

Abstract
Background and purpose
Animals subjected to an inflammatory insult with lipopolysaccharide (LPS) at the time of stroke are predisposed to develop a detrimental autoimmune response to myelin basic protein (MBP). In this study, we sought to determine whether other inflammatory stimuli could similarly invoke central nervous system (CNS) autoimmunity and whether these detrimental autoimmune responses occurred to antigens other than MBP.
Methods
Male Lewis rats underwent 3 h middle cerebral artery occlusion (MCAO) and received intraperitoneal injections of LPS, staphylococcal enterotoxin B (SEB), lipoteichoic acid (LTA) or saline at the time of reperfusion. Behavioral tests were performed at set time intervals after MCAO and animals were sacrificed at 1 month to analyze the immune response to MBP, neuron specific enolase (NSE) and proteolipid protein (PLP).
Results
Lymphocytes from SEB treated animals were highly reactive to all tested CNS antigens, but treatment with LPS was most likely to lead to a Th1(+) response. A Th1(+) response to MBP, NSE or PLP in spleen was associated with worse outcome, although the response to NSE was most predictive of poor outcome. Animals with a cell mediated autoimmune response to either MBP or NSE in spleen had a concomitant humoral response to these antigens.
Conclusions
These data show that LPS, but not other inflammatory stimuli, increase the likelihood of developing a detrimental autoimmune response to an array of brain antigens.
Keywords: Stroke, Toll-like receptor, Autoimmune, LPS, LTA, SEB, MBP, NSE, PLP, Th1, Fractalkine
Introduction
Pathogen-associated molecular patterns (PAMPs) are molecular motifs that are highly conserved within a class of microbes. PAMPS are capable of initiating the innate immune response through activation of toll-like receptors (TLRs) [1].
Lipopolysaccharide (LPS), a component of the Gram-negative bacterial cell, is a prototypical PAMP and a potent activator of the innate immune response; this effect of LPS is mediated through TLR4 [2]. In previous studies we showed that animals subjected to a systemic inflammatory insult with LPS at the time of stroke develop a deleterious autoimmune response to the central nervous system (CNS) antigen myelin basic protein (MBP) [3]. Several recent studies demonstrate a role for TLR4 in potentiating cerebral ischemic injury in experimental stroke [4–6].
Infections are common following stroke and are associated with worse outcome [7, 8]. The risk of infection is highly associated with stroke severity [7], and patients with the most severe strokes tend to be cared for in the intensive care unit setting. The detrimental effects of LPS on stroke outcome are therefore clinically relevant. It is not just Gram-negative bacteria that cause infections, however, so in the current study we sought to determine whether activation of TLR2 (using lipoteichoic acid [LTA], a component of the Gram-positive cell wall) would similarly induce CNS autoimmunity and worsen outcome in an animal model of severe stroke. We also assessed the immune response to other CNS antigens, namely neuron specific enolase (NSE) and proteolipid protein (PLP). Finally, the effect of the bacterial superantigen staphylococcal enterotoxin B (SEB), an antigen non-specific activator of T cells, on stroke outcome and the development of post-ischemic autoimmunity was assessed.
Methods
Animals
Experiments were approved by the Institution's Animal Care and Use Committee. Male Lewis rats (250–300 g) were used for all studies.
Middle Cerebral Artery Occlusion (MCAO)
Anesthesia was induced with 5% and maintained with 1.5% isoflurane. After midline neck incision, the right common carotid, internal carotid and pterygopalatine arteries were ligated. A monofilament suture (4.0) was inserted into the common carotid artery and advanced into the internal carotid artery to occlude the origin of middle cerebral artery (MCA) [9]. Animals were maintained at normothermia during surgery and reperfused 3 h after MCAO. In sham-operated animals, the suture was inserted into the carotid artery but not advanced. Rectal temperature and body weight were assessed at set time intervals. Animals were sacrificed 1 month after surgery.
Administration of Immunologically Active Substances
All immunologically active substances were purchased from Sigma and administered intraperitoneally at a dose of 1 mg/kg (mixed as 1 mg/ml) 3 h after MCAO (at reperfusion) or sham surgery. Animals were treated with endotoxin-free saline (“controls”, 1 ml/kg; n = 21), LPS (from Escherichia coli, serotype 026:B6; n = 30), SEB (from Staphylococcus aureus, strain #S6; n = 20) or LTA (from S. aureus, strain # DSM20233; n = 30); doses were chosen based on prior experience and literature review [3, 10, 11]. Sham surgery was performed on saline (n = 4), LPS (n = 5), SEB (n = 4) and LTA (n = 6) treated animals.
Neurological Outcome
Neurological outcome was assessed at set time points. Tests included a modification of the Bederson scale (0 = no deficit, 1 = holds forepaw in flexed posture, 2 = inability to resist lateral push, 3 = circling, 4 = agitated circling, 5 = stupor), performance on the rotarod and performance on the foot fault test [12, 13]. Only animals that had a neurological score of at least 3 were included in the study. For the rotarod, animals were trained prior to surgery until they could remain on the rotating rod at 5 rpm for 100 s; following surgery, the longest time animals could remain on the rotarod before falling (100 s maximum) was recorded (using the best of 3 trials). For the foot fault test, rats were placed on a wire grid for 3 min and the number of times the affected front paw slipped through the grid per total number of steps taken was recorded.
