

Abstract
Striatum is one of the brain regions that are highly sensitive to transient cerebral ischemia. Most of the striatal neurons die shortly after ischemia but interneurons including large aspiny (LA) neurons survive the same insult. Previous studies have shown that inhibitory synaptic transmission is enhanced in LA neurons after ischemia. The present study is aimed at revealing the mechanisms underlying this phenomenon. Immunohistochemical studies and western blotting were performed to examine the expression of glutamic decarboxylase (GAD), the key enzyme in the synthesis of GABA, in the striatum. GAD65 expression and the number of GAD67-positive cells were increased after ischemia. GAD67-positive cells in the striatum co-expressed GAD65 after ischemia. The increase of GAD67-positive cells did not result from neurogenesis. Doublelabeling of GAD67 and SOM indicates that some of the GAD67-positive cells are from the phenotypic shift of pre-existing somatostatin (SOM)-containing GABAergic interneurons after ischemia. Facilitation of inhibitory synaptic transmission by muscimol, a specific GABAA receptor agonist, increased the number of survived cells in the striatum after ischemia. Altogether, these data suggest that GAD expression is increased in the striatum after ischemia, which might contribute to the facilitated inhibitory synaptic transmission and the consequent survival of LA neurons.
Keywords: BrdU, excitotoxicity, GABA, GABAergic interneurons, global ischemia
Introduction
The selective vulnerability of specific brain regions and cell types to ischemic insults has been well established (Pulsinelli, 1985). In the dorsolateral striatum, medium spiny (MS) neurons, which function as the GABAergic projection neurons, die roughly one day following a 25-30 min transient forebrain ischemia (Pulsinelli et al., 1982). Interestingly, most interneurons in the striatum are resistant to ischemia (Chesselet et al., 1990). The interneurons are divided into two classes based on the neurotransmitter they produce: large aspiny (LA) cholinergic interneurons and GABAergic interneurons. GABAergic interneurons are further characterized into parvalbumin (PV)-containing interneurons, calretinin-containing, and lastly, those that co-localize nitric oxide synthase (NOS), somatostatin (SOM), and neuropeptide Y (NPY) (Kawaguchi et al., 1995).
The mechanisms for this differential vulnerability in the striatum remain unclear. Previous studies have shown that LA neurons and MS neurons respond differentially to excitatory inputs after ischemia. For the activation of ionotropic glutamate receptors, larger depolarizations occur in MS neurons than in LA neurons (Calabresi et al., 1998). For the activation of metabotropic glutamate receptors, membrane depolarization and calcium accumulation occur in MS neurons but not in LA neurons (Calabresi et al., 1999). Ischemia-induced long-term potentiation (LTP) occurs in MS neurons but not in LA neurons (Calabresi et al., 2002). Moreover, excitatory synaptic transmission is depressed in LA neurons (Pang et al., 2002) while enhanced in MS neurons after ischemia (Zhang et al., 2006).
We recently reported that inhibitory synaptic transmission is enhanced in LA neurons while depressed in MS neurons after transient cerebral ischemia. The enhancement of inhibitory synaptic transmission in LA neurons is mediated by the increased release of GABA (Li et al., 2009). It is known that LA neurons receive inhibitory inputs from MS neurons (Bolam et al., 1986). With the massive degeneration of MS neurons after ischemia, how could the presynaptic release increase? To answer this question, we studied glutamate decarboxylase (GAD), the rate-limiting enzyme in the synthesis of GABA. GAD expression is closely related to GABAergic activity. Increased expression of GAD has been considered as a sign of GABAergic hyperactivity (Lindefors, 1993) while decreased expression is associated with reduced GABA synthesis (Mason et al., 2001). In addition, considering the differential changes of inhibitory synaptic transmission in MS neurons and LA neurons, we examined whether facilitation of inhibitory synaptic transmission by muscimol could protect striatal neurons against ischemic damage.
Materials and Methods
Male Wistar rats of 200-250 g (Charles River Laboratories, Wilmington, MA) were used in the present study. Experimental protocols were institutionally approved in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. We made efforts to minimize both the suffering and number of animals used.
Animal models of transient forebrain ischemia
Transient forebrain ischemia was induced by four-vessel occlusion with modifications (Ren et al., 1997). To produce uniform blood glucose levels, rats were fasted overnight. Rats were anesthetized with a mixture of 1-2% halothane in 33% oxygen and 66% nitrogen via a nasal mask during the surgery. The silicon tubing (0.025” I.D., 0.047” O.D.) was placed underneath each common carotid artery and passed through the two holes in a small Teflon button. A loop was formed loosely around each common carotid artery with a plastic cylinder and a suture line was tied at the end of the loop to allow subsequent occlusion of these vessels. Then, the two vertebral arteries were electrocauterized. A temperature probe (0.025” diameter, Physitemp, Clifton, NJ) was placed underneath the skull and the brain temperature was maintained at 36.5 °C with a heating lamp (BAT-10, Physitemp, Clifton, NJ). A glass microelectrode filled with 2 M NaCl was advanced 3.0 mm below the dura mater into the striatum. Occlusion was performed by pulling the silicon tubing through the cylinder to compress the artery against the Teflon button. Ischemic depolarizations (DC potential shifts from 0 mV to about -20 mV), which occurred approximately 2-3 min after the occlusion, was recorded by a neuroprobe amplifier (Model 1600, A-M systems, Carlsborg, WA). The occlusion lasted for 20 min. Cerebral blood flow resumed immediately upon release of the tubing but it took about 1-2 min for the DC potential to return to 0 mV.
