

Abstract
Alzheimer's disease (AD) is a significant and growing health problem in the aging population. Although definitive mechanisms of pathogenesis remain elusive, genetic and histological clues have implicated the proteins presenilin (PS) and tau as key players in AD development. PS mutations lead to familial AD, and although tau is not mutated in AD, tau pathology is a hallmark of the disease. Axonal transport deficits are a common feature of several neurodegenerative disorders and may represent a point of intersection of PS and tau function. To investigate the contribution of wild-type, as opposed to mutant, tau to axonal transport defects in the context of presenilin loss, we used a mouse model postnatally deficient for PS (PS cDKO) and expressing wild-type human tau (WtTau). The resulting PS cDKO;WtTau mice exhibited early tau pathology and axonal transport deficits that preceded development of these phenotypes in WtTau or PS cDKO mice. These deficits were associated with reduced neurotrophin signaling, defective learning and memory and impaired synaptic plasticity. The combination of these effects accelerated neurodegeneration in PS cDKO;WtTau mice. Our results strongly support a convergent role for PS and tau in axonal transport and neuronal survival and function and implicate their misregulation as a contributor to AD pathogenesis.
Introduction
Alzheimer's disease (AD) is a significant and growing health problem in the aging population. Although definitive mechanisms of pathogenesis remain elusive, genetic and histological clues have implicated the proteins presenilin (PS) and tau as key players in AD development. Studies of familial AD have identified 178 mutations in the presenilin 1 (PS1) gene and 14 mutations in its homolog, presenilin 2 (PS2). Both through its role as the catalytic subunit of γ-secretase and via γ-secretase-independent mechanisms, presenilin can affect multiple cellular systems that mediate neuronal function and survival (for review, see Parks and Curtis, 2007). Unlike presenilin, tau is not known to be mutated in AD. Instead, aberrant phosphorylation of wild-type tau leads to its aggregation into neurofibrillary tangles, a pathological hallmark of AD. This hyperphosphorylation has also been shown to interfere with tau's normal physiological role of binding and stabilizing microtubules, leading to impairments in axonal transport (for review, see Mi and Johnson, 2006).
Defective axonal transport has been demonstrated in multiple neurodegenerative diseases, including AD, Huntington's disease, and Parkinson's disease (for review, see Higuchi et al., 2002). Tau and presenilin have both been implicated in decreased trafficking in AD, suggesting a common pathway toward neurodegeneration. Loss of presenilin in vitro and expression of PS1 familial AD mutations in vivo impair fast axonal transport through dysregulation of GSK3β, a prominent tau kinase that also phosphorylates the motor protein kinesin to promote its release of cargo vesicles (Pigino et al., 2003; Lazarov et al., 2007). Transgenic mouse models of a human tauopathy also exhibit reduced rates of axonal transport (Zhang et al., 2004; Ittner et al., 2008). This phenomenon has been attributed to the propensity of these tau mutants to aggregate into filaments, leading to premature GSK3β-mediated release of kinesin from its cargoes, as with presenilin loss or mutation (LaPointe et al., 2009).
The overlap of presenilin and tau function with regard to axonal transport led us to hypothesize that conditions favoring tau pathology and presenilin loss of function would promote greater inhibition of axonal trafficking through combinatorial effects. We chose presenilin conditional double knock-out (PS cDKO) mice to study postnatal effects of complete presenilin loss to avoid the confounding effects of residual PS function in mutant models (Feng et al., 2004). These mice develop forebrain-specific knock-out of PS1 on a PS2 −/− background, thus preventing PS2 compensation. We bred these animals with mice neuronally expressing 4-repeat human tau (WtTau), which is considered the most pathology-prone of the 6 wild-type human isoforms (Kimura et al., 2007). The resulting PS cDKO;WtTau mice exhibited early tau pathology and axonal transport deficits that preceded development of these phenotypes in WtTau or PS cDKO mice. These deficits were associated with reduced neurotrophin signaling, defective learning and memory, impaired synaptic plasticity, and accelerated neurodegeneration.
Materials and Methods
Mouse models.
All mice in this study were on a C57BL/6J background and were homozygous for both the exon 4-floxed PS1 allele (PS1fl) described by Feng et al. (2001) and the conventional PS2 knock-out described by Donoviel et al. (1999). These mice have been previously compared with nontransgenic animals and were phenotypically wild-type in all experiments performed; they are therefore considered equivalent to wild-type controls in this study (Feng et al., 2004). Transgenic mice expressing the longest isoform of human tau (WtTau) and CaMKIIα-Cre mice have been described previously (Tsien et al., 1996; Kimura et al., 2007). All genotypes in this study were generated by breeding Cre; PS1fl/fl; PS2−/− mice with WtTau; PS1fl/fl; PS2−/− mice to yield the following: PS1fl/fl; PS2−/− (PS2 KO), PS1fl/fl; PS2−/−; WtTau (WtTau), Cre; PS1fl/fl; PS2−/− (PS cDKO), and Cre; PS1fl/fl; PS2−/−; WtTau (PS cDKO; WtTau). Mice were housed 2–5 per cage with ad libitum access to food and water in a room with a 12 h light/dark cycle. All procedures were performed in accordance with NIH guidelines and with the approval of the Baylor College of Medicine Institutional Animal Care and Use Committee.
Histology and immunohistochemistry.
Mice anesthetized with Avertin (2,2,2-tribromoethanol in 2-methyl-2-butanol, 0.4 mg/kg, i.p., Sigma) underwent transcardial perfusion with PBS containing heparin (10 U/ml) and fixation with 4% paraformaldehyde (PFA) for 5 min. Brains (n = 3/genotype) were dissected and postfixed in 30% sucrose in 4% PFA overnight, then transferred to 30% sucrose in PBS, all at 4°C. Frozen brains were sliced into 30 μm floating sections using a sliding microtome (Leica SM2000R) and stored in cryoprotectant solution (0.1 m PBS, pH 7.2, containing 30% sucrose, 30% ethylene glycol, and 1% polyvinylpyrrolidone) at −20°C until further use. For tau immunostaining, endogenous peroxidase activity was blocked and tissue was permeabilized with 3% hydrogen peroxide in Tris-buffered saline (TBS) with 0.5% Triton X-100 for 30 min at room temperature (RT). Sections were then blocked in 5% nonfat dry milk in TBS with 0.4% Triton X-100 for 1 h at RT, then incubated in primary antibody (CP13, 1:5000; MC1, 1:200; generous gifts from Dr. Peter Davies, Albert Einstein College of Medicine, New York, NY) in the milk blocking solution at 4°C overnight. After four 5 min washes with TBS with 0.05% Triton X-100, sections were incubated in biotinylated goat anti-mouse secondary antibody (1:1000, Vector Labs) in 20% Superblock (Pierce Protein Research Products, Thermo Fischer Scientific) in TBS with 0.05% Triton X-100 for 2 h at RT. Sections were again washed 4 × 5 min with TBS with 0.05% Triton X-100, then incubated in avidin-biotin-horseradish peroxidase complex (ABC) reagent (R.T.U. Vectastain Universal Elite ABC kit, Vector Labs, PK-7200) for 1 h at RT. After three 5 min washes with TBS with 0.05% Triton X-100, staining was visualized by incubation in diaminobenzidine (DAB) with hydrogen peroxide (DAB Substrate Kit for Peroxidase, Vector Labs, SK-4100) at RT for 2 min. Sections were then transferred to TBS, mounted onto slides, and then dried overnight before coverslip mounting. Images were acquired on a Zeiss Axioskop 2 Plus upright microscope with Axio Cam MRC camera using Axiovision 3.1 software. For Nissl staining, frozen brain sections were first mounted onto gelatin-coated slides and dried overnight. The tissue was dehydrated by serial passage through 70%, 95%, and 100% ethanol and lipids were removed by immersion in xylene. Sections were then rehydrated and incubated in 0.1% cresyl violet at room temperature for 10 min, followed by a water rinse and differentiation in 95% ethanol until desired staining was observed. Stained tissue was dehydrated in 100% ethanol and cleared by xylene before mounting coverslips with DPX mounting medium (Sigma). Images were acquired on an Olympus IX50 inverted microscope with Photometrics CoolSNAP ES camera using MetaMorph 6.0 software.