ELISPOT Assay
Animals were sacrificed 1 month after MCAO/sham surgery and mononuclear cells (MNCs) isolated from the entire forebrain and spleen using previously described methods [3, 14]. MNCs were cultured (1 × 105 cells/well) for 48 h in 96-well plates (MultiScreen®-IP; Millipore) in media alone or in media supplemented with MBP (50 μg/ml; Sigma), NSE (10 μg/ml; Sigma) or PLP 139-151 (10 μg/ml; ANASPEC). All experiments were performed in triplicate. Antigen-specific secretion of IFN-γ (number of spots with antigen above number of spots in media alone) was used as an indicator of the Th1 response; antigen-specific secretion of TGF-β1 (number of spots with antigen above number of spots in medial alone) was used as an indicator of the Treg response. Spots were counted by two independent investigators blinded to treatment status and aided by a semi-automated software system (Metamorph®). Results are expressed as the ratio of the increase in the number of antigen-specific IFN-γ secreting cells to the ratio of the increase in the number of antigen-specific TGF-β1 secreting cells. Based on previous ELISPOT data from our laboratory, the lower interquartile range for this ratio in animals sensitized to MBP (by injection in complete Freund's adjuvant) was 1.48 [15]. We thus considered animals to have a Th1(+) response to the antigen of interest if the ratio of the IFN:TGF response was ≥1.48.
Enzyme-Linked ImmunoSorbent Assays (ELISAs)
Blood was collected by cardiac puncture at the time of sacrifice; serum was stored at −80° until use. Fractalkine (CX3CL1) concentrations were assessed using a commercially available kit (R&D Systems). Titers of IgG antibodies specific for MBP, NSE, and PLP were measured using indirect ELISA; data are presented as relative optical density units.
Statistics
Data are displayed as mean ± standard error of the mean (SEM). Categorical data were evaluated using the χ2-test statistic. Parametric data were compared using the t-test or analysis of variance (ANOVA) with post-hoc Dunnett's test. Significance was set at P < 0.05.
Results
Effects of LPS, LTA and SEB Treatment
Mortality rates were 18/30 (60%) in LPS treated, 2/20 (10%) in SEB treated, 12/30 (40%) in LTA treated and 6/22 (27%) in saline treated animals (P = 0.003). With the exception of one SEB treated animal that died between weeks 3 and 4 of an unknown cause, all deaths occurred within 48 h of MCAO and were felt to be related to herniation. There were no deaths among sham-operated animals.
Body temperatures were similar following MCAO, except at 6 h when the rectal temperature was higher in LPS than saline treated animals (Fig. 1a). Animals treated with LPS tended to have consistently higher (worse) neurological scores than saline treated animals over the first week of the study (although neurological scores were highest in LTA treated animals at 1 month after MCAO; Fig. 1b). Changes in body weight differed significantly among treatment groups (P = 0.036), with LTA treated animals regaining the least and SEB regaining the most weight (Fig. 1c); these changes, however, did not differ significantly from saline treated animals. Behavioral outcome was also similar among experimental groups with no differences in performance on the foot fault test (Fig. 1d) or on the rotarod (Fig. 1e). There were no differences in the change in body weight, temperature or neurological outcome of LPS, LTA, SEB or saline treated sham-operated rats (data not shown).
Fig. 1.

Effect of LPS, SEB and LTA administration on outcome. Animals treated with LPS had higher temperatures 6 h after MCAO (a), but temperatures were otherwise similar between treatment groups. Treatment with LPS was associated with higher (worse) neurological scores at multiple time points after MCAO, but treatment with LTA was associated with the worst scores 1 month after MCAO (b). There were significant differences in the degree of weight loss following MCAO, but the degree of weight loss was not significantly different from that of saline treated animals (c). Performance on the foot fault test (d) and the rotarod (e) did not differ between treatment groups. * P < 0.05, ** P < 0.01 compared to saline treatment using ANOVA with post-hoc Dunnet's
Animals treated with LPS had higher circulating titers of MBP antibodies (0.487 ± 0.073 optical units) and NSE antibodies (0.181 ± 0.056 optical units) than control animals (0.254 ± 0.041 optical units; P = 0.002 and 0.0173 ± 0.008 optical units; P = 0.001, respectively). There were no significant differences in the titers of these antibodies among SEB, LTA and saline treated animals. PLP antibody titers were similar among all treatment groups.
Immune Responses to Brain Antigens
MNCs isolated from the forebrain of LPS (15.5 × 106 ± 1.3 × 106; P = 0.03), SEB (18.1 × 106 ± 2.3 × 106; P = 0.009) and LTA (16.3 × 106 ± 4.5 × 106; P = 0.08) treated animals were greater in number than in control animals (8.9 × 106 ± 1.6 × 106). Splenocyte numbers were similar in all treatment groups. SEB treated animals had the most robust responses to the tested antigens (MBP, NSE and PLP) in both brain (Fig. 2a) and spleen (Fig. 2b). These responses, however, were similarly robust for IFN-γ and TGF-β1, such that there was no difference in the ratio of the relative increase in the number of antigen-specific IFN-γ secreting cells to the relative increase in the number of antigen-specific TGF-β1 secreting cells (the “Th1 response”) to MBP, NSE or PLP between SEB and saline treated animals.
Fig. 2.