Immunohistochemistry
Under deep anesthesia, rats were transcardially perfused with heparinized saline followed by 4% paraformaldehyde in 0.15 M NaPBS (Ph=7.4). Brains were removed and post fixed in the same fixative at 4 °C overnight. They were then cryoprotected in 30% sucrose in 0.15 M NaPBS for at least 48 h. Brains were frozen in the embedding medium (O.C.T., Sakura Finetek, Torrance, CA) on dry ice for at least half an hour before they were transferred to the cryostat. Coronal sections of 30 μm were cut and kept in the cryoprotective buffer at -20 °C.
All sections from the control and ischemic animals within the same experimental group were processed together under the same condition. Sections incubated without the primary antibodies remained unstained and served as negative controls. Free floating sections processed for ABC reactions were rinsed in 0.01 M KPBS three times and then incubated in the blocking serum (5% normal serum in 0.01 M KPBS with 0.5% Triton X-100) for 1 h at room temperature. They were incubated with the primary antibodies in the blocking serum at 4 °C overnight. After washes, they were incubated with the secondary antibodies for 1 h at room temperature. The secondary antibodies used were Biotinylated horse anti-mouse IgG (1:100; Vector, Burlingame, CA) and goat anti-rabbit IgG (Vector). Sections were further incubated in ABC (Avidin-Biotin Complex) solutions (Vector) for 1 h at room temperature. Next, the immunohistochemical products were detected by 3’, 3’-diaminobenzidine tetrahydrochloride (DAB, Sigma, St. Louis, MO). Sections were then mounted on glass slides, dehydrated, and cover slipped.
For immunofluorescence studies, the procedures were the same as above except different secondary antibodies tagged with Fluorescein (1:100, Vector) or Texas Red (1:100, Vector) were used. Sections were counterstained with 4’, 6’-diamidino-2-phenylindole-dihydrochloride (DAPI) (Sigma). They were mounted on slides and cover slipped with Vectashield (Vector) for observation.
The primary antibodies used were rabbit anti-bromodeoxyuridine (BrdU) (1:100, Roche, Indianapolis, IN), rabbit anti-glutamate decarboxylase65 (GAD65) (1:1000, Millipore, Temecula, CA), mouse anti-synaptophysin (1:100, Millipore), mouse anti-GAD67 (1:2000, Millipore), mouse anti-vesicular glutamate transporter2 (vGLUT2) (1:1000, Millipore), goat anti-doublecortin (DCX) (1:250, Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit anti-somatostatin (SOM) (1:50, Santa Cruz Biotechnology).
For the immunostaining of BrdU, the following steps were performed. Sections were treated with 2 N HCl for 1 h at 37 °C to denature DNA. They were then immersed in 0.1 M borate buffer (Ph=8.5) twice for 10 min to neutralize the acid. Endogenous peroxidase activity was quenched by incubating sections in 1% H2O2 in KPBS for 15 min. After washes, they were incubated with the blocking serum and then the anti-BrdU antibodies.
Confocal images were acquired using a Zeiss LSM510 microscope.
Western blotting
Control rats and rats at one day and three days after ischemia were sacrificed and brain slices containing striata were cut at the thickness of 600 μm similar to previously described (Li et al., 2009). Striata were dissected out and stored on the dry ice before being manually homogenized using ice-cold radio immunoprecipitation assay (RIPA) buffer (10 mg tissue/100 ml buffer, Boston Bioproducts, Worcestere, MA) supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN). After brief sonication, the lysates were centrifuged at 14,000 g for 20 min at 4 °C. Supernatants were collected for further analysis. Protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA) was used to determine the protein concentration. The final concentration of the protein sample was adjusted to 1 μg/ml in the 2×SDS gel-loading buffer (Invitrogen, Carlsbad, CA). Protein samples were denatured by boiling before being loaded (20 μg) on the 10% SDS-polyacrylamide gel. After separation on the gel, the proteins were transferred to nitrocellulose membranes (Millipore, Bedford, MA). The membranes were then blocked with 0.1% bovine serum albumin in Tris-buffered saline containing 0.1% Triton-X for 1 h at room temperature. They were then incubated either in the mouse anti-actin (1:100,000, Sigma) or the rabbit anti-GAD65 (1:6,000, Millipore) antibodies in the blocking buffer at 4 °C overnight. After washes, the membranes were incubated in the secondary antibodies: peroxidase labeled anti-mouse (1:30,000, Vector) or anti-rabbit (1:100,000, Vector) IgG in the blocking buffer for 1 h at room temperature. The corresponding bands were detected with the enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ) and visualized by exposing the membrane to autoradiography films (Fuji, Tokyo, Japan). To provide semi-quantitative analysis, three samples of each experimental group were loaded and underwent Western blotting. Band densitometry analysis of the membrane was performed using scanned images of non-saturated immunoblot films, using NIH ImageJ 1.37 analysis software. Pixel intensities of the bands obtained in each experiment were normalized using β-actin signals, then calculated as a percentage of the mean of three control bands in the same membrane (Lei et al., 2008).
BrdU administration
BrdU (Sigma) was dissolved in saline (10 mg/ml). Control rats and rats at one day and three days after ischemia received intraperitoneal BrdU injections at the dose of 50 mg/kg every 8 hours for three days before they were sacrificed.
Muscimol administration and sample preparation for paraffin embedding
Muscimol (Enzo Life Sciences International, Plymouth Meeting, PA) was dissolved in saline (0.5 mg/ml). At 30 min before ischemia, muscimol (1mg/kg) (Costa et al., 2004; Xu et al., 2008) was given to the rats intraperitoneally. One day after ischemia, the rats were deeply anesthetized with the Ketaset and then transcardially perfused with 0.01 M NaPBS (pH=7.4) followed by the fixative (10% acetic acid, 10% formaldehyde (37%) and 80% methanol). The brain stayed in situ overnight. The next day, the brain was removed and post fixed in 4% paraformaldehyde for several days before it was transferred to 70% ethanol for 1-2 days. Then they were sent for paraffin embedding by a specialist.