Manganese-enhanced magnetic resonance imaging.
The use of manganese-enhanced magnetic resonance imaging (MEMRI) to measure in vivo axonal transport has been described previously (Smith et al., 2007). Mn2+ was administered by nasal lavage of 4 μl (2 μl per nostril) of 0.75 g/ml MnCl2 solution to a mouse anesthetized by 5% isoflurane. The mouse was then allowed to recover for 45 min on a heating pad. Following the recovery period, the mouse was again anesthetized with 5% isoflurane before placement in the prone position in a modified Bruker mouse holder. From this point through imaging, the mouse was maintained on 2–3% isoflurane in 100% O2; appropriate isoflurane levels were determined by respiration rate (target: 40–60 breaths/min). Respiration rate was measured by a pressure pad placed beneath the animal, and temperature was monitored using a rectal probe. Temperature was maintained at 37°C throughout imaging using an air heating system (SA Instruments). A 9.4T, Bruker Avance Biospec Spectrometer, 21 cm bore horizontal scanner with a 35 mm volume resonator was used for all image acquisition described in this article. For MEMRI experiments, imaging parameters were as follows: repetition time (TR) = 500 ms; echo time (TE) = 10.2 ms; field of view (FOV) = 3.0 cm; slice thickness = 1 mm; matrix = 128 × 128; number of excitations = 2; number of cycles = 15. All images were acquired, and MEMRI images were analyzed, using Paravision software, version 4 (Bruker). Two different cohorts were imaged at 2 months (PS2 KO n = 5, WtTau n = 4, PS cDKO n = 6, PS cDKO;WtTau n = 6) and 6 months (PS2 KO n = 7, WtTau n = 6, PS cDKO n = 7, PS cDKO;WtTau n = 9).
To calculate transport rates, a region of interest (ROI) was designated exactly as in Smith et al. (2007). The ROI was located in an axial slice 1 mm anterior to the posterior aspect of the olfactory bulb. Exact placement was determined by identifying the vertical midpoint of the olfactory bulb and positioning the ROI at this height at the bulb's periphery; this region contains the olfactory neuronal layer at its widest point. The ROI therefore represents a single fascicle of axons originating from olfactory sensory neurons in the nasal epithelium (Akins and Greer, 2006). To normalize for overall changes in signal intensity between images, the signal intensity of this ROI was normalized to a separate, larger ROI within the facial muscle. Normalized signal intensity was then measured for each cycle and the values plotted against time. The axonal transport rate was calculated as the slope of the resulting regression line, and data for each genotype were represented as the mean ± SEM. Data were compared between genotypes by one-way ANOVA with Newman–Keuls posttests using Prism software, version 4 (GraphPad Software).
Synaptosome fractionation.
To isolate synaptosome fractions, fresh mouse hippocampi were homogenized in ice-cold buffer (0.32 m sucrose in 10 mm HEPES-NaOH, pH 7.4) containing Complete Protease Inhibitor Cocktail (Roche Diagnostics) with a glass-Teflon homogenizer (10 strokes at 900 rpm). The homogenate was then centrifuged at 4°C in a JA-20 rotor (Beckman-Coulter) at 2500 rpm for 30 min. The resulting supernatant was recentrifuged at 7600 rpm for 30 min to yield a crude synaptosome pellet. This pellet was then resuspended in ice-cold homogenization buffer and lysed hypo-osmotically with 5 mm HEPES, pH 7.4, containing Complete Protease Inhibitor Cocktail. The resuspended synaptosomes were then homogenized with 10 strokes of a glass-Teflon homogenizer at 900 rpm and shaken for 15 min at 4°C. The lysed synaptosomes were then pelleted by centrifugation at 4°C in a JA-17 rotor (Beckman-Coulter) at 10,000 rpm for 30 min and stored at −80°C for future use.
Western blot analysis.
For analysis of synaptosome fractions (n = 3/genotype), pellets were resuspended in homogenization buffer. For all other immunoblotting experiments (n = 4/genotype), samples were prepared from quick-frozen hippocampus by homogenization in ice-cold modified RIPA buffer (50 mm Tris, pH 7.4; 150 mm NaCl; 1% NP-40; 1 mm EDTA; 0.25% sodium deoxycholate) with phosphatase and protease inhibitors. Following 3 × 10 pulses of sonication, homogenates were centrifuged at 10,500 × g for 10 min at 4°C and supernatants collected. Protein concentrations of all samples were determined using a detergent-compatible (DC) colorimetric protein assay (Bio-Rad) before dilution in 2× loading buffer. Samples were boiled at 95°C for 5 min before loading. Sample loading was as follows: 5 μg/well for synaptosome quality control Western blots; 15 μg/well for BDNF and CaMKII synaptosome Western blots; 35 μg/well for c-Myc and APPc Western blots; and 50 μg/well for Erk, Akt, and total BDNF and CaMKII Western blots. For BDNF Western blots, samples were run on a 12% SDS-polyacrylamide gel at 100 mV for 2 h; all other proteins were resolved on a 9% gel under the same conditions. Proteins were transferred onto a nitrocellulose membrane (Bio-Rad) in transfer buffer (0.05 m Tris, 0.04 m glycine, 20% methanol, 0.01% SDS) for 1 h at a constant 100 mV, with current between 150 and 200 mA. BDNF membranes were blocked in 5% nonfat dry milk in Superblock overnight at 4°C. All other membranes were blocked in 5% nonfat dry milk in 0.1% Tween 20 (Sigma) in TBS (TBST) for 1 h at room temperature. Membranes were incubated in primary antibody in blocking solution overnight at 4°C. The following antibodies were used: PHF-1, 1:500 (a generous gift from Dr. Peter Davies, Albert Einstein College of Medicine, New York), AT8, 1:500 (Pierce Endogen, Thermo Fisher Scientific), Tau 5, 1:1000 (Millipore Bioscience Research Reagents), γ-tubulin 1:10,000 (Sigma), APPc 1:2000 (Sigma), c-Myc (9E10) 1:1000 (Santa Cruz Biotechnology), BDNF (N-20) 1:500 (Santa Cruz Biotechnology), synaptotagmin 1:500 (a generous gift from Dr. Louis Reichardt, University of California, San Francisco, School of Medicine, San Francisco, CA), NMDAR1, 1:5000 (Millipore Bioscience Research Reagents), Erk1/2, 1:1000 (Cell Signaling Technology), phospho-Erk1/2 (Thr202/Tyr204) 1:1000 (Cell Signaling Technology), Akt 1:1000 (Cell Signaling Technology), phospho-Akt 1:1000 (Cell Signaling Technology), CaMKII 1:500 (Santa Cruz Biotechnology). Following primary incubation, membranes were washed 3 × 10 min with TBST, then incubated in species-specific goat secondary antibody (1:5000, horseradish peroxidase-conjugated anti-mouse or anti-rabbit, Vector Labs) in blocking solution. Membranes were again washed 3 × 10 min with TBST, then bands were visualized using ECL chemiluminescence reagent (GE Healthcare Life Sciences). Band density was calculated using Scion Image software. Band densities normalized to the respective loading controls were then compared with data for PS2 KO mice and represented as percentage of control ± SEM. Data were compared between genotypes by one-way ANOVA with Newman–Keuls posttests using Prism software, version 4 (GraphPad Software).