Antigen-specific IFN-γ and TGFβ-1 immune responses in brain and spleen. Treatment with SEB resulted in marked activation of the immune response, with an increase in the number of antigen-specific cells producing both cytokines (IFN-γ and TGFβ-1) in brain (a) and spleen (b). * P < 0.05, ** P < 0.01 compared to saline treatment using ANOVA with post-hoc Dunnet's
The degree of the Th1 response to MBP, NSE and PLP among MNCs isolated from brain was similar for LPS, SEB, LTA and saline treated animals; a more robust Th1 response, however, was seen to MBP and NSE in the splenocytes from LPS treated animals (Fig. 3). Moreover, the proportion of animals evidencing a Th1(+) response to MBP, NSE or PLP in spleen was greater in LPS treated animals; there were no differences in the proportion of animals evidencing a Th1(+) response to any of these antigens as detected in the MNCs isolated from brain (Table 1).
Fig. 3.
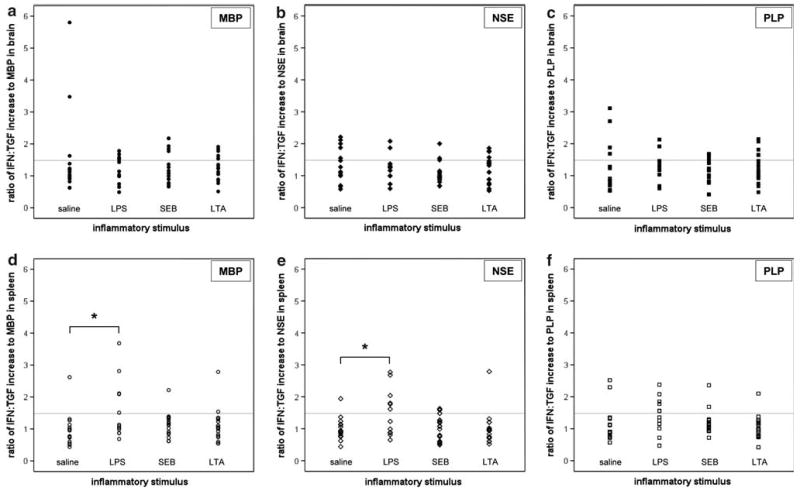
Th1 responses to MBP, NSE and PLP; individual animal data. Individual animal data are presented and depict the ratio of the relative increase in the number of cells responding to MBP, NSE and PLP with the secretion of IFN-γ to that responding with the secretion of TGFβ-1 among mononuclear cells isolated from brain (a–c) and spleen (d–f). A value ≥1.48 (gray line) is considered indicative of a Th1 response. * P < 0.05 compared to saline treatment using ANOVA with post-hoc Dunnet's
Table 1.
Proportion of animals developing an immune response to brain antigens by treatment status
| Stimulus: | None N = 15 |
LPS N = 12 |
SEB N = 18 |
LTA N = 18 |
P | ||||
|---|---|---|---|---|---|---|---|---|---|
| Th1(−) | Th1(+) | Th1(−) | Th1(+) | Th1(−) | Th1(+) | Th1(−) | Th1(+) | ||
| Brain | |||||||||
| MBP | 12 (81%) | 3 (19%) | 8 (67%) | 4 (33%) | 13 (72%) | 5 (28%) | 12 (67%) | 6 (33%) | NS |
| NSE | 10 (67%) | 5 (33%) | 10 (83%) | 2 (17%) | 15 (83%) | 3 (17%) | 13 (72%) | 5 (28%) | NS |
| PLP | 11 (73%) | 4 (27%) | 10 (83%) | 2 (17%) | 14 (78%) | 4 (22%) | 13 (72%) | 5 (28%) | NS |
| Spleen | |||||||||
| MBP | 14 (93%) | 1 (7%) | 7 (58%) | 5 (42%) | 16 (89%) | 2 (11%) | 17 (94%) | 1 (6%) | 0.025 |
| NSE | 14 (93%) | 1 (7%) | 6 (50%) | 6 (50%) | 17 (94%) | 1 (6%) | 14 (78%) | 4 (22%) | 0.011 |
| PLP | 13 (87%) | 2 (13%) | 6 (50%) | 6 (50%) | 17 (94%) | 1 (6%) | 16 (89%) | 2 (11%) | 0.010 |
Given that the differences in the immune response were more evident in spleen, data addressing the effect of the immune response on outcome will focus on the splenic response. Neurological scores were consistently higher (worse) among animals with a Th1(+) response to MBP, NSE or PLP in spleen (Fig. 4). Animals with a Th1(+) response to NSE and PLP in spleen had more weight loss than those without a Th1(+) response to these antigens (Fig. 5). A Th1(+) response to NSE in spleen was predictive of worse performance on the foot fault test both early (3 days) and late (1 month) after MCAO (Fig. 6b), while a Th1(+) response to PLP in spleen was predictive of worse performance only at 1 month after MCAO (Fig. 6c). In addition, the degree of the Th1 response to NSE and PLP correlated with performance on the foot fault test, with those animals displaying a more robust response making more errors (r = 0.335, P = 0.021 for NSE and r = 0.343, P = 0.018 for PLP). And for animals with a Th1(+) response to NSE in spleen, higher titers of NSE antibodies also correlated with worse performance on the foot fault test (r = 0.828, P = 0.006). Performance on the rotarod was also associated with the immune response to CNS antigens; animals evidencing a Th1(+) response to MBP, NSE or PLP had shorter latencies to fall than animals without such an immune response (Fig. 7a–c). Further, there was an inverse relationship between the time spent on the rotarod and the degree of the Th1 response to MBP (r = −0.304, P = 0.021), NSE (r = −0.396, P = 0.002) and PLP (r = −0.277, P = 0.037).