Quantification analysis of the terminals and survived cells in the striatum
Quantification analysis was performed by an investigator who was blinded to the treatments using the software ImageJ. Images were captured from dorsolateral and/or dorsomedial striatum from three interaural planes corresponding to 10.6, 8.7, and 8.08 mm (Paxinos & Watson. The rat brain: in stereotaxic coordinates). Four sections from each plane were used.
To analyze the optical density (OD) of GAD65 and vGLUT2-positive terminals, images were captured in black and white 8-bit grayscale under 100× using a CCD camera. The density was determined in fixed regions measuring 1×1 mm2 that was kept constant across all the images. The density in the white matter, such as corpus collosum and striatal fibers, was subtracted as the background. The final numeric value represented the OD of the terminals in each mm2. To obtain the information of terminal number, size, and intensity, images were taken in black and white 8-bit grayscale under 400×. Thresholding was performed to distinguish the object terminals from the background. After precise thresholding, the minimum and maximum size of the terminals was set to occlude the objects clearly not of interest (Liu et al., 2006).
To count the GAD67-positive cells in the striatum, images were taken under 200× magnificantion. In the dorsolateral striatum, the GAD67-positive cells showing one cell body and two or more processes were counted. For the quantification analysis of survived cells,coronal sections of 10 μm were cut after paraffin embedding. Dorsolateral and dorsomedial striatum were randomly selected in each hemisphere of a section under 200× after hematoxylin & eosin (HE) staining. Dead cells showed shrunken nuclei while survived cells remained normal morphology. We assessed ischemic neuronal damage by counting survived cells in each captured field (800×600 μm).
Statistics
A mean was calculated for each animal and a total mean±SEM was determined for each group. One-way ANOVA was applied by using the software Statview (Statview, Abacus Concepts, Cary, NC). Changes were considered significant if P<0.05.
Results
GAD65 expression is increased in the striatum after transient cerebral ischemia
There are two isoforms of GAD in the striatum, GAD65 and GAD67. GAD65 is abundant in the terminals of MS neurons. GAD67 is strongly expressed in the GABAergic interneurons, with the exception of SOM-containing GABAergic interneurons. Normally, GAD67 cannot be detected in SOM-containing GABAergic interneurons although GABA immunoreactivity can be detected in their terminals (Kawaguchi et al., 1995). Since LA neurons receive inhibitory inputs from MS neurons (Bolam et al., 1986), we first examined the immunoreactivity of GAD65 on the striatal slices after ABC reaction. The OD of GAD65-positive terminals in the dorsolateral striatum showed a significant increase from 25.31±5.94 in control (n=6), to 69.01±2.79 at one day (n=6; P<0.05), and 69.36±14.21 at three days after ischemia (n=6; P<0.05; Fig. 1A). There were no significant differences in OD between one day and three days after ischemia.
Fig. 1.

The immunoreactivity of GAD65 is increased in the striatum after transient cerebral ischemia. A. Representative images of GAD65 immunostaining from the dorsolateral striata of control (n=5), one day (n=6), and three days (n=6) after ischemia. The insets show GAD65-positive puncta at higher magnifications (1000×). Scale bar in the insets, 5 μm. The upper panel of the histogram shows a significant increase in the OD of GAD65 immunoreactivity after ischemia. Images were taken under 100×. Scale bar, 100 μm. B. Representative images of GAD65 (green) after immunofluorescence staining from the dorsolateral striata of control, one day, and three days after ischemia. The middle and lower panel of the histogram show a significant increase in the number and intensity of GAD65-positive puncta after ischemia. Images were taken under 400×. Scale bar, 10 μm. C. Confocal images of GAD65 (green), synaptophysin (red), and the merged image of GAD65 and synaptophysin after doublelabeling immunofluorescence from the dorsolateral striatum. GAD65-positive puncta were found overlapping, or in close apposition with synaptosphysin. Scale bar, 1 μm. * P<0.05, ** P<0.01, compared to the control.
We also did immunofluorescence labeling of GAD65. The number and intensity of GAD65-positive puncta were estimated in the dorsolateral region of the striatum. After ischemic insults, the number of GAD65-positive puncta significantly increased (Fig. 1B). For the control group, the number was 554.27±109.26/field (n=6). One day after ischemia, it was 2077.17±341.5/field (n=6; P<0.05). Three days after ischemia, it was 2494.94±294.81/field (n=6; P<0.05; Fig. 1, middle panel of histogram). No significant difference in the number of GAD67-positive puncta was observed between one day and three days after ischemia.
The intensity of the GAD65-positive puncta was significantly increased at three days after ischemia. It was 60.41±1.75 in the control (n=6), 64.02±0.33 at one day (n=6; P>0.05), and 69.97±3.74 at three days after ischemia (n=6; P<0.05; Fig. 1, lower panel of histogram).
To confirm that GAD65-positive puncta were representative of GABAergic presynaptic terminals, we performed immunofluorescence double labeling of GAD65 and synaptophysin, a marker for presynaptic terminals. Confocal images demonstrated that GAD65-positive puncta were overlapped, or in close apposition with synaptophysin (Fig. 1C), suggesting that the most, if not all, of the GAD65-positive puncta are synaptic terminals.