Associative fear conditioning.
PS2 KO (n = 14), WtTau (n = 14), PS cDKO (n = 11), and PS cDKO;WtTau (n = 16) male and female mice were tested at 6 months of age. Testing was performed as in Spencer et al. (2006), with the exception that freezing was measured automatically using the FreezeFrame/FreezeView monitoring system (San Diego Instruments) to control testing parameters and collect freezing data with baseline thresholds set just above the level of motion detected in an empty chamber. Training began with a 2 min exploration period in the test chamber (reported as “immediate freezing”), followed by two pairings of conditioned stimulus (CS) (30 s of 80 db white noise) and unconditioned stimulus (US) (2 s 0.7 mA foot shock) separated by 2 min. Twenty-four hours after training, mice were placed back in the training chamber for 5 min to test contextual memory. One hour after context testing, cued memory was tested by introducing the mice for 3 min to the training chamber after visual, tactile, and olfactory conditions were altered, then playing the CS for 3 min. The cued response was reported as the difference between freezing during the CS and freezing during the acclimation period. Datasets for immediate freezing, context freezing, and cued freezing were analyzed separately by one-way ANOVA with Newman–Keuls posttests using Prism software, version 4 (GraphPad Software).
Slice preparation and field EPSP recordings.
Horizontal hippocampal slices (400 μm) were prepared from 6- to 7-month-old PS2 KO [n = 13 for long-term potentiation (LTP), 30 for paired-pulse], PS cDKO (n = 14 for LTP, 21 for paired-pulse), and PS cDKO;WtTau (n = 11 for LTP, 32 for paired-pulse) mice using a vibratome (Global Medical Instrumentation, series 1000) and ice-cold cutting artificial CSF (aCSF) containing the following (in mm): 110 sucrose, 60 NaCl, 3 KCl, 7 MgCl, 28 NaHCO3, 1.25 NaH2PO4, 5 d-glucose, 0.5 CaCl2. Slices were incubated for 1 h in room-temperature recording aCSF (in mm): 125 NaCl, 2.5 KCl, 1 MgCl, 25 NaHCO3, 1.25 NaH2PO4, 10 d-glucose, 2 CaCl2 consistently bubbled with 95%O2/5%CO2.Slices were equilibrated for 20–30 min in recording aCSF, at 31 ± 0.5°C before electrode (2–4 mΩ, filled with recording aCSF) placement in area CA1. Schaffer collaterals were stimulated by a bipolar electrode. Basal synaptic transmission was monitored at 0.05 Hz. In paired-pulse experiments, field EPSPs (fEPSPs) were elicited by two-pulse stimulation with interpulse intervals ranged from 10 to 200 ms. Facilitation was evaluated by calculating the ratio P2/P1: where P2 is the fEPSP amplitude elicited by the second stimulus and P1 is the fEPSP amplitude elicited by the first stimulus. Hippocampal LTP was induced using a theta burst protocol (TBS) consisting of 10 bursts at 5 Hz, and each burst consisting of four pulses at 100 Hz, with a pulse width of 0.05 ms. Stimulation strength was set to provide fEPSPs with an amplitude of ∼30% of the maximum. fEPSPs were filtered at 2 kHz, digitized at 20 kHz, recorded with Clampex 9 and analyzed with Clampfit software (Molecular Devices) and OriginPro 7.5 (OriginLab).
Three-dimensional magnetic resonance imaging.
Equipment and mouse handling were as described in the MEMRI experiments. PS2 KO, PS cDKO, and PS cDKO;WtTau mice were imaged at 2 months (PS2 KO n = 3, PS cDKO n = 2, PS cDKO;WtTau n = 2) and 6 months (PS2 KO n = 3, PS cDKO n = 4, PS cDKO;WtTau n = 2). Images were acquired using a 3D rapid acquisition with relaxed enhancement (RARE) protocol using the following parameters: RARE factor = 8; TR = 2000 ms; TE = 45.2 ms; FOV = 30 mm × 30 mm × 30 mm; matrix size = 128 × 128 × 128; number of averages = 2. Brain regions (cerebral cortex, ventricles, and cerebellum) were manually labeled within each slice and volumes were analyzed using AMIRA software (Mercury Computer Systems) (Redwine et al., 2003). Volumes for each region were compared separately across genotypes by one-way ANOVA with Newman–Keuls posttests using Prism software, version 4 (GraphPad Software).
Results
Loss of presenilin leads to increased tau phosphorylation and pathological conformation changes in both the dentate gyrus and septal nuclei of mice expressing wild-type human tau
Presenilin conditional double knock-out (PS cDKO) mice exhibit tau hyperphosphorylation as early as 9 months of age, a finding that likely results from loss of presenilin's inhibition of certain tau kinases (Saura et al., 2004). We chose to explore this phenotype further by breeding PS cDKO mice with mice expressing the longest isoform of wild-type human tau (WtTau), which serves as a better substrate for aberrant phosphorylation than endogenous mouse tau. The littermate PS2 knock-out animals produced from the crosses (the details of which can be found in Materials and Methods) were used as controls. These mice have been previously compared with nontransgenic animals and were phenotypically wild-type in all experiments performed; they are therefore considered equivalent to wild-type in this study (Feng et al., 2004). We used immunoblotting to confirm expression of c-Myc-tagged WtTau and accumulation of the C-terminal fragment (CTF) of the amyloid precursor protein (APP), which indicates loss of γ-secretase function. WtTau expression and APP CTF accumulation were observed in their corresponding genotypes within the olfactory bulb, hippocampus, and cortex but were absent in the cerebellum, all of which indicate successful forebrain targeting of genetic modifications in PS and tau (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). We hypothesized that these PS cDKO;WtTau mice would develop tau pathology earlier than PS2 single knock-out (PS2 KO), WtTau, and PS cDKO controls. As predicted, while control genotypes did not exhibit tau hyperphosphorylation by 6 months of age, PS cDKO;WtTau mice demonstrated pathological alterations in tau in certain brain regions. Using the CP13 antibody to detect phosphorylation of serine 202 and MC-1 to detect pretangle conformational changes in tau, we observed distinct immunoreactivity with both antibodies in the polymorphic and granule cell layers of the dentate gyrus of the hippocampus in these mice (Fig. 1 A). The lateral and medial septal nuclei also exhibited a high number of CP13- and MC-1-positive neurons (Fig. 1 B). Both antibodies characteristically stained the somatodendritic compartments of the neurons, indicating pathological localization of tau (Fig. 1 A,B, high magnification). The recognition of these neurons by MC-1 also reflects pathological changes in tau conformation as seen in human AD brains. These findings were specific to the hippocampus, as the striatum and cortex did not display CP13- or MC-1-positive neurons in any of the genotypes tested (supplemental Figure S2, available at www.jneurosci.org as supplemental material).