Fig. 4.

Neurological scores as a function of the immune response following MCAO. For animals with a Th1(+) response to MBP, NSE or PLP in spleen, the neurological scores were higher (worse) following MCAO (a–c). * P < 0.05, ** P < 0.01 using the t-test
Fig. 5.

Change in weight as a function of the immune response following MCAO. A Th1(+) response to MBP in spleen was not associated with a differential in weight gain after MCAO (a). A Th1(+) immune response to NSE or PLP in spleen was associated with less weight gain after stroke (b, c), especially for those animals with a Th1 response to PLP. * P < 0.05 using the t-test
Fig. 6.

Immunological outcome and foot fault performance. A Th1(+) response to MBP in spleen did not affect performance on the foot fault test after MCAO (a). Animals evidencing a Th1(+) response to NSE (b) or PLP (c) in spleen made more errors on the foot fault test 1 month after MCAO. * P < 0.05, ** P < 0.01 using the t-test
Fig. 7.

Immunological outcome and rotarod performance. Animals exhibiting a Th1(+) response to MBP (a), NSE (b) or PLP (c) in spleen performed less well on the rotarod (with shorter latency to fall) after stroke. * P < 0.05, ** P < 0.01 using the t-test
Of the 9 animals with a Th1(+) response to MBP in spleen, 7 also evidenced a Th1(+) response to NSE and 5 evidenced a Th1(+) response to PLP in spleen (Fig. 8a). There were 5 animals that evidenced a Th1(+) response to all tested antigens, 3 of which received LPS (P = 0.089). The 5 animals with a Th1(+) response to all antigens performed considerably worse on the rotarod than other animals (10.5% ± 3.0% vs. 59.3% ± 5.6%; P < 0.001); there was also a trend toward worse performance on the foot fault test (32.2% ± 8.7% vs. 19.5% ± 2.1%; P = 0.065). In addition, these animals with cellular immune responses to MBP, NSE and PLP had higher MBP antibody titers (P = 0.029).
Fig. 8.

Overlap in the immune response to antigen in brain and spleen among individual animals. In both spleen (a) and brain (b), there were 5 animals that evidenced a Th1(+) response to all 3 antigens studied. There was little overlap in the Th1(+) response to MBP (c) or PLP (e) in spleen and brain, while the overlap for NSE was more substantial (d)
The immune response in brain was not as predictive of outcome following MCAO as the response in spleen, but animals with a Th1(+) response to MBP in brain performed worse on the rotarod at 1 month (data not shown). A Th1(+) response to NSE in brain was also associated with worse performance on the rotarod 1 month after MCAO and more errors on the foot fault test 2 weeks after MCAO (data not shown). The robustness of this cellular immune response to NSE in brain was also inversely correlated to performance on the rotarod (r = −0.345, P = 0.009).
Of the 18 animals with a Th1(+) response to MBP in brain, 8 also evidenced a Th1(+) response to NSE in brain and 10 evidenced a Th1(+) response to PLP in brain; there were 5 animals that evidenced a Th1(+) response to all 3 antigens (Fig. 8b). The 5 animals with Th1(+) responses to all 3 antigens made more errors on the foot fault test (15.3% ± 1.2% vs. 21.2% ± 2.3%; P = 0.030). Interestingly, animals with a Th1(+) response to MBP, NSE and PLP in brain had lower titers of MBP (P = 0.039) and NSE (P < 0.001), but not PLP, antibodies.
Among the 18 animals with a Th1(+) response to MBP in brain, only 2 (11%) evidenced a Th1(+) response to MBP in spleen (Fig. 8c). Among the 15 animals with a Th1(+) response to NSE in brain, 6 (40%) also evidenced a Th1(+) response to NSE in spleen (Fig. 8d). And for the 15 animals with a Th1(+) response to PLP in brain, only 2 (13%) had evidence of a Th1(+) response to PLP in spleen (Fig. 8e). Animals with a Th1(+) response to MBP or NSE in both brain and spleen performed worse on the rotarod (P < 0.001 and P = 0.017, respectively). Surprisingly, antibody titers to NSE were significantly lower in animals with a Th1(+) response to this antigen in both brain and spleen (P < 0.001).
Among sham-operated animals, there were no differences in the cellular or humoral responses to MBP, PLP or NSE among LPS, SEB, LTA and saline treated animals.
Fractalkine
Serum fractalkine concentrations were 526.5 ± 55.3 pg/ml in control animals, 813.7 ± 142.0 (P = 0.087) in LPS treated, 689.4 ± 83.0 pg/ml (NS) in SEB treated and 776.9 ± 54.9 (P = 0.035) in LTA treated animals. There was an inverse correlation between fractalkine concentrations and performance on the rotarod (r = −0.370; P = 0.026).