The pathological process in ischemic tissues might facilitate the penetration of the antibodies. Therefore, the increase of immunoreactivity of GAD65 might not stem from the increase of protein expression after ischemia but the easier access of the antibodies as compared with the control tissue. To address this issue, we examined the immunoreactivity of vGLUT2 in the striatum and GAD67 in the hippocampus after ischemia. VGLUT2 is located on the glutamatergic terminals from the thalamus that synapse on LA neurons (Fremeau et al., 2001; Lapper and Bolam, 1992). If the increased immunoreactivity of GAD65 is due to the increased accessibility of antibodies, the immunoreactivity of vGLUT2 and GAD67 might also increase after ischemia. The results showed that there is no significant increase in vGLUT2 immunoreactivity in the striatum after ischemia (Supplementary Fig. 1A and B). Three days after ischemia, there is a decreased immunoreactivity of GAD67 in the dentate gyrus of the hippocampus as compared to the control (Supplementary Fig. 1C). These results suggest that the increased penetration of antibodies does not contribute to the increased immunoreactivity of GAD65.
To examine if the increased immunoreactivity of GAD65 reflected the increased expression level of GAD65 after ischemia, we performed western blotting. The relative density of the bands for GAD65 increased significantly after ischemia. For the control group, it was 100±16.93% (n=4). One day after ischemia, it was 289.86±37.85% (n=4; P<0.05). Three days after ischemia, it was 284.86±61.70% (n=4; P<0.05; Fig. 2).
Fig 2.
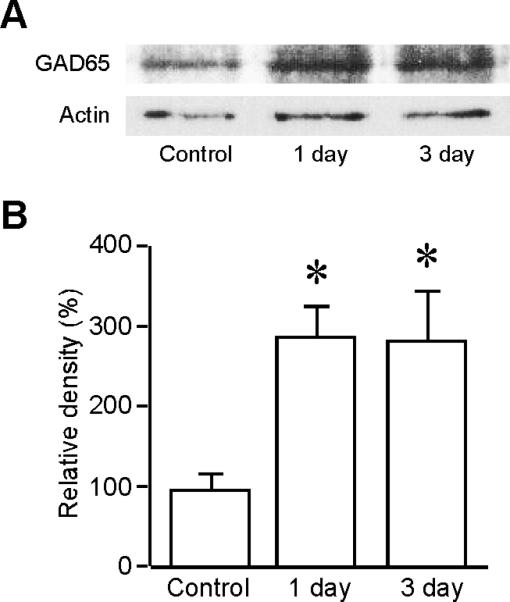
GAD65 expression is increased after ischemia. A. Western blotting was performed on the striatal lysates from the control rats (n=3), rats at one day (n=3), and three days (n=3) after ischemia. Antibodies against GAD65 and β-Actin were used. β-Actin is the loading control. Blots are representative of four independent experiments. B. Quantification analysis of the relative density of GAD65 in control and after ischemia. * P<0.05, compared to the control.
The above data demonstrated that GAD65 expression is enhanced in the striatum after ischemia. The increased expression of GAD65 highly suggests that presynaptic GABA synthesis is increased after ischemia.
GAD67-positive cells co-express GAD65 after ischemia
GABAergic interneurons survive transient cerebral ischemia (Chesselet et al., 1990). In the present study, the number of GAD67-positive cells in the striatum was significantly increased after ischemia (Fig. 3A-D). A cell count of the dorsolateral striatum yielded a control mean value of 8.67±1.69/field (n=6), a value of 21.69±1.92/field (n=6; P < 0.01) at one day, and 22.18±2.77/field (n=6; P < 0.01) at three days after ischemia. To examine if the survived GABAergic interneurons play a role in the increased expression of GAD65, we performed double-labeling immunofluorescence of GAD65 and GAD67. No expression of GAD65 was found in the cell bodies of control GAD67-positive cells (Fig. 3E1-E3). After ischemia, we found GAD67-positive cells co-labeled GAD65 in their soma (Fig. 3F1-F3). This suggests that GAD67-positive cells contribute to the increased GAD65 immunoreactivity in the striatum after ischemia.
Fig. 3.

The number of GAD67-positive cells is increased in the striatum after ischemia. Representative images of GAD67 immunostaining from the striata of control (A), one day after ischemia (B), and three days after ischemia (C). Images were taken under 200×. Scale bar, 100 μm. The insets show the GAD67-positive cells by the arrowhead under higher magnification (400×). Only cells showing one cell body and two or more processes were counted. Scale bar in the insets, 10 μm. D. Histogram shows that the number of GAD67-positive cells is increased in the striatum at one day (n=6) and three days (n=6) after ischemia compared to the control (n=5). ** P<0.01, compared to the control. E1-E3. Control GAD67-positive cells did not express GAD65. Images of immunofluorescence staining of GAD67 (red) (E1), GAD65 (green) (E2), and the merged GAD67 and GAD65 (E3) in the control. F1-F3. GAD67-positive cells expressed GAD65 after ischemia. Images of immunofluorescence staining of GAD67 (F1), GAD65 (F2), and the merged GAD67 and GAD65 (F3) after ischemia. Images were taken under 400×. Scale bar, 10 μm.
The origin of GAD67-positive cells after ischemia
The above results demonstrated that the number of GAD67-positive cells was increased in the striatum after ischemia. There are at least two possible origins of these GAD67-positive cells. One possibility is that they are derived either from precursors in the subventricular zone (SVZ) (Doetsch et al., 1999) or from precursors in the striatum (Bedard et al., 2002). The other possibility is that they are from the mature striatal neurons that undergo phenotypic shift after ischemia.
We labeled BrdU, a thymidine analog that is incorporated into the DNA of dividing cells, and in particular, the precursor cells during S phase of the cell cycle. In control animals, there were no BrdU-positive cells within the striatum (Fig. 4B). BrdU-positive cells were seen in the SVZ of control rats, validating the merit of this antibody (Fig. 4A). At one day following ischemic injury, only a small number of BrdU-positive cells were seen within the striatum (Fig. 4C). At three days following ischemic injury, there was a marked increase of BrdU-positive cells (Fig. 4D).
Fig. 4.

