.; Astruc D. Chem. Rev. 2004, 104, 293–346. [DOI] [PubMed] [Google Scholar]
- Medintz I. L.; Uyeda H. T.; Goldman E. R.; Mattoussi H. Nat. Mater. 2005, 4, 435–446. [DOI] [PubMed] [Google Scholar]
- Na H. B.; Song I. C.; Hyeon T. Adv. Mater. 2009, 21, 2133–2148. [Google Scholar]
- Pankhurst Q. A.; Connolly J.; Jones S. K.; Dobson J. J. Phys. D: Appl. Phys. 2003, 36, R167–R181. [Google Scholar]
- Gao J. H.; Gu H. W.; Xu B. Acc. Chem. Res. 2009, 42, 1097–1107. [DOI] [PubMed] [Google Scholar]
- Sun C.; Lee J. S. H.; Zhang M. Q. Adv. Drug Deliv. Rev. 2008, 60, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiseh O.; Gunn J. W.; Zhang M. Q. Adv. Drug Deliv. Rev. 2010, 62, 284–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong U.; Teng X. W.; Wang Y.; Yang H.; Xia Y. N. Adv. Mater. 2007, 19, 33–60. [Google Scholar]
- Lu A. H.; Salabas E. L.; Schuth F. Angew. Chem., Int. Ed. 2007, 46, 1222–1244. [DOI] [PubMed] [Google Scholar]
- Gao J. H.; Xu B. Nano Today 2009, 4, 37–51. [Google Scholar]
- Brown M. A.; Semelka R. C.. MRI: Basic Principles and Applications, 3rd ed.; Wiley: Hoboken, NJ, 2005. [Google Scholar]
- Yan G.; Robinson L.; Hogg P. Radiography 2007, 13, e5−e19. [Google Scholar]
- Kim J.; Piao Y.; Hyeon T. Chem. Soc. Rev. 2009, 38, 372–390. [DOI] [PubMed] [Google Scholar]
- Bulte J. W. M.; Kraitchman D. L. NMR Biomed. 2004, 17, 484–499. [DOI] [PubMed] [Google Scholar]
- Modo M. M. J.; Bulte J. W. M.. Molecular and Cellular MR Imaging, 1st ed.; CRC Press: Boca Raton, FL, 2007. [Google Scholar]
- Tanaka Y.; Maenosono S. J. Magn. Magn. Mater. 2008, 320, L121–L124. [Google Scholar]
- Josephson L.; Tung C. H.; Moore A.; Weissleder R. Bioconjugate Chem. 1999, 10, 186–191. [DOI] [PubMed] [Google Scholar]
- Kang H. W.; Josephson L.; Petrovsky A.; Weissleder R.; Bogdanov A. Bioconjugate Chem. 2002, 13, 122–127. [DOI] [PubMed] [Google Scholar]
- Sun S. H. Adv. Mater. 2006, 18, 393–403. [Google Scholar]
- Nakaya M.; Kanehara M.; Teranishi T. Langmuir 2006, 22, 3485–3487. [DOI] [PubMed] [Google Scholar]
- Jeyadevan B.; Hobo A.; Urakawa K.; Chinnasamy C. N.; Shinoda K.; Tohji K. J. Appl. Phys. 2003, 93, 7574–7576. [Google Scholar]
- Takahashi M.; Ogawa T.; Hasegawa D.; Jeyadevan B. J. Appl. Phys. 2005, 97, 10J307. [Google Scholar]
- Yan Q. Y.; Purkayastha A.; Kim T.; Kroger R.; Bose A.; Ramanath G. Adv. Mater. 2006, 18, 2569–2573. [Google Scholar]
- Saita S.; Maenosono S. Chem. Mater. 2005, 17, 6624–6634. [Google Scholar]
- Sun S. H.; Murray C. B.; Weller D.; Folks L.; Moser A. Science 2000, 287, 1989–1992. [DOI] [PubMed] [Google Scholar]
- Chen M.; Liu J. P.; Sun S. J. Am. Chem. Soc. 2004, 126, 8394–8395. [DOI] [PubMed] [Google Scholar]
- Nandwana V.; Elkins K. E.; Poudyal N.; Chaubey G. S.; Yano K.; Liu J. P. J. Phys. Chem. C 2007, 111, 4185–4189. [Google Scholar]
- Howard L. E. M.; Nguyen H. L.; Giblin S. R.; Tanner B. K.; Terry I.; Hughes A. K.; Evans J. S. O. J. Am. Chem. Soc. 2005, 127, 10140–10141. [DOI] [PubMed] [Google Scholar]
- Nguyen H. L.; Howard L. E. M.; Stinton G. W.; Giblin S. R.; Tanner B. K.; Terry I.; Hughes A. K.; Ross I. M.; Serres A.; Evans J. S. O. Chem. Mater. 2006, 18, 6414–6424. [Google Scholar]
- Delalande M.; Marcoux P. R.; Reiss P.; Samson Y. J. Mater. Chem. 2007, 17, 1579–1588. [Google Scholar]
- Gu H. W.; Ho P. L.; Tsang K. W. T.; Yu C. W.; Xu B. Chem. Commun. 2003, 1966–1967. [DOI] [PubMed] [Google Scholar]
- Gu H. W.; Ho P. L.; Tsang K. W. T.; Wang L.; Xu B. J. Am. Chem. Soc. 2003, 125, 15702–15703. [DOI] [PubMed] [Google Scholar]
- Maenosono S.; Saita S. IEEE Trans. Magn. 2006, 42, 1638–1642. [Google Scholar]