Animals with a Th1(+) response to NSE in brain had higher serum concentrations of fractalkine than those without (857.6 ± 77.6 pg/ml vs. 649.1 ± 42.0; P = 0.021); neither a Th1(+) response to MBP nor PLP in brain affected the circulating concentration of fractalkine. Fractalkine concentrations were also higher in animals with a Th1(+) response to MBP, NSE or PLP in spleen; for individual animals with a Th1(+) response to all 3 antigens, fractalkine concentrations were markedly elevated (1039.7 ± 20.7 pg/ml vs. 681.7 ± 39.0 pg/ml; P < 0.001).
There were no differences among fractalkine concentrations in sham-operated animals among LPS, SEB, LTA and saline treated animals.
Discussion
Infections are common following stroke and are associated with worse outcome [7, 8]. Typical infections include pneumonias, which are caused by both Gram-positive and Gram-negative bacteria, and urinary tract infections, which are generally caused by Gram-negative bacteria [8]. LPS is a component of the Gram-negative bacterial cell wall and LTA a component of the Gram-positive cell wall. Both LPS and LTA are examples of PAMPs capable of initiating the innate immune response through activation of TLRs; LPS mediates its effects through TLR4 while LTA mediates its effects through TLR2 [1].
Multiple lines of evidence suggest that activation of the TLRs provides an important link between innate and adaptive immunity [16, 17]. In the current study, administration of LPS itself was not associated with worse outcome at 1 month after MCAO, arguing that it is the predisposition to the development of an autoimmune response to CNS antigens (MBP, NSE, PLP) in LPS treated animals that results in the poor outcome. Administration of LTA (a TLR2 agonist), on the other hand, was associated with worse outcomes (higher neurological scores and less weight gain) following MCAO but did not predispose to the development of a Th1(+) response to any of the CNS antigens studied. This observation also argues against the possibility that LPS exacerbated the initial brain injury that predisposed to the generation of an immune response to brain. Further, we showed in a prior study that infarct size was similar in LPS and saline treated animals [3]. It is known, however, that lymphocytes function better at higher temperatures, suggesting that one of the possible mechanisms by which LPS predisposes to CNS autoimmune responses following stroke is by enhancing lymphocytic responses during times of blood–brain barrier (BBB) compromise [18].
For a lymphocyte to become activated to a specific antigen, it must encounter that antigen in context of the major histocompatibility class II (MHC II) molecule and receive an additional costimulatory signal [19]. In brain, microglia can express MHC II and act as antigen presenting cells (APCs) [20, 21]. LPS and LTA are both capable of inducing APCs in the brain and the periphery to express the costimulatory signals needed for lymphocyte activation [22–24]. LPS, however, can also stimulate T cells directly through TLR4 to affect their responses [25]. And despite the fact that TLR2 stimulation activates dendritic cells, recent studies suggest that stimulation of TLR2 induces T regulatory responses and protects against autoimmunity [26]. These facts may help to explain why TLR4 activation appears to be a more potent link between the innate and adaptive immune response than TLR2 activation [27–29].
SEB is a “superantigen” derived from S. aureus; superantigens are capable of activating T cells in the absence of antigen through interaction with MHC II and the β chain of the T cell receptor [30]. Since early studies implicated superantigens as an important link to autoimmune disease, we hypothesized that administration of SEB would be associated with development of an autoimmune response in our stroke model [31]. More recent data, however, suggest that superantigens may actually induce immunological tolerance [32]. Despite robust lymphocyte activation following SEB treatment in the current study, SEB did not predispose to the development of a Th1(+) response to the CNS antigens examined or worsen outcome. These findings, in concert, suggest that it may be activation of the innate immune response through TLR4 that is crucial for the development of detrimental autoimmunity.
T cell responses are characterized by the cytokines produced upon antigen stimulation; Th1 responses are characterized by antigen-induced secretion of IFN-γ and are important for cell mediated immunity. Regulatory T cell responses (Treg), on the other hand, serve to limit the immune response and are characterized by antigen-specific secretion of either IL-10 or TGFβ-1 [33]. We previously showed that the nature of the immune response to MBP following stroke predicts outcome [3, 34]. In the current study, these findings have been extended to show that Th1(+) responses to NSE and PLP also predict poor outcome; further, the peripheral immune response was more predictive of poor outcome than the central immune response. Surprisingly, we did not see a great deal of overlap in the immune response to a given antigen in brain and spleen among individual animals. Equally surprising was the fact that animals evidencing a Th1(+) response to MBP and NSE in brain tended to have a decrease in circulating antibodies to these antigens, an observation not previously documented in the context of other CNS inflammatory diseases. There are ample data, however, to suggest that the immune response in the brain and periphery is differentially regulated [35, 36].
The fact that a detrimental immune response could be detected to all of the antigens studied (MBP, NSE, PLP) might argue that these responses reflect a fundamental perturbation of the immune system that is associated with worse outcome and argue against a pathological role for the response. Our experiments cannot directly support or refute that an antigen-specific response to brain antigens leads to the worse outcome, an issue that will need to be addressed in future studies. That stroke can lead to a cellular immune response directed toward brain antigens, however, is demonstrated in patients within weeks of stroke onset; MBP and PLP specific T cells are found in the peripheral circulation of patients, with increased numbers of these autoreactive T cells identified in their cerebrospinal fluid [37]. In this clinical study, there was no attempt to correlate the immune response to MBP or PLP with stroke outcome, and no data were provided regarding the contribution of infection (and hence, TLR stimulation) to the development of these cellular immune responses. Other clinical studies have demonstrated the presence of a humoral response to a variety of brain antigens following stroke, including neurofilaments and portions of the N-methyl-d-aspartate (NMDA) receptor [38, 39]. Whether or not these circulating antibodies are pathologic is unknown, but multiple studies demonstrate the presence of an immune response to such antigens in a variety of post-infectious autoimmune diseases, as well as in chronic neurodegenerative diseases [40–45]. Under normal circumstances, these antibodies would be excluded from the brain; during periods of BBB compromise, however, the antibodies could gain reach the parenchyma and have potentially detrimental effects.