The increased GAD67-positive cells after ischemia are from the SOM-containing cells in the striatum but not from precursors. A & B. BrdU-positive cells were observed in the SVZ but not in the striatal parenchyma in the control. Representative images of BrdU immunostaining from the SVZ (A) and the striatal parenchyma (B) of the control rat. C & D. More BrdU-positive cells were observed in the striatal parenchyma after ischemia. Representative images of BrdU immunostaining from the striatal parenchyma at one day (C) and three days (D) after ischemia. Images were taken under 200×. Scale bar, 20 μm. E1-E3. GAD67-positive cells did not express BrdU after ischemia. Images of immunofluorescence staining of GAD67 (green) (E1), BrdU (red) (E2), and the merged GAD67 and BrdU (E3). F1-F3. GAD67-positive cells did not express DCX after ischemia. Images of immunofluorescence staining of GAD67 (red) (F1), DCX (green) (F2), and the merged GAD67 and DCX (F3). Images were taken under 400×. Scale bar, 10 μm. G1-G3. SOM-containing cells did not express GAD67 in the control. Images of immunofluorescence staining of GAD67 (green) (G1), SOM (red) (G2), and the merged GAD67 and SOM (G3) in the control. H1-H3. SOM-containing cells expressed GAD67 after ischemia. Images of immunofluorescence staining of GAD67 (green) (H1), SOM (red) (H2), and the merged GAD67 and SOM (H3) after ischemia. In this picture, one SOM-containing cell showed GAD67 staining after ischemia. Images were taken under 400×. Scale bar, 10 μm.
To answer the question whether the GAD67-positive cells observed after ischemia were BrdU-positive, we did double-labeling immunofluorescence of GAD67 and BrdU. No double-labeling of GAD67 and BrdU was observed, indicating that the GAD67-positive cells are not newly generated (Fig. 4E1-E3).
To examine whether the GAD67-positive cells were migrated from the SVZ, we did double-labeling immunofluorescence of GAD67 and doublecortin (DCX). DCX is a microtubule-associated protein associated with migrating neurons and serves as a principal marker for immature neuronal phenotypes (Francis et al., 1999). No double-labeling of DCX and GAD67 was observed (Fig. 4F1-F3).
To examine whether the increased number of GAD67-positive cells were from the SOM-containing GABAergic interneurons, we double labeled GAD67 and SOM in control and ischemic rats. In control rats, SOM-containing cells did not label GAD67 (Fig. 4G1-G3). However, after ischemia, we observed that some of the GAD67-positive cells co-labeled SOM (Fig. 4H1-H3).
The above results demonstrated that GAD67-positive cells observed after ischemia are not precursors and they are not migrated from the SVZ. After ischemia, SOM-containing GABAergic interneurons undergo phenotypic shift and express GAD67.
Muscimol protects striatal cells against ischemic injury
Previous studies indicated that muscimol enhances inhibitory synaptic transmission in LA neurons after ischemia through the activation of presynaptic GABAA receptors (Li et al., 2009). The above results indicate that GAD expression is increased in the striatum after ischemia, which might contribute to the enhancement of inhibitory synaptic transmission. To answer the question whether facilitation of inhibitory synaptic transmission could protect striatal cells against ischemia, the rats were injected intraperitoneally with muscimol (1mg/kg) (Costa et al., 2004; Xu et al., 2008) 30 min before the ischemic insults. We then examined neuronal injury by counting survived cells in the striatum.
Compared to the ischemic rats without muscimol treatment, the number of survived cells in the striatum was significantly increased in the ischemic rats treated with muscimol (Fig. 5A). For the sham-control group, the number was 344.5±1.45/field (n=12). For the ischemia group, the number of survived cells was 85.12±14.67/field (n=6). For the muscimol treatment group, the number was 171.44±29.54/field (n=9, P<0.05). This was mainly contributed by the increase of survived cells in the dorsomedial striatum (ischemia group: 101.18±15.30/field, n=6; muscimol treatment group: 224.22±36.78/field, n=9; P<0.05). The number of survived cells in the dorsolateral striatum showed no significant changes with muscimol treatment (ischemia group: 69.07±14.59/field, n=6; muscimol treatment group: 118.67±25.40/field, n=9; P=0.16; Fig. 5B). We concluded that muscimol could protect striatal cells against ischemic injury.
Fig. 5.

Muscimol application increases the number of survived cells in the striatum after ischemia. A. Representative images of HE staining after paraffin embedding. Images were taken from dorsolateral (DL) and dorsomedial (DM) striatal slices (10 μm) under 200×. Rats were subjected to either transient cerebral ischemia or ischemia plus muscimol intraperitoneal injections 30 min before the ischemic insults. Scale bar, 50 μm. B. Histograms show there was a significant increase of survived cells in the muscimol treatment group (n=9) compared to the non-treatment group (n=6) after ischemia. * P<0.05, compared to the ischemia group.
Discussion
The present study provides evidence that GAD65 expression and the number of GAD67-positive cells are increased in the striatum after transient cerebral ischemia. GAD67-positive cells contribute to the increased GAD65 expression after ischemia. The increased GAD67-positive cells do not result from neurogenesis. Doublelabeling of GAD67 and SOM indicates that some of the GAD67-positive cells are from the phenotypic shift of some SOM-containing GABAergic interneurons after ischemia. Muscimol application increases the number of survived cells in the striatum after ischemia.
Increased GAD expression in the striatum after transient cerebral ischemia
It is known that changes in GAD expression are closely associated with its biochemical activities (Segovia et al., 1990), the rate of GABA synthesis (Mason et al., 2001), and electrophysiological properties (Gonzalez-Hernandez et al., 2002). GAD expression and its activity vary in diseases. Changes in GAD expression have been intensively studied following lesions in the dopaminergic systems (Lindefors, 1993).