Figure 1.
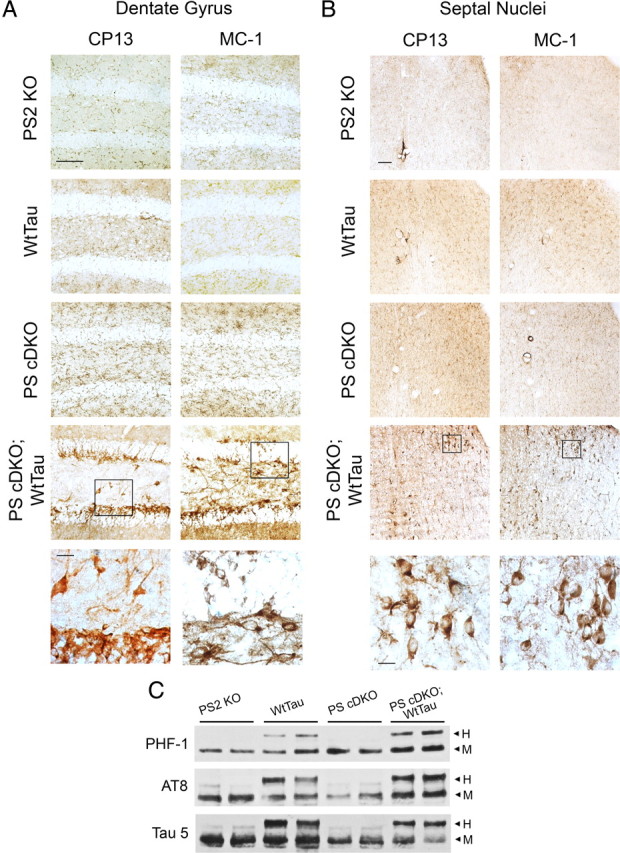
Tau pathology in 6-month-old PS cDKO;WtTau mice. A, Representative images from the dentate gyrus showing CP13 (left) and MC-1 (right) immunostaining of PS2 KO, WtTau, PS cDKO, and PS cDKO;WtTau mice. B, Representative images of CP13 and MC-1 immunostaining of a region including both the dorsolateral septal nucleus (upper right of each panel) and the medial septum (lower left of each panel). High-magnification insets show characteristic somatodendritic immunoreactivity in PS cDKO;WtTau neurons. Scale bars, 10 μm for inset images, 50 μm for all others. C, Representative Western blots of phosphorylated (PHF-1 and AT8) and total (Tau 5) tau in hippocampal tissue homogenates dissected from 6-month-old mice show increased tau phosphorylation in PS cDKO;WtTau animals. H, human transgenic tau; M, mouse endogenous tau.
Western blotting of dissected hippocampus from 6-month-old mice revealed an increase in immunoreactivity for AT8, which recognizes phosphorylation of serine 202 and threonine 205, and PHF-1, which detects phosphorylated serine 396 and 404, in PS cDKO;WtTau animals (Fig. 1 C). Similar experiments performed in dissected septum did not reveal differences between genotypes, most likely due to the small number of CP13- and MC-1-positive neurons in that area (data not shown). The AT8 results corroborate our findings that CP13 immunoreactivity, and therefore ser202 phosphorylation, is increased in the dentate gyrus of these mice. Increased PHF-1 site phosphorylation suggests a progression toward paired-helical filament development and represents the furthest degree of tau pathology detected in our studies. Consistent with this assessment, Campbell-Switzer silver stain did not reveal any neurofibrillary tangles in any of the genotypes at this age (data not shown), indicating that late-stage pathology is not yet present.
The expression of WtTau in PS cDKO mice leads to a reduction in anterograde axonal transport rates
Because tau hyperphosphorylation can impair axonal trafficking, we used Mn2+-enhanced magnetic resonance imaging (MEMRI) to measure the efficiency of fast axonal transport. This method takes advantage of multiple useful features of manganese: 1) Mn2+ ions are paramagnetic and thus can serve as contrast agents for MRI; 2) as a calcium (Ca2+) analog, Mn2+ exploits the cellular machinery responsible for uptake and vesicular packaging of Ca2+ to be effectively introduced to the neuron and its transport system; and 3) Mn2+ moves anterogradely along microtubules via fast axonal transport, allowing measurements of its synaptic accumulation to reflect overall transport rates (Mendonça-Dias et al., 1983; Burnett et al., 1984; Geraldes et al., 1986; Cory et al., 1987; Fornasiero et al., 1987; Sloot and Gramsbergen, 1994; Tindemans et al., 2003; Cross et al., 2008; Minoshima and Cross, 2008). We chose to measure trafficking in the olfactory system because it provides a convenient and pathologically relevant site for measurement of Mn2+ transport; ions can be administered noninvasively via nasal lavage and olfactory deficits have been well documented in patients with Alzheimer's disease (Mesholam et al., 1998; Peters et al., 2003).
Figure 2 A shows representative images from 6-month-old PS2 KO and PS cDKO;WtTau mice at the first and last time points with outlined areas emphasizing signal enhancement. We tested PS2 KO, WtTau, PS cDKO, and PS cDKO;WtTau mice at 2 and 6 months of age and found that mice of all genotypes exhibited normal rates of transport at the younger age, indicating that the system is not developmentally impaired (Fig. 2 B). At 6 months, however, PS cDKO;WtTau mice displayed a reduction (∼50%, p < 0.001) in the rate of axonal transport compared with controls (Fig. 2 C). This reduced rate was remarkably consistent between PS cDKO;WtTau animals (Fig. 2 D). Interestingly, neither WtTau nor PS cDKO mice showed deficits at this age, providing evidence for a genetic interaction between presenilin and tau.
Figure 2.

Age-dependent reduction in axonal transport rates of PS cDKO;WtTau mice. A, Representative MEMRI color map images from PS2 KO controls and PS cDKO;WtTau mice at 6 months of age showing signal enhancement (emphasized by rectangular outline) between the first image (left) and the last image (right) of the series. B, Axonal transport rates are normal in 2-month-old PS2 KO, WtTau, PS cDKO, and PS cDKO;WtTau mice. C, Axonal transport rates at 6 months are selectively reduced in PS cDKO;WtTau mice. D, Linear regression of all data points from each genotype of 6-month-old mice. Given slopes are graphically represented and statistically analyzed in C). ΔSI/t = change in normalized signal intensity over time = rate of axonal transport. ***p < 0.001.
PS cDKO;WtTau mice exhibit reductions in synaptic brain-derived neurotrophic factor levels and Erk activation
To investigate the consequences of decreased axonal transport, we next examined brain-derived neurotrophic factor (BDNF) levels in the hippocampus. BDNF synthesis and anterograde trafficking occur within the hippocampus, but some hippocampal BDNF is also received anterogradely from the entorhinal cortex (Conner et al., 1997; Caleo and Cenni, 2004). BDNF is reduced in the brains of patients with AD, and recent work has shown that administration of BDNF into the entorhinal cortex can improve cell signaling, cognition, and neuronal survival in various mammalian models of AD through transport and signaling to the hippocampus (Murer et al., 2001; Gauthier et al., 2004; Nagahara et al., 2009). We therefore sought to determine whether the observed axonal transport deficit in PS cDKO;WtTau mice would cause a decrease in BDNF trafficking to the hippocampus, thus impairing its downstream signaling. Although total BDNF levels were unchanged in the hippocampi of PS2 KO, WtTau, PS cDKO, and PS cDKO;WtTau mice at 6 months of age, synaptosomal BDNF was decreased in PS cDKO;WtTau mice (Fig. 3 A,B, p < 0.01), indicating a reduction in localization of BDNF to the synapse. Synaptotagmin undergoes anterograde transport through a mechanism distinct from that of BDNF and therefore served as both a loading control for the synaptosome fraction and a separate confirmation of the specific change in BDNF synaptic localization. The quality of synaptosomal fractionation was determined by Western blots for enrichment of the synaptic proteins PSD-95 and NMDA receptor 1 (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 3.