In the current study, administration of LPS was associated with a transient worsening of neurological function as assessed by the neurological score (Fig. 1b). Given that this effect was short lived, it can be most easily attributed to the “sickness behavior” associated with LPS administration [46]. Our data also show that the immune responses to MBP, NSE or PLP differentially affect the measures of behavioral outcome; the neurological score, the foot fault test and the rotarod all assess discrete aspects of neurological function. Given that MBP, NSE and PLP are differentially expressed by cells within the CNS, it seems reasonable that an immune response directed specifically against one of these antigens could result in distinct functional impairments. Future studies should address the effect these autoimmune responses on learning and “cognitive” outcomes.
Fractalkine (CX3CL1) is a chemokine that is highly expressed on neurons and endothelial cells; the fractalkine receptor (CX3CR1) is expressed on microglia and mononuclear cells (especially natural killer cells) [47, 48]. Fractalkine is cleaved from the surface of neurons during injury and is highly chemotactic for monocytes [49]. Experimental studies show increased expression of fractalkine following stroke and improved stroke outcome in transgenic animals that do not express fractalkine or its receptor [50–52]. Additionally, fractalkine appears to be useful as a surrogate marker of inflammation in immune mediated diseases [53–55]. In a previous study we showed that circulating fractalkine concentrations were higher in animals with a Th1(+) response to MBP [34]. In the current study, a similar elevation in fractalkine concentrations was seen in animals with a Th1(+) response to NSE and PLP (as well as MBP); the elevation in circulating fractalkine concentrations was particularly striking among the 5 animals that developed a Th1(+) response to all 3 antigens in spleen. While animals treated with LPS in our study did not evidence higher concentrations of fractalkine in sera at 1 month following MCAO, LPS is known to upregulate fractalkine and amplify the Th1 response [56]. It is thus possible that an increase in fractalkine following LPS treatment contributed to the development of the Th1 response. Whether fractalkine predisposes to worse outcome or is merely associated with worse outcome, animals with higher fractalkine concentrations perform less well on the rotarod than those with lower concentrations. To our knowledge, fractalkine concentrations have not been assessed in patients with stroke; our findings suggest that circulating fractalkine may be a useful surrogate marker of inflammation in stroke.
In summary, administration of the TLR4 agonist LPS in the immediate post-stroke period is associated with the development of a detrimental immune response to brain antigens; this immune response has both a cellular and humoral component. Whether this immune response leads to worse outcome or is just associated with worse outcome needs to be addressed in future studies, as does the dependence of TLR4 in the development of this response. Our findings may help to explain why infection is associated with worse outcome from stroke and suggest that infection with Gram-negative organisms would more adversely affect outcome than infection with Gram-positive organisms.
Acknowledgments
This work was supported by grants from the National Institutes of Neurological Disorders and Stroke (NINDS) (1RO1NS056457).
References
- 1.Netea MG, van der Graaf C, Van der Meer JW, Kullberg BJ. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J Leukoc Biol. 2004;75:749–55. doi: 10.1189/jlb.1103543. [DOI] [PubMed] [Google Scholar]
- 2.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 3.Becker KJ, Kindrick DL, Lester MP, Shea C, Ye ZC. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J Cereb Blood Flow Metab. 2005;25:1634–44. doi: 10.1038/sj.jcbfm.9600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomyinduced neurodegeneration. Neurobiol Dis. 2008;31:33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Caso JR, Pradillo JM, Hurtado O, Leza JC, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke. 2008;39:1314–20. doi: 10.1161/STROKEAHA.107.498212. [DOI] [PubMed] [Google Scholar]
- 6.Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–14. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 7.Aslanyan S, Weir CJ, Diener HC, Kaste M, Lees KR. Pneumonia and urinary tract infection after acute ischaemic stroke: a tertiary analysis of the GAIN International trial. Eur J Neurol. 2004;11:49–53. doi: 10.1046/j.1468-1331.2003.00749.x. [DOI] [PubMed] [Google Scholar]