We observed increased expression of GAD in the striatum after transient cerebral ischemia. As to the origin of this increase, there are two possibilities. One is that it is originated from the terminals of the survived MS neurons or the MS neurons that are not severely damaged. The majority of the striatal neurons are MS neurons (97.7%) (Rymar et al., 2004). Pulsinelli et al. found that twenty-minute of transient forebrain ischemia will induce neuronal damage in 50% of the striatal populations after 72 h of survival (Pulsinelli et al., 1982). Most of the functional neurons after ischemia, especially the ones in the dorsomedial striatum (Pulsinelli et al., 1982), might contribute to the increased expression of GAD65. The other possibility is that the survived GABAergic interneurons (GAD67-positive) express GAD65 after ischemia. We observed GAD65 expression in the soma of GAD67-positive cells, highly suggesting GABAergic interneurons contribute to the increased GAD65 expression. It has been suggested that GABA synthesized by GAD67 is mainly found in the tonically active neurons and mostly provides a pool of GABA for general metabolic activity. GABA synthesized by GAD65 is mainly found in phasically active neurons and more likely to be involved in synaptic transmission (Martin and Rimvall, 1993). Although they are localized on two different chromosomes, GAD65 and 67 share strong sequence similarity (Bu et al., 1992). After ischemia, the loss of MS neurons suggests inhibitory synaptic transmission will be greatly weakened. Due to the strong sequence similarity, it is highly possible that ischemic insults could induce gene regulation and convert GAD67 to GAD65 in GAD67-positive cells to compensate for the massive loss of GAD65-positive MS neurons. We also observed that muscimol protected striatal cells when given 30 min before ischemia. It is speculated that this neuroprotection is mediated by the presynaptic GABAA receptors, rather than by the postsynaptic GABAA receptors, since postsynaptic response is depressed in LA neurons after ischemia (Li et al., 2009). Muscimol treatment significantly increased the number of survived cells in the dorsomedial striatum. One explanation is that because more neurons in the dorsolateral striatum die after ischemia, GABAergic strength should be significantly weakened after ischemia, whereas more neurons survived in the dorsomedial striatum (Pulsinelli et al., 1982). Therefore, the dorsomedial region has stronger responses to the muscimol application and enhances GABAergic strength.
Phenotypic shift of striatal cells after transient cerebral ischemia
Two hypotheses were made for the increased number of GAD67-positive cells after ischemia. One is that these cells are generated de novo either from the SVZ or from the stem cells within the striatum. Precursor cells exist in the striatum after focal ischemia (Arvidsson et al., 2002; Yamashita et al., 2006), transient global ischemia (Tonchev et al., 2005), and hypoxic/ischemic brain injury in neonatal rats (Yang et al., 2008). Precursors in the SVZ migrate to the striatum after ischemic injury (Arvidsson et al., 2002; Yamashita et al., 2006). New neurons are indeed generated de novo throughout adult life in the monkey striatum (Bedard et al., 2002). In addition, there is evidence showing stroke-induced neurogenesis in the human brain (Jin et al., 2006). However, the lack of double-labeling of BrdU and GAD67 disproves this hypothesis. From Hou et al's work, GAD67 positive newborn neurons were detected in the striatum after ischemia (Hou et al., 2008). The discrepancy might stem from: 1) The animal models in these two studies are different. We used global ischemia model while a focal ischemia model was used in the study mentioned above. There are reports showing that precursor cells differentiate into mature striatal neurons after focal ischemia (Arvidsson et al., 2002; Yamashita et al., 2006). However, few neurogenesis was reported in the striatum after transient global ischemia (Tonchev et al., 2005). 2) The experimental time points are different. We observed GAD67 cells at one day and three days after ischemia while they did it at 4 weeks. It is well known that adult neurogenesis varies significantly according to the triggering factors and the time of observation. Further studies are needed to determine whether newborn GAD67 cells could be observed at later time points after global ischemia and in early time points in focal ischemia.
The other hypothesis for the increased number of GAD67-positive cells after ischemia is that the survived striatal cells change their phenotype and express GAD67 after ischemia. In the striatum, phenotypic shift has been reported after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism, in which dopaminergic cells are from GABAergic interneurons (Tande et al., 2006). After colchicine treatment, SOM-containing GABAergic interneurons express GAD67 (Kubota et al., 1993). We demonstrated that SOM-containing cells, which are GAD67-negative in the control condition (Kawaguchi et al., 1995), co-expressed GAD67 after ischemia. This observation supports the phenotypic shift of SOM-containing neurons to GAD67-positive cells after ischemia. On the other hand, despite an almost three-fold increase in the number of GAD67-positive cells after ischemia, fewer than 10 cells were double positive for GAD67 and SOM in each single section, which indicates that SOM-positive cells contributed to the increased number of GAD67-positive cells but were not the only source for such change. One other possible source is that the GAD67 levels were low (below the detectable levels) in some of the GABAergic interneurons in control animals, but were significantly increased and become detectable after ischemia. Our studies suggest that expression of the normally quiescent GAD67 gene in SOM-containing GABAergic neurons could be triggered under certain conditions including ischemia. Whether this phenotypic shift is a temporary response to ischemic insults or has any functional impacts on the post-ischemic striatum remains to be determined.