- Seehra M. S.; Singh V.; Dutta P.; Neeleshwar S.; Chen Y. Y.; Chen C. L.; Chou S. W.; Chen C. C. J. Phys. D: Appl. Phys. 2010, 43, 145001–145007. [Google Scholar]
- Maenosono S.; Suzuki T.; Saita S. J. Magn. Magn. Mater. 2008, 320, L79–L83. [Google Scholar]
- Gao J. H.; Liang G. L.; Cheung J. S.; Pan Y.; Kuang Y.; Zhao F.; Zhang B.; Zhang X. X.; Wu E. X.; Xu B. J. Am. Chem. Soc. 2008, 130, 11828–11833. [DOI] [PubMed] [Google Scholar]
- Chou S. W.; Shau Y. H.; Wu P. C.; Yang Y. S.; Shieh D. B.; Chen C. C. J. Am. Chem. Soc. DOI: 10.1021/ja1035013, Publication Date (Web) June 24, 2010. [Google Scholar]
- Gao J.; Liang G.; Zhang B.; Kuang Y.; Zhang X.; Xu B. J. Am. Chem. Soc. 2007, 129, 1428–1433. [DOI] [PubMed] [Google Scholar]
- Xu C. J.; Yuan Z. L.; Kohler N.; Kim J. M.; Chung M. A.; Sun S. H. J. Am. Chem. Soc. 2009, 131, 15346–15351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole R.; van Schooneveld M. M.; Hilhorst J.; Donega C. D.; Hart D. C.; van Blaaderen A.; Vanmaekelbergh D.; Meijerink A. Chem. Mater. 2008, 20, 2503–2512. [Google Scholar]
- Jung C. W.; Jacobs P. Magn. Reson. Imaging 1995, 13, 661–674. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for details.
- Wang L. J.; Wang Z. G.; Frank T. G.; Brown S. I.; Chudek S. A.; Cuschieri A. Nanomedicine−UK 2009, 4, 305–315. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia M. A.; McNeil S. E. Nat. Nanotechnol. 2007, 2, 469–478. [DOI] [PubMed] [Google Scholar]
- Lewinski N.; Colvin V.; Drezek R. Small 2008, 4, 26–49. [DOI] [PubMed] [Google Scholar]
- Gupta A. K.; Gupta M. Biomaterials 2005, 26, 1565–1573. [DOI] [PubMed] [Google Scholar]
- Skotland T.; Sontum P. C.; Oulie I. J. Pharmaceut. Biomed. 2002, 28, 323–329. [DOI] [PubMed] [Google Scholar]
- Arbab A. S.; Wilson L. B.; Ashari P.; Jordan E. K.; Lewis B. K.; Frank J. A. NMR Biomed. 2005, 18, 383–389. [DOI] [PubMed] [Google Scholar]
- Conner S. D.; Schmid S. L. Nature 2003, 422, 37–44. [DOI] [PubMed] [Google Scholar]
- Kirkham M.; Parton R. G. Biochim. Biophys. Acta 2005, 1745, 273–286. [DOI] [PubMed] [Google Scholar]
- Xu C.; Yuan Z.; Kohler N.; Kim J.; Chung M. A.; Sun S. J. Am. Chem. Soc. 2009, 131, 15346–15351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton R. R.; Wilmet S.; Legssyer R.; Ward R. J. J. Inorg. Biochem. 2002, 91, 9–18. [DOI] [PubMed] [Google Scholar]
- Bilensoy E. Exp. Opin. Drug Deliv. 2010, 7, 795–809. [DOI] [PubMed] [Google Scholar]
- Han G.; Mokari T.; Ajo-Franklin C.; Cohen B. E. J. Am. Chem. Soc. 2008, 130, 15811–15813. [DOI] [PubMed] [Google Scholar]
- Mei B. C.; Susumu K.; Medintz I. L.; Mattoussi H. Nat. Protoc. 2009, 4, 412–423. [DOI] [PubMed] [Google Scholar]
- Arbab A. S.; Bashaw L. A.; Miller B. R.; Jordan E. K.; Lewis B. K.; Kalish H.; Frank J. A. Radiology 2003, 229, 838–846. [DOI] [PubMed] [Google Scholar]
- Arbab A. S.; Yocum G. T.; Wilson L. B.; Parwana A.; Jordan E. K.; Kalish H.; Frank J. A. Mol. Imaging 2004, 3, 24–32. [DOI] [PubMed] [Google Scholar]
- Dausend J.; Musyanovych A.; Dass M.; Walther P.; Schrezenmeier H.; Landfester K.; Mailander V. Macromol. Biosci. 2008, 8, 1135–1143. [DOI] [PubMed] [Google Scholar]
- Raffa V.; Ciofani G.; Vittorio O.; Riggio C.; Cuschieri A. Nanomedicine 2010, 5, 89–97. [DOI] [PubMed] [Google Scholar]
- Lee D. C.; Mikulec F. V.; Pelaez J. M.; Koo B.; Korgel B. A. J. Phys. Chem. B 2006, 110, 11160–11166. [DOI] [PubMed] [Google Scholar]
- Thomson T.; Terris B. D.; Toney M. F.; Raoux S.; Baglin J. E. E.; Lee S. L.; Sun S. J. Appl. Phys. 2004, 95, 6738–6740. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.