- 8.Harms H, Prass K, Meisel C, et al. Preventive antibacterial therapy in acute ischemic stroke: a randomized controlled trial. PLoS ONE. 2008;3:e2158. doi: 10.1371/journal.pone.0002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 10.Huang W, Koller LD. Superantigen activation and kinetics of cytokines in the Long-Evans rat. Immunology. 1998;95:331–8. doi: 10.1046/j.1365-2567.1998.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterjee PK, Zacharowski K, Cuzzocrea S, et al. Lipoteichoic acid from Staphylococcus aureus reduces renal ischemia/reperfusion injury. Kidney Int. 2002;62:1249–63. doi: 10.1111/j.1523-1755.2002.kid580.x. [DOI] [PubMed] [Google Scholar]
- 12.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–6. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 13.Hernandez TD, Schallert T. Seizures and recovery from experimental brain damage. Exp Neurol. 1988;102:318–24. doi: 10.1016/0014-4886(88)90226-9. [DOI] [PubMed] [Google Scholar]
- 14.Becker K, Kindrick D, McCarron R, Hallenbeck J, Winn R. Adoptive transfer of myelin basic protein-tolerized splenocytes to naive animals reduces infarct size: a role for lymphocytes in ischemic brain injury? Stroke. 2003;34:1809–15. doi: 10.1161/01.STR.0000078308.77727.EA. [DOI] [PubMed] [Google Scholar]
- 15.Becker KJ. Sensitization and tolerization to brain antigens in stroke. Neuroscience. 2009;158:1090–7. doi: 10.1016/j.neuroscience.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasare C, Medzhitov R. Toll-like receptors and acquired immunity. Semin Immunol. 2004;16:23–6. doi: 10.1016/j.smim.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Toubi E, Shoenfeld Y. Toll-like receptors and their role in the development of autoimmune diseases. Autoimmunity. 2004;37:183–8. doi: 10.1080/08916930410001704944. [DOI] [PubMed] [Google Scholar]
- 18.Huang YH, Haegerstrand A, Frostegard J. Effects of in vitro hyperthermia on proliferative responses and lymphocyte activity. Clin Exp Immunol. 1996;103:61–6. doi: 10.1046/j.1365-2249.1996.00932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croft M, Dubey C. Accessory molecule and costimulation requirements for CD4 T cell response. Crit Rev Immunol. 1997;17:89–118. doi: 10.1615/critrevimmunol.v17.i1.40. [DOI] [PubMed] [Google Scholar]
- 20.Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–2. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- 21.Kato H, Kogure K, Liu XH, Araki T, Itoyama Y. Progressive expression of immunomolecules on activated microglia and invading leukocytes following focal cerebral ischemia in the rat. Brain Res. 1996;734:203–12. [PubMed] [Google Scholar]
- 22.Hathcock KS, Laszlo G, Pucillo C, Linsley P, Hodes RJ. Comparative analysis of B7-1 and B7-2 costimulatory ligands: expression and function. J Exp Med. 1994;180:631–40. doi: 10.1084/jem.180.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang-Shieh YF, Yeh KY, Wei IH, et al. Responses of microglia in vitro to the gram-positive bacterial component, lipoteichoic acid. J Neurosci Res. 2005;82:515–24. doi: 10.1002/jnr.20663. [DOI] [PubMed] [Google Scholar]
- 24.Lee SJ, Lee S. Toll-like receptors and inflammation in the CNS. Curr Drug Targets Inflamm Allergy. 2002;1:181–91. doi: 10.2174/1568010023344698. [DOI] [PubMed] [Google Scholar]
- 25.Zanin-Zhorov A, Tal-Lapidot G, Cahalon L, et al. Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J Immunol. 2007;179:41–4. doi: 10.4049/jimmunol.179.1.41. [DOI] [PubMed] [Google Scholar]
- 26.Manicassamy S, Ravindran R, Deng J, et al. Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med. 2009;15:401–9. doi: 10.1038/nm.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kerfoot SM, Long EM, Hickey MJ, et al. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol. 2004;173:7070–7. doi: 10.4049/jimmunol.173.11.7070. [DOI] [PubMed] [Google Scholar]
- 28.Kato N, Fujii Y, Agata N, et al. Experimental murine model for autoimmune myocarditis using Klebsiella pneumoniae O3 lipopolysaccharide as a potent immunological adjuvant. Autoimmunity. 1993;14:231–6. doi: 10.3109/08916939309077370. [DOI] [PubMed] [Google Scholar]
- 29.Zaccone P, Fehervari Z, Blanchard L, Nicoletti F, Edwards CK, 3rd, Cooke A. Autoimmune thyroid disease induced by thyroglobulin and lipopolysaccharide is inhibited by soluble TNF receptor type I. Eur J Immunol. 2002;32:1021–8. doi: 10.1002/1521-4141(200204)32:4<1021::AID-IMMU1021>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 30.Proft T, Fraser JD. Bacterial superantigens. Clin Exp Immunol. 2003;133:299–306. doi: 10.1046/j.1365-2249.2003.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedman SM, Tumang JR, Crow MK. Microbial superantigens as etiopathogenic agents in autoimmunity. Rheum Dis Clin North Am. 1993;19:207–22. [PubMed] [Google Scholar]
- 32.Ivars F. Superantigen-induced regulatory T cells in vivo. Chem Immunol Allergy. 2007;93:137–60. doi: 10.1159/000100862. [DOI] [PubMed] [Google Scholar]
- 33.Romagnani S. Regulation of the T cell response. Clin Exp Allergy. 2006;36:1357–66. doi: 10.1111/j.1365-2222.2006.02606.x. [DOI] [PubMed] [Google Scholar]