Decreased synaptosomal BDNF levels and Erk1/2 activation in the hippocampus of 6-month-old PS cDKO;WtTau animals. A, Representative Western blots of total BDNF and BDNF from synaptosome fractions (BDNF). B, Quantification of normalized BDNF band densities relative to control (PS2 KO). C, Representative Western blots of total and phospho-Erk1/2. D, Quantification of normalized Erk1/2 band densities relative to control. Loading controls were as follows: synaptotagmin (Syt) for synaptosomal BDNF and γ-tubulin for all others. *p < 0.05, **p < 0.01.
BDNF binds to its receptor, Tropomycin-related kinase B (TrkB), at the synapse to initiate downstream signaling through three main pathways: the phosphatidylinositol 3-kinase (PI3K)-Akt pathway, the phospholipase Cγ1 (PCγ1)-Ca2+ pathway, and the Ras-mitogen-activated protein kinase (MAPK)-Erk1/2 pathway (Kaplan and Miller, 2000). Total levels of Erk were unchanged in all genotypes at 6 months of age, but activation of Erk through phosphorylation was specifically reduced in PS cDKO;WtTau mice (Fig. 3 C,D, p < 0.05). Preliminary immunoblotting experiments did not detect changes in expression or activation of certain members of the PI3K or PCγ1 pathways (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), but the possibility of altered signaling cannot be excluded. These data suggest that impaired transport of BDNF to the synapse leads to reduced downstream signaling through Erk.
Additional expression of WtTau worsens contextual memory deficits in PS cDKO mice at 6 months of age
Because of Erk's well established role in learning and memory, we used the conditioned fear paradigm to test associative memory in 6-month-old mice. Impairments in contextual memory have been reported in PS cDKO mice as early as 2 months of age, and cued learning deficits have also been observed in these animals at 12 months of age (Saura et al., 2004; Chen et al., 2008). The percentage of time the mice spent frozen during the initial phase of training, before exposure to conditioned and unconditioned stimuli, was unchanged across genotypes, suggesting that baseline activity and anxiety levels were similar in all tested animals (Fig. 4 A, Immediate). Consistent with previous studies, we observed a significant decrease (∼40%, p < 0.01) in freezing of PS cDKO mice during the context test, reflecting a defect in hippocampus-dependent associative memory. Interestingly, PS cDKO; WtTau mice exhibited an even larger impairment in contextual memory (∼65%, p < 0.001 vs control), which was significantly worse than that of PS cDKO animals (Fig. 4 A, Context Test, p < 0.05 vs PS cDKO). We did not detect a difference in freezing of 6-month-old mice of any genotype during the cued test, indicating that amygdala-mediated learning pathways were functioning properly and that contextual memory deficits were likely due to hippocampus-specific defects (Fig. 4 A, Cued Test).
Figure 4.

Hippocampal-dependent memory and synaptic plasticity impairments in PS cDKO;WtTau mice at 6 months of age. A, Freezing behaviors during various phases of fear conditioning. Immediate freezing occurs during the initial exploratory period of training. Context test freezing represents the amount of time the mice froze during second exposure to the training chamber 24 h after training. Normalized cued test freezing is the difference between freezing during second exposure to the conditioned stimulus (CS) and freezing before CS in a novel chamber 24 h after training. B, Following 10 min of baseline recording, theta burst stimulation (TBS) was delivered to the Schaffer collateral pathway. When compared with PS2 KO controls, PS cDKO;WtTau mice demonstrate significantly impaired LTP induction and maintenance at all time points tested; however, the difference between PS cDKO;WtTau and PS cDKO mice reaches significance only at the initial 3 and last 40 min. No significant difference is observed between PS cDKO and PS2 KO mice. Insets (a–c), Example fEPSP traces taken before and after TBS from PS2 KO, PS cDKO, and PS cDKO;WtTau mice, respectively (calibration, 1 mV, 5 ms). C, Increased paired-pulse ratio in PS cDKO;WtTau mice at 10 and 20 ms interpulse intervals indicating altered presynaptic function in the hippocampus of PS cDKO;WtTau mice. *p < 0.05, **p < 0.01, ***p < 0.001.
Presynaptic alteration underlies impaired Schaffer collateral LTP in PS cDKO;WtTau mice
In addition to being important modulators of learning and memory, BDNF and Erk play critical roles in LTP (Ying et al., 2002). We therefore examined field Schaffer collateral LTP in hippocampal brain slices of 6-month-old PS2 KO, PS cDKO and PS cDKO;WtTau mice. Potentiation induced by theta-burst stimulation (TBS) was significantly reduced and remained lower in PS cDKO;WtTau mice compared with PS2 KO controls (p < 0.01), but we found no significant differences between PS cDKO and PS2 KO animals (Fig. 4 B). In contrast, the difference in LTP induction and maintenance between PS cDKO;WtTau and PS cDKO mice only reached significance during the first three and last 40 min (p < 0.05). To determine whether the deficit in LTP was related to presynaptic or postsynaptic defects, we conducted a paired-pulse protocol in which the ratio between the mean amplitudes of the second fEPSP over the first fEPSP (P2/P1 ratio) is inversely related to the initial release probability (Dobrunz and Stevens, 1997). In general, the smaller the probability of release to the first pulse, the more facilitated the response to the second pulse. We observed a significantly increased paired-pulse ratio in PS cDKO;WtTau mice compared with PS2 KO controls at the shortest interpulse intervals tested (Fig. 4 C, p < 0.05), suggesting a reduced initial release probability in the PS cDKO;WtTau animals. Thus, altered presynaptic function may underlie the impaired Schaffer collateral LTP observed in PS cDKO;WtTau mice.
Expression of WtTau exacerbates cortical neurodegeneration in PS cDKO mice
PS cDKO mice are known to exhibit atrophy of the cerebral cortex by 6 months of age (Feng et al., 2004; Saura et al., 2004; Chen et al., 2008). Given our findings that the addition of WtTau can impair axonal transport, neurotrophin signaling, contextual memory, and LTP in PS cDKO mice, we hypothesized that these deficits could impact neuronal survival in this mouse model with proven susceptibility to cellular insults. We chose three-dimensional magnetic resonance imaging (3D MRI) to evaluate neurodegeneration because this method presents an opportunity to measure the volume of entire brain regions in vivo, eliminating the inconsistencies associated with histological processing. While our primary region of interest was the cerebral cortex, we also looked for the compensatory enlargement of ventricles that accompanies loss of cortical tissue. The volume of the cerebellum was analyzed as a negative control. Because no neuronal loss has been observed in WtTau mice up to 22 months of age, we elected to omit this group (Kimura et al., 2007). The other three genotypes were studied at 2 months of age to control for developmental differences in brain volumes and at 6 months to determine whether the aforementioned impairments at this age have an effect on neuron loss.