The proliferation of progenitor cells brings us hope for the repair of the damaged brain after ischemic injury. In fact, precursor cells differentiate into mature striatal neurons from 30 to 90 days after focal ischemia (Arvidsson et al., 2002; Yamashita et al., 2006). However, few neurogenesis was reported in the striatum up to 79 days after transient global ischemia (Tonchev et al., 2005). Gliogenesis occurs shortly after transient global ischemia (Pforte et al., 2005). Moreover, BrdU is incorporated into dying neurons after hypoxic/ischemic brain injury (Kuan et al., 2004) but not observed in the model of transient global ischemia (Pforte et al., 2005; Tonchev et al., 2005). The present study observed BrdU-positive cells in the striatum at one day and three days after transient global ischemia. Together with previous studies, we speculate that most of the BrdU-positive cells are for gliogenesis but not neurogenesis at such early time points and they are not dying cells.
Conclusions
Taken together, we found a compensatory increase of GAD expression in the striatum after transient global ischemia. The increased expression of GAD and the consequent hyperactivity of GABAergic transmission might counterbalance excitotoxicity and contribute to the selective survival of striatal neurons after ischemia, which is confirmed by muscimol's protective effects.
Supplementary Material
Supplementary Fig. 1. The immunostaining of vGLUT2 is not changed in the striatum and the immunostaining of GAD67 in the dentate gyrus is decreased after ischemia. A. Representative images of vGLUT2 immunostaining from the striata of control (n=5), one day (n=6), and three days (n=6) after ischemia. Images were taken under 100×. Scale bar, 100 μm. The insets show vGLUT2-positive puncta at higher magnifications (1000×). Scale bar in the insets, 5 μm. B. Group data shows that there was no increase in the OD of vGLUT2 immunoreactivity after ischemia. C. Representative images of GAD67 immunostaining from the dentate gyrus of hippocampus show a decrease of GAD67 immunoreactivity at three days after ischemia compared to that of the control. Images were taken under 400×. Scale bar, 40 μm.
Acknowledgements
This work was supported by NINDS/NIH Grant NS38053 and AHA 0655747Z to Zao C. Xu. Zhigang Lei and Yan Li are the recipients of AHA Fellowships (0526007Z, 0825810G, and 0710027Z). We thank Drs Xiang Gao and Jinhui Chen for their assistance on confocal imaging, Yiwen Ruan on immunohistochemistry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Bedard A, Cossette M, Levesque M, Parent A. Proliferating cells can differentiate into neurons in the striatum of normal adult monkey. Neurosci Lett. 2002;328:213–216. doi: 10.1016/s0304-3940(02)00530-x. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Ingham CA, Izzo PN, Levey AI, Rye DB, Smith AD, Wainer BH. Substance P-containing terminals in synaptic contact with cholinergic neurons in the neostriatum and basal forebrain: a double immunocytochemical study in the rat. Brain Res. 1986;397:279–289. doi: 10.1016/0006-8993(86)90629-3. [DOI] [PubMed] [Google Scholar]
- Bu DF, Erlander MG, Hitz BC, Tillakaratne NJ, Kaufman DL, Wagner-McPherson CB, Evans GA, Tobin AJ. Two human glutamate decarboxylases, 65-kDa GAD and 67-kDa GAD, are each encoded by a single gene. Proc Natl Acad Sci U S A. 1992;89:2115–2119. doi: 10.1073/pnas.89.6.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Bernardi G. Metabotropic glutamate receptors and cell-type-specific vulnerability in the striatum: implication for ischemia and Huntington's disease. Exp Neurol. 1999;158:97–108. doi: 10.1006/exnr.1999.7092. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Sancesario G, Gubellini P, Marfia GA, Bernardi G. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington's disease. Ann Neurol. 1998;43:586–597. doi: 10.1002/ana.410430506. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Saulle E, Centonze D, Pisani A, Marfia GA, Bernardi G. Post-ischaemic long-term synaptic potentiation in the striatum: a putative mechanism for cell type-specific vulnerability. Brain. 2002;125:844–860. doi: 10.1093/brain/awf073. [DOI] [PubMed] [Google Scholar]
- Chesselet MF, Gonzales C, Lin CS, Polsky K, Jin BK. Ischemic damage in the striatum of adult gerbils: relative sparing of somatostatinergic and cholinergic interneurons contrasts with loss of efferent neurons. Exp Neurol. 1990;110:209–218. doi: 10.1016/0014-4886(90)90032-n. [DOI] [PubMed] [Google Scholar]
- Costa C, Leone G, Saulle E, Pisani F, Bernardi G, Calabresi P. Coactivation of GABA(A) and GABA(B) receptor results in neuroprotection during in vitro ischemia. Stroke. 2004;35:596–600. doi: 10.1161/01.STR.0000113691.32026.06. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–256. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr., Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hernandez T, Barroso-Chinea P, Perez de la Cruz MA, Valera P, Dopico JG, Rodriguez M. Response of GABAergic cells in the deep mesencephalic nucleus to dopaminergic cell degeneration: an electrophysiological and in situ hybridization study. Neuroscience. 2002;113:311–321. doi: 10.1016/s0306-4522(02)00186-0. [DOI] [PubMed] [Google Scholar]
- Hou SW, Wang YQ, Xu M, Shen DH, Wang JJ, Huang F, Yu Z, Sun FY. Functional integration of newly generated neurons into striatum after cerebral ischemia in the adult rat brain. Stroke. 2008;39:2837–2844. doi: 10.1161/STROKEAHA.107.510982. [DOI] [PubMed] [Google Scholar]