- 34.Gee JM, Kalil A, Thullbery M, Becker KJ. Induction of immunologic tolerance to myelin basic protein prevents central nervous system autoimmunity and improves outcome after stroke. Stroke. 2008;39:1575–82. doi: 10.1161/STROKEAHA.107.501486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hofstetter HH, Targoni OS, Karulin AY, Forsthuber TG, Tary-Lehmann M, Lehmann PV. Does the frequency and avidity spectrum of the neuroantigen-specific T cells in the blood mirror the autoimmune process in the central nervous system of mice undergoing experimental allergic encephalomyelitis? J Immunol. 2005;174:4598–605. doi: 10.4049/jimmunol.174.8.4598. [DOI] [PubMed] [Google Scholar]
- 36.Muhallab S, Lidman O, Weissert R, Olsson T, Svenningsson A. Intra-CNS activation by antigen-specific T lymphocytes in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2001;113:202–11. doi: 10.1016/s0165-5728(00)00438-0. [DOI] [PubMed] [Google Scholar]
- 37.Wang WZ, Olsson T, Kostulas V, Hojeberg B, Ekre HP, Link H. Myelin antigen reactive T cells in cerebrovascular diseases. Clin Exp Immunol. 1992;88:157–62. doi: 10.1111/j.1365-2249.1992.tb03056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bornstein NM, Aronovich B, Korczyn AD, Shavit S, Michaelson DM, Chapman J. Antibodies to brain antigens following stroke. Neurology. 2001;56:529–30. doi: 10.1212/wnl.56.4.529. [DOI] [PubMed] [Google Scholar]
- 39.Dambinova SA, Khounteev GA, Izykenova GA, Zavolokov IG, Ilyukhina AY, Skoromets AA. Blood test detecting autoantibodies to N-methyl-d-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin Chem. 2003;49:1752–62. doi: 10.1373/49.10.1752. [DOI] [PubMed] [Google Scholar]
- 40.Dale RC, Candler PM, Church AJ, Wait R, Pocock JM, Giovannoni G. Neuronal surface glycolytic enzymes are autoantigen targets in post-streptococcal autoimmune CNS disease. J Neuroimmunol. 2006;172:187–97. doi: 10.1016/j.jneuroim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 41.Fillit HM, Kemeny E, Luine V, Weksler ME, Zabriskie JB. Antivascular antibodies in the sera of patients with senile dementia of the Alzheimer's type. J Gerontol. 1987;42:180–4. doi: 10.1093/geronj/42.2.180. [DOI] [PubMed] [Google Scholar]
- 42.Jankovic BD, Horvat J, Djordjijevic D, Ramah A, Fridman V, Spahic O. Brain-associated autoimmune features in heroin addicts: correlation to HIV infection and dementia. Int J Neurosci. 1991;58:113–26. doi: 10.3109/00207459108987188. [DOI] [PubMed] [Google Scholar]
- 43.Braus BK, Hauck SM, Amann B, et al. Neuron-specific enolase antibodies in patients with sudden acquired retinal degeneration syndrome. Vet Immunol Immunopathol. 2008;124:177–83. doi: 10.1016/j.vetimm.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 44.Ikeda Y, Maruyama I, Nakazawa M, Ohguro H. Clinical significance of serum antibody against neuron-specific enolase in glaucoma patients. Jpn J Ophthalmol. 2002;46:13–7. doi: 10.1016/s0021-5155(01)00455-5. [DOI] [PubMed] [Google Scholar]
- 45.Jankovic BD, Djordjijevic D. Differential appearance of autoantibodies to human brain S100 protein, neuron specific enolase and myelin basic protein in psychiatric patients. Int J Neurosci. 1991;60:119–27. doi: 10.3109/00207459109082042. [DOI] [PubMed] [Google Scholar]
- 46.Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am. 2009;29:247–64. doi: 10.1016/j.iac.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–30. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 48.Umehara H, Bloom E, Okazaki T, Domae N, Imai T. Fractalkine and vascular injury. Trends Immunol. 2001;22:602–7. doi: 10.1016/s1471-4906(01)02051-8. [DOI] [PubMed] [Google Scholar]
- 49.Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20:RC87. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tarozzo G, Campanella M, Ghiani M, Bulfone A, Beltramo M. Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Eur J Neurosci. 2002;15:1663–8. doi: 10.1046/j.1460-9568.2002.02007.x. [DOI] [PubMed] [Google Scholar]
- 51.Soriano SG, Amaravadi LS, Wang YF, et al. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002;125:59–65. doi: 10.1016/s0165-5728(02)00033-4. [DOI] [PubMed] [Google Scholar]
- 52.Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–21. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- 53.Kastenbauer S, Koedel U, Wick M, Kieseier BC, Hartung HP, Pfister HW. CSF and serum levels of soluble fractalkine (CX3CL1) in inflammatory diseases of the nervous system. J Neuroimmunol. 2003;137:210–7. doi: 10.1016/s0165-5728(03)00085-7. [DOI] [PubMed] [Google Scholar]
- 54.Matsunawa M, Isozaki T, Odai T, et al. Increased serum levels of soluble fractalkine (CX3CL1) correlate with disease activity in rheumatoid vasculitis. Arthritis Rheum. 2006;54:3408–16. doi: 10.1002/art.22208. [DOI] [PubMed] [Google Scholar]
- 55.Yajima N, Kasama T, Isozaki T, et al. Elevated levels of soluble fractalkine in active systemic lupus erythematosus: potential involvement in neuropsychiatric manifestations. Arthritis Rheum. 2005;52:1670–5. doi: 10.1002/art.21042. [DOI] [PubMed] [Google Scholar]
- 56.Fraticelli P, Sironi M, Bianchi G, et al. Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J Clin Invest. 2001;107:1173–81. doi: 10.1172/JCI11517. [DOI] [PMC free article] [PubMed] [Google Scholar]