As expected, the volumes of all brain regions were comparable across genotypes at 2 months of age, demonstrating normal brain development in the absence of postnatal presenilin expression (Fig. 5 A). As previously reported, PS cDKO mice exhibited a reduction in cortical volume at 6 months of age, which was accompanied by a compensatory increase in ventricular volume (Fig. 5 B). Interestingly, PS cDKO;WtTau mice displayed even greater cortical atrophy at this age, supporting a role for tau in this neurodegenerative phenotype (Fig. 5 B,C). At both ages, cerebellar volumes remained stable across genotypes, indicating that changes in cortical and ventricular volumes resulted specifically from the CaMKIIα-regulated genetic modifications in PS cDKO and PS cDKO;WtTau mice.
Figure 5.
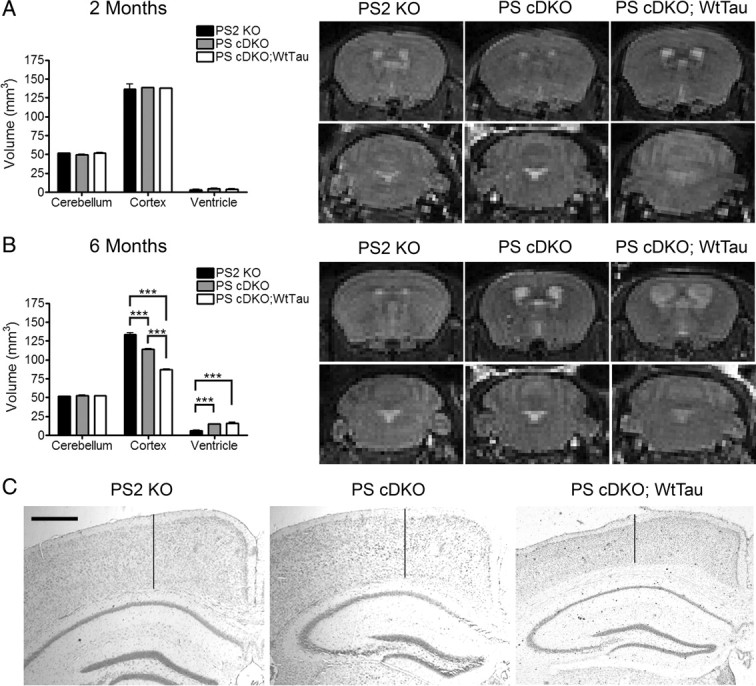
Increased cortical neurodegeneration in PS cDKO;WtTau compared with PS cDKO mice. A, Volumes of cerebellum, cerebral cortex, and ventricles are normal in 2-month-old PS2 KO, PS cDKO, and PS cDKO;WtTau mice. Quantification of volumes (left) and representative 3D MRI images (right) show no obvious differences in structures between genotypes. B, Cerebral cortical atrophy of PS cDKO mice is exacerbated in PS cDKO;WtTau animals at 6 months. Quantification of volumes (left) and representative 3D MRI images (right) show a decrease in cortical volume with compensatory increase in ventricular volume (top) in PS cDKO mice and, to a further degree, in PS cDKO;WtTau mice. No change in cerebellar volumes was detected (bottom). C, Representative images from cresyl violet (Nissl)-stained frozen brain sections demonstrate cortical atrophy in 6-month-old PS cDKO and PS cDKO;WtTau mice as quantified in panel 5B. Vertical lines are provided to assist with visual assessment of cortical thickness at sites that are comparable between sections. Scale bar, 500 μm, ***p < 0.001.
Discussion
Although a large body of evidence supports some level of interaction between presenilin and tau in AD pathogenesis, the precise contributions of each have yet to be fully ascertained. Specifically, the role of wild-type human tau has been largely passed over in favor of study of mutant tau-expressing mouse models, which more readily develop tau pathology but do not accurately reflect the genetics of tau in AD. Here we have shown that expression of WtTau in PS cDKO mice can impair multiple cellular functions that are significant to the development of AD (Fig. 6). In these mice, tau hyperphosphorylation and pathological conformation were detected as early as 6 months of age in neurons of the septal nuclei of the basal forebrain and the dentate gyrus of the hippocampus. We determined that anterograde axonal transport was impaired specifically in PS cDKO;WtTau mice at this time point, suggesting a role for tau hyperphosphorylation and/or aggregation in this deficit. In the hippocampus, reduced synaptosomal BDNF in the context of normal levels of total BDNF provided additional support for the hypothesis that axonal transport is impaired in areas with tau pathology. The finding that Erk activation was also diminished in the hippocampus of PS cDKO;WtTau mice implied that this level of synaptic BDNF reduction was sufficient to decrease its downstream signaling. Deficient BDNF/Erk signaling had important consequences for contextual learning and memory, and long-term potentiation as well, as impairments in both were observed to a greater degree in PS cDKO mice expressing WtTau. Finally, the combination of these deficits led to an increase in cortical atrophy in PS cDKO;WtTau mice when compared with PS cDKO mice, suggesting that multiple mechanisms underlie the neurodegeneration in these mice, and possibly in AD.
Figure 6.

Model of neuronal dysfunction and neurodegeneration in PS cDKO;WtTau mice. Presenilin loss has been shown previously to increase tau phosphorylation, reduce TrkB receptor maturation and synaptic localization, and result in defective Ca2+ homeostasis (dotted lines; see Discussion for references). In combination with presenilin loss, expression of WtTau accelerates tau hyperphosphorylation, leading to impaired anterograde axonal transport of synaptic proteins such as BDNF. This decrease in BDNF at the synapse reduces downstream signaling, which, along with loss of presenilin, can directly affect neuronal survival. In addition, specific reduction of Erk phosphorylation/activation impairs hippocampal-dependent memory and long-term potentiation, phenotypes to which presenilin loss likely contributes through mechanisms outside the scope of this study. Boxed results were obtained from in vivo studies, with the exception of LTP.
The presence of tau pathology in the septal nuclei and dentate gyrus of PS cDKO;WtTau mice could have several implications for neuronal function in the hippocampus. Septal neurons are known to project to the dentate gyrus in addition to other hippocampal and extrahippocampal regions. Lesion of septal cholinergic neurons or pharmacological inhibition of septohippocampal cholinergic signaling impairs hippocampal-dependent learning and memory, a deficit that can be rescued by administration of acetylcholine to the hippocampus (for review, see Parent and Baxter, 2004). This corresponds with the loss of cholinergic neurons in AD, which results in memory impairment that can be partially allayed by treatment with acetylcholinesterase inhibitors (Kása et al., 1997; Seltzer, 2006). In addition, pharmacological inhibition of cholinergic signaling by scopolamine has been shown to reduce protein levels of BDNF in the rodent hippocampus (Kotani et al., 2006). Although we did not detect a change in total levels of BDNF in the hippocampus, tau phosphorylation of a subset of neurons in the septohippocampal pathway would perturb the system to a significantlys lesser degree than global inhibition of cholinergic signaling. This limited tau phosphorylation also explains our inability to detect with Western blots the tau pathology observed with immunohistochemistry. Ultimately, the finding that interruption of cholinergic signaling can lead to reductions in downstream BDNF levels provides support for our model that disruption of septohippocampal signaling through tau hyperphosphorylation leads to reduced synaptic BDNF in the hippocampus. Furthermore, strong involvement of this pathway in hippocampal-dependent learning and memory implies a potential mechanism for the specific contextual memory deficit we observed in PS cDKO;WtTau mice.