- Jin K, Wang X, Xie L, Mao XO, Zhu W, Wang Y, Shen J, Mao Y, Banwait S, Greenberg DA. Evidence for stroke-induced neurogenesis in the human brain. Proc Natl Acad Sci U S A. 2006;103:13198–13202. doi: 10.1073/pnas.0603512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Schloemer AJ, Lu A, Burns KA, Weng WL, Williams MT, Strauss KI, Vorhees CV, Flavell RA, Davis RJ, Sharp FR, Rakic P. Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci. 2004;24:10763–10772. doi: 10.1523/JNEUROSCI.3883-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Mikawa S, Kawaguchi Y. Neostriatal GABAergic interneurones contain NOS, calretinin or parvalbumin. Neuroreport. 1993;5:205–208. doi: 10.1097/00001756-199312000-00004. [DOI] [PubMed] [Google Scholar]
- Lapper SR, Bolam JP. Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience. 1992;51:533–545. doi: 10.1016/0306-4522(92)90293-b. [DOI] [PubMed] [Google Scholar]
- Lei Z, Deng P, Xu ZC. Regulation of Kv4.2 channels by glutamate in cultured hippocampal neurons. J Neurochem. 2008;106:182–192. doi: 10.1111/j.1471-4159.2008.05356.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Lei Z, Xu ZC. Enhancement of inhibitory synaptic transmission in large aspiny neurons after transient cerebral ischemia. Neuroscience. 2009;159:670–681. doi: 10.1016/j.neuroscience.2008.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindefors N. Dopaminergic regulation of glutamic acid decarboxylase mRNA expression and GABA release in the striatum: a review. Prog Neuropsychopharmacol Biol Psychiatry. 1993;17:887–903. doi: 10.1016/0278-5846(93)90018-n. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, El-Hayek YH, Gomez E, Zukin RS, Fehlings MG, Wan Q. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci. 2006;26:5309–5319. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DL, Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain. J Neurochem. 1993;60:395–407. doi: 10.1111/j.1471-4159.1993.tb03165.x. [DOI] [PubMed] [Google Scholar]
- Mason GF, Martin DL, Martin SB, Manor D, Sibson NR, Patel A, Rothman DL, Behar KL. Decrease in GABA synthesis rate in rat cortex following GABA-transaminase inhibition correlates with the decrease in GAD(67) protein. Brain Res. 2001;914:81–91. doi: 10.1016/s0006-8993(01)02778-0. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Deng P, Ruan YW, Xu ZC. Depression of fast excitatory synaptic transmission in large aspiny neurons of the neostriatum after transient forebrain ischemia. J Neurosci. 2002;22:10948–10957. doi: 10.1523/JNEUROSCI.22-24-10948.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pforte C, Henrich-Noack P, Baldauf K, Reymann KG. Increase in proliferation and gliogenesis but decrease of early neurogenesis in the rat forebrain shortly after transient global ischemia. Neuroscience. 2005;136:1133–1146. doi: 10.1016/j.neuroscience.2005.08.043. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res. 1985;63:29–37. doi: 10.1016/S0079-6123(08)61973-1. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Ren Y, Li X, Xu ZC. Asymmetrical protection of neostriatal neurons against transient forebrain ischemia by unilateral dopamine depletion. Exp Neurol. 1997;146:250–257. doi: 10.1006/exnr.1997.6525. [DOI] [PubMed] [Google Scholar]
- Rymar VV, Sasseville R, Luk KC, Sadikot AF. Neurogenesis and stereological morphometry of calretinin-immunoreactive GABAergic interneurons of the neostriatum. J Comp Neurol. 2004;469:325–339. doi: 10.1002/cne.11008. [DOI] [PubMed] [Google Scholar]
- Segovia J, Tillakaratne NJ, Whelan K, Tobin AJ, Gale K. Parallel increases in striatal glutamic acid decarboxylase activity and mRNA levels in rats with lesions of the nigrostriatal pathway. Brain Res. 1990;529:345–348. doi: 10.1016/0006-8993(90)90849-7. [DOI] [PubMed] [Google Scholar]
- Tande D, Hoglinger G, Debeir T, Freundlieb N, Hirsch EC, Francois C. New striatal dopamine neurons in MPTP-treated macaques result from a phenotypic shift and not neurogenesis. Brain. 2006;129:1194–1200. doi: 10.1093/brain/awl041. [DOI] [PubMed] [Google Scholar]
- Tonchev AB, Yamashima T, Sawamoto K, Okano H. Enhanced proliferation of progenitor cells in the subventricular zone and limited neuronal production in the striatum and neocortex of adult macaque monkeys after global cerebral ischemia. J Neurosci Res. 2005;81:776–788. doi: 10.1002/jnr.20604. [DOI] [PubMed] [Google Scholar]
- Xu J, Li C, Yin XH, Zhang GY. Additive neuroprotection of GABA A and GABA B receptor agonists in cerebral ischemic injury via PI-3K/Akt pathway inhibiting the ASK1-JNK cascade. Neuropharmacology. 2008;54:1029–1040. doi: 10.1016/j.neuropharm.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ninomiya M, Hernandez Acosta P, Garcia-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, You Y, Levison SW. Neonatal hypoxic/ischemic brain injury induces production of calretinin-expressing interneurons in the striatum. J Comp Neurol. 2008;511:19–33. doi: 10.1002/cne.21819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Deng P, Li Y, Xu ZC. Enhancement of excitatory synaptic transmission in spiny neurons after transient forebrain ischemia. J Neurophysiol. 2006;95:1537–1544. doi: 10.1152/jn.01166.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. The immunostaining of vGLUT2 is not changed in the striatum and the immunostaining of GAD67 in the dentate gyrus is decreased after ischemia. A. Representative images of vGLUT2 immunostaining from the striata of control (n=5), one day (n=6), and three days (n=6) after ischemia. Images were taken under 100×. Scale bar, 100 μm. The insets show vGLUT2-positive puncta at higher magnifications (1000×). Scale bar in the insets, 5 μm. B. Group data shows that there was no increase in the OD of vGLUT2 immunoreactivity after ischemia. C. Representative images of GAD67 immunostaining from the dentate gyrus of hippocampus show a decrease of GAD67 immunoreactivity at three days after ischemia compared to that of the control. Images were taken under 400×. Scale bar, 40 μm.