The development of tau pathology and impaired axonal transport in PS cDKO;WtTau mice are likely mediated by changes in the activity of tau kinases. Levels of p25, a potent activator of the tau kinase Cdk5, have been shown to increase in PS cDKO mice by 9 months of age (Saura et al., 2004). Interestingly, loss of presenilin has also been shown to increase GSK3β kinase activity, leading to GSK3β-mediated phosphorylation of the motor protein kinesin to promote its release from its membrane-bound cargoes during anterograde axonal transport (Morfini et al., 2002; Pigino et al., 2003). GSK3β misregulation due to presenilin loss could also lead to tau phosphorylation, suggesting two possible mechanisms for kinase-mediated axonal transport defects in PS cDKO mice: directly through kinesin phosphorylation, and/or indirectly through tau phosphorylation. Because PS cDKO mice did not demonstrate impairment of axonal transport in our studies, tau phosphorylation appears to exert a more profound effect on trafficking than kinesin phosphorylation. Other kinases that phosphorylate tau could also be involved, and it is possible that a combination of small increases in multiple kinases, rather than large increases in a single kinase's activity, could be responsible for the tau phenotypes observed in our study.
While most neurotrophins undergo primarily retrograde axonal transport from the synapse to the cell body, anterograde transport of BDNF is seen in multiple pathways within the brain, including septal and cortical projections to the hippocampus and intrahippocampal circuits (for review, see Schindowski et al., 2008). Along with our observation that synaptic localization of BDNF is reduced, this supports the hypothesis that the defects in axonal transport detected in the olfactory system of PS cDKO;WtTau mice might extend to other brain regions, notably the hippocampus. Decreased Erk signaling in the hippocampus and functional deficits in hippocampal-dependent memory and synaptic plasticity in PS cDKO;WtTau animals further support this notion. In addition to a role for impaired axonal transport in reduced BDNF/Erk signaling, presenilin loss may contribute to this phenotype through other mechanisms. Loss of presenilin in primary neurons results in decreased maturation and membrane localization of TrkB, the primary receptor for BDNF (Naruse et al., 1998). This reduction in mature TrkB at the synaptic membrane could exacerbate an Erk signaling impairment caused by reduced synaptic BDNF. Although PS cDKO mice did not exhibit a statistically significant reduction in Erk activation, a downward trend was observed in these mice, supporting the existence of more than one mechanism underlying this phenotype. In fact, this could be the case with each of the phenotypes exhibited by PS cDKO mice that were exacerbated in PS cDKO;WtTau animals (i.e., contextual memory deficits and neurodegeneration).
Although LTP defects were not clearly demonstrated in PS cDKO mice, a trend toward reduced potentiation did exist. As with Erk signaling, other effects of presenilin loss could be contributing to this phenotype. LTP and other measures of synaptic plasticity require Ca2+ signaling, which is regulated by presenilin and is defective in familial AD mutants or PS-deficient mice (for review, see Mattson, 2010). A recent study genetically and pharmacologically dissected the role of presenilin in Schaffer collateral neurotransmitter release and found that presynaptic loss of presenilin impairs theta-burst-induced LTP through impairment of calcium release (Zhang et al., 2009). This corresponds with our observation that in addition to LTP, paired-pulse facilitation is altered in PS cDKO;WtTau mice, indicating a presynaptic defect. Although these alterations in Ca2+ release could also affect Mn2+, our administration of saturating doses of MnCl2 and the lack of axonal transport impairment in PS cDKO mice make it unlikely that such a phenomenon occurred in our study. Our inability to detect LTP deficits in PS cDKO mice could be explained by differences in experimental setup or variation in mouse strains and transgenes between our study and previous reports. Because of the trend toward a decrease in PS cDKO vs PS2 KO mice in our present study, however, it is possible that a statistically significant difference would be observed with an increased sample size or a modified stimulation protocol.
Together, our data support a combined role for presenilin and tau in AD pathogenesis. Substantial evidence supports the notion that familial AD mutations in presenilin genes primarily result in PS loss of function, particularly with regard to γ-secretase cleavage of Notch, cadherins, and APP (Chen et al., 2002; Moehlmann et al., 2002; Wiley et al., 2005). In PS mutants, the cleavage of APP generates β-amyloid species in a ratio that favors the pathogenic Aβ42 over Aβ40, but this could result from decreased production of Aβ40, not excess Aβ42 (for review, see De Strooper, 2007). Presenilin mutations can also impair γ-secretase-independent functions, leading to defects in Wnt signaling and Ca2+ homeostasis (for review, see Shen and Kelleher, 2007). This evidence supports the use of PS cDKO mice to study the roles of presenilin and tau in AD pathogenesis. We have established that loss of presenilin alone results in a phenotype that can be enhanced and even expanded by the presence of wild-type human tau, leading to impairment of axonal trafficking, neurotrophin signaling, learning and memory, and synaptic plasticity, all of which culminate in neurodegeneration. These findings provide evidence for the convergence of multiple pathways in the progression of AD and indicate that subtle differences in protein composition (i.e., human vs mouse tau) can dramatically affect cellular processes governing neuronal function and survival.
Footnotes
This work was supported by grants from the National Institutes of Health (NIH) (AG20670 and NS40039 to H.Z., and AG29977 to R.G.P.). E.P. was a trainee of NIH Training Grant T32 AG000183. We thank Drs. S. Sisodia (University of Chicago, Chicago, IL) and S. Tonegawa (Massachusetts Institute of Technology, Cambridge, MA) for the PS1 floxed mice and the CaMKIIα-Cre transgenic line, respectively. We are grateful to N. Aithmitti and X. Chen for expert technical support and members of the Zheng laboratory for constructive discussions. We acknowledge C. Spencer and the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center Neurobehavioral Core (HD24064) for their assistance.
References
- Akins MR, Greer CA. Cytoskeletal organization of the developing mouse olfactory nerve layer. J Comp Neurol. 2006;494:358–367. doi: 10.1002/cne.20814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett KR, Goldstein EJ, Wolf GL, Sen S, Mamourian AC. The oral administration of MnCl2: a potential alternative to IV injection for tissue contrast enhancement in magnetic resonance imaging. Magn Reson Imaging. 1984;2:307–314. doi: 10.1016/0730-725x(84)90197-8. [DOI] [PubMed] [Google Scholar]
- Caleo M, Cenni MC. Anterograde transport of neurotrophic factors: possible therapeutic implications. Mol Neurobiol. 2004;29:179–196. doi: 10.1385/mn:29:2:179. [DOI] [PubMed] [Google Scholar]
- Chen F, Gu Y, Hasegawa H, Ruan X, Arawaka S, Fraser P, Westaway D, Mount H, St George-Hyslop P. Presenilin 1 mutations activate gamma 42-secretase but reciprocally inhibit epsilon-secretase cleavage of amyloid precursor protein (APP) and S3-cleavage of notch. J Biol Chem. 2002;277:36521–36526. doi: 10.1074/jbc.M205093200. [DOI] [PubMed] [Google Scholar]
- Chen Q, Nakajima A, Choi SH, Xiong X, Tang YP. Loss of presenilin function causes Alzheimer's disease-like neurodegeneration in the mouse. J Neurosci Res. 2008;86:1615–1625. doi: 10.1002/jnr.21601. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory DA, Schwartzentruber DJ, Mock BH. Ingested manganese chloride as a contrast agent for magnetic resonance imaging. Magn Reson Imaging. 1987;5:65–70. doi: 10.1016/0730-725x(87)90485-1. [DOI] [PubMed] [Google Scholar]
- Cross DJ, Flexman JA, Anzai Y, Maravilla KR, Minoshima S. Age-related decrease in axonal transport measured by MR imaging in vivo. Neuroimage. 2008;39:915–926. doi: 10.1016/j.neuroimage.2007.08.036. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:141–146. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Miller MW, Ware CB, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin-1 and presenilin-2. Proc Natl Acad Sci U S A. 2004;101:8162–8167. doi: 10.1073/pnas.0402733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornasiero D, Bellen JC, Baker RJ, Chatterton BE. Paramagnetic complexes of manganese(II), iron(III), and gadolinium(III) as contrast agents for magnetic resonance imaging. The influence of stability constants on the biodistribution of radioactive aminopolycarboxylate complexes. Invest Radiol. 1987;22:322–327. doi: 10.1097/00004424-198704000-00008. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Geraldes CF, Sherry AD, Brown RD, 3rd, Koenig SH. Magnetic field dependence of solvent proton relaxation rates induced by Gd3+ and Mn2+ complexes of various polyaza macrocyclic ligands: implications for NMR imaging. Magn Reson Med. 1986;3:242–250. doi: 10.1002/mrm.1910030207. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Lee VM, Trojanowski JQ. Tau and axonopathy in neurodegenerative disorders. Neuromolecular Med. 2002;2:131–150. doi: 10.1385/NMM:2:2:131. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Fath T, Ke YD, Bi M, van Eersel J, Li KM, Gunning P, Götz J. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc Natl Acad Sci U S A. 2008;105:15997–16002. doi: 10.1073/pnas.0808084105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kása P, Rakonczay Z, Gulya K. The cholinergic system in Alzheimer's disease. Prog Neurobiol. 1997;52:511–535. doi: 10.1016/s0301-0082(97)00028-2. [DOI] [PubMed] [Google Scholar]
- Kimura T, Yamashita S, Fukuda T, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N, Takashima A. Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J. 2007;26:5143–5152. doi: 10.1038/sj.emboj.7601917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani S, Yamauchi T, Teramoto T, Ogura H. Pharmacological evidence of cholinergic involvement in adult hippocampal neurogenesis in rats. Neuroscience. 2006;142:505–514. doi: 10.1016/j.neuroscience.2006.06.035. [DOI] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Pigino G, Gadadhar A, Chen X, Robinson J, Ho H, Brady ST, Sisodia SS. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease-linked mutant presenilin 1. J Neurosci. 2007;27:7011–7020. doi: 10.1523/JNEUROSCI.4272-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. ER calcium and Alzheimer's disease: in a state of flux. Sci Signal. 2010;3:pe10. doi: 10.1126/scisignal.3114pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonça-Dias MH, Gaggelli E, Lauterbur PC. Paramagnetic contrast agents in nuclear magnetic resonance medical imaging. Semin Nucl Med. 1983;13:364–376. doi: 10.1016/s0001-2998(83)80048-8. [DOI] [PubMed] [Google Scholar]
- Mesholam RI, Moberg PJ, Mahr RN, Doty RL. Olfaction in neurodegenerative disease: a meta-analysis of olfactory functioning in Alzheimer's and Parkinson's diseases. Arch Neurol. 1998;55:84–90. doi: 10.1001/archneur.55.1.84. [DOI] [PubMed] [Google Scholar]
- Mi K, Johnson GV. The role of tau phosphorylation in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2006;3:449–463. doi: 10.2174/156720506779025279. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Cross D. In vivo imaging of axonal transport using MRI: aging and Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2008;35(Suppl 1):S89–S92. doi: 10.1007/s00259-007-0707-8. [DOI] [PubMed] [Google Scholar]
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci U S A. 2002;99:8025–8030. doi: 10.1073/pnas.112686799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Parent MB, Baxter MG. Septohippocampal acetylcholine: involved in but not necessary for learning and memory? Learn Mem. 2004;11:9–20. doi: 10.1101/lm.69104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AL, Curtis D. Presenilin diversifies its portfolio. Trends Genet. 2007;23:140–150. doi: 10.1016/j.tig.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Peters JM, Hummel T, Kratzsch T, Lötsch J, Skarke C, Frölich L. Olfactory function in mild cognitive impairment and Alzheimer's disease: an investigation using psychophysical and electrophysiological techniques. Am J Psychiatry. 2003;160:1995–2002. doi: 10.1176/appi.ajp.160.11.1995. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redwine JM, Kosofsky B, Jacobs RE, Games D, Reilly JF, Morrison JH, Young WG, Bloom FE. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: a magnetic resonance microscopy and stereologic analysis. Proc Natl Acad Sci U S A. 2003;100:1381–1386. doi: 10.1073/pnas.242746599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Schindowski K, Belarbi K, Buée L. Neurotrophic factors in Alzheimer's disease: role of axonal transport. Genes Brain Behav. 2008;7(Suppl 1):43–56. doi: 10.1111/j.1601-183X.2007.00378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer B. Cholinesterase inhibitors in the clinical management of Alzheimer's disease: importance of early and persistent treatment. J Int Med Res. 2006;34:339–347. doi: 10.1177/147323000603400401. [DOI] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloot WN, Gramsbergen JB. Axonal transport of manganese and its relevance to selective neurotoxicity in the rat basal ganglia. Brain Res. 1994;657:124–132. doi: 10.1016/0006-8993(94)90959-8. [DOI] [PubMed] [Google Scholar]
- Smith KD, Kallhoff V, Zheng H, Pautler RG. In vivo axonal transport rates decrease in a mouse model of Alzheimer's disease. Neuroimage. 2007;35:1401–1408. doi: 10.1016/j.neuroimage.2007.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Serysheva E, Yuva-Paylor LA, Oostra BA, Nelson DL, Paylor R. Exaggerated behavioral phenotypes in Fmr1/Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X-related proteins. Hum Mol Genet. 2006;15:1984–1994. doi: 10.1093/hmg/ddl121. [DOI] [PubMed] [Google Scholar]
- Tindemans I, Verhoye M, Balthazart J, Van Der Linden A. In vivo dynamic ME-MRI reveals differential functional responses of RA- and area X-projecting neurons in the HVC of canaries exposed to conspecific song. Eur J Neurosci. 2003;18:3352–3360. doi: 10.1111/j.1460-9568.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain [see comments] Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Wiley JC, Hudson M, Kanning KC, Schecterson LC, Bothwell M. Familial Alzheimer's disease mutations inhibit gamma-secretase-mediated liberation of beta-amyloid precursor protein carboxy-terminal fragment. J Neurochem. 2005;94:1189–1201. doi: 10.1111/j.1471-4159.2005.03266.x. [DOI] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Higuchi M, Yoshiyama Y, Ishihara T, Forman MS, Martinez D, Joyce S, Trojanowski JQ, Lee VM. Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J Neurosci. 2004;24:4657–4667. doi: 10.1523/JNEUROSCI.0797-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopolous V, Wines-Samuelson M, Zhang D, Dragatsis I, Sudhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]