

Abstract
DNA methylation is a vital component of the epigenetic machinery that orchestrates changes in multiple genes and helps regulate gene expression in all known vertebrates. We evaluated immunoreactivity for two markers of DNA methylation and eight methylation maintenance factors in entorhinal cortex layer II, a region exhibiting substantial Alzheimer's disease (AD) pathology in which expression changes have been reported for a wide variety of genes. We show, for the first time, neuronal immunoreactivity for all 10 of the epigenetic markers and factors, with highly significant decrements in AD cases. These decrements were particularly marked in PHF1/PS396 immunoreactive, neurofibrillary tangle-bearing neurons. In addition, two of the DNA methylation maintenance factors, DNMT1 and MBD2, have been reported also to interact with ribosomal RNAs and ribosome synthesis. Consistent with these findings, DNMT1 and MBD2, as well as p66α, exhibited punctate cytoplasmic immunoreactivity that co-localized with the ribosome markers RPL26 and 5.8s rRNA in ND neurons. By contrast, AD neurons generally lacked such staining, and there was a qualitative decrease in RPL26 and 5.8s rRNA immunoreactivity. Collectively, these findings suggest epigenetic dysfunction in AD-vulnerable neurons.
Keywords: epigenetics, DNA methylation, Alzheimer's disease, neuron, ribosome
1. Introduction
Gene expression in the Alzheimer's disease (AD) brain has been shown to be altered in a wide variety of reports (Robinson et al., 1994, Loring et al., 2001, Dunckley et al., 2006, Weeraratna et al., 2007, Liang et al., 2008a, Liang et al., 2008b, Liang et al., 2008c), including a recent large-scale expression array study of single cell laser-captured entorhinal cortex layer II neurons (Dunckley et al., 2006). Multiple physiologic and molecular pathways are affected, including energy metabolism (Liang et al., 2008c) inflammation (Loring et al., 2001, Weeraratna et al., 2007) and abberant cell cycle events (Arendt, 2000, Bowser and Smith, 2002), among others. Although individual pathogenic factors such as amyloid β peptide (Aβ) and tau phosphorylation are clearly critical links, no over-arching principle to explain the consistency, extent, and breadth of the gene expression, physiologic, and molecular changes reported in AD has received consensus acceptance. Epigenetic mechanisms such as histone modification (McLachlan et al., 1984), binding of non-histone proteins, and DNA methylation (Adcock et al., 2007, Suzuki and Bird, 2008) are capable of modulating coordinate expression of large numbers of genes across many different pathways, and may therefore warrant investigation for their potential role in AD pathogenesis.
DNA methylation is a highly conserved process that has been implicated in many different modalities of gene expression. The factors responsible for the methylation process are a family of DNA methyltransferases that have been shown to catalyze the transfer of a methyl group to single-stranded DNA using S-adenosyl methionine as the methyl donor. The recognition sequence for the mammalian DNA methyltransferase is relatively invariant, with nearly all cytosine methylations occurring on 5‘-C-p-G-3‘ (CpG) (Bird, 1986, Bird, 1992). There are four known active DNA methyltransferases in mammals, DNMT1, DNMT2, DNMT3A and DNMT3B. Of these, DNMT1 is the most abundant in mammalian cells. DNMT1 has been reported to be a key player in maintaining methylation in somatic cells, and loss of this enzyme has been shown to lead to nuclear disorganization, increased histone acetylation, and apoptosis (Chan et al., 2001, Fan et al., 2001, Jackson et al., 2004, Milutinovic et al., 2004, Espada et al., 2007).
Once methylation has occurred, methylation stability is maintained by the binding of specific complexes, MeCP1, to methylated regions of DNA. MeCP1 is not bound directly to methylated DNA, but rather to a single methyl-CpG-binding domain protein, MBD2. The resulting MeCP1/MBD2 complex is composed of 10 known proteins that include the complete nucleosome remodeling and histone deacetylase (NuRD) core, as well as MBD2. This group of proteins, in conjunction with CDK2AP1 (Doc1), make up a complex capable of nucleosome remodeling and histone deacetylation (Feng and Zhang, 2001, Feng and Zhang, 2003).
Because methylation and methylation maintenance factors can orchestrate changes in expression of a wide range of genes (Ashraf and Ip, 1998, Nan et al., 1998, Fujita et al., 1999, Ng et al., 1999, Feng and Zhang, 2001, Adcock et al., 2007, Suzuki and Bird, 2008), we hypothesized that alterations in methylation and methylation stability might provide an over-arching mechanism that could help explain expression differences in the thousands of genes that are reportedly altered in AD (Robinson et al., 1994, Loring et al., 2001, Dunckley et al., 2006, Liang et al., 2007, Weeraratna et al., 2007, Liang et al., 2008a, Liang et al., 2008b, Liang et al., 2008c). Here, we report highly significant decrements in immunoreactivity for two markers of DNA methylation and eight DNA methylation maintenance factors in AD neurons of entorhinal cortex layer II, one of the most consistently vulnerable brain regions to AD pathology (Braak, Braak and Bohl, 1993; Kordower et al., 2001).
2. Methods
2.1 Subjects and brain samples
Brain tissue was obtained through the Sun Health Research Institute Brain and Body Donation Program (Sun City, AZ). Specimens were collected under IRB-approved protocols and informed consents that permitted use of the samples for research by the investigators. Cases included in the study had received antemortem evaluation by board-certified neurologists and neuropsychologists, as well as postmortem evaluation by a board-certified neuropathologist. Evaluations and diagnostic criteria followed consensus guidelines for National Institute on Aging Alzheimer's Disease Centers. Diagnoses of patient condition included AD (N = 20) and cognitively and neurologically normal for age (ND) (N = 20). AD patients taking conventional AD therapeutics were not excluded from the study. At expiration, subject ages ranged from 60 to 97 years old, with a mean of 79.9 ± 1.3 years. Postmortem intervals for the subjects averaged 2 h 40 min ± 12 min. Subject age, gender, and postmortem interval were well matched between the experimental groups, and there were no significant between-groups differences on any of these variables. Because neurons of entorhinal cortex layer II, which form clusters or “islands”, are among the earliest and most consistently impacted neurons in AD brain (26), this brain region was the focus for all experiments.
2.2 Immunohistochemistry
Temporal cortex was sliced axially into 1-cm thick slabs, immersion fixed for 48h in buffered 4% paraformaldehyde at 4°C, washed extensively in phosphate buffer (PB), and cryoprotected in ethylene glycol and glycerol. The slabs were then sectioned at 40 μm a freezing cryostat. Free-floating sections at the level of the entorhinal cortex were stored in freezing solution (glycol/glycerol/PB) at -20°C until required for experiments. Tissue sections used for bright field microscopy were immunoreacted using the avidin-biotin complex/diaminobenzidine (DAB) method. Briefly, tissues were washed 2X, blocked in 1% hydrogen peroxide for 45 min, washed 3X, blocked in 3% bovine serum albumin (BSA) for 1 h, washed 2X, and incubated at 4°C overnight in primary antibody solutions containing 0.25% BSA. Unless otherwise stated, all washes were with 1X PBS Triton (PBST). Available information about the antibodies is given in Table 1. After incubation with primary antibody, sections were washed 3X, incubated in biotinylated, species-specific secondary antibodies (Vector) for 2 h, washed 3X, and incubated in avidin-biotin complex (Pierce) for 1 h. Following incubation with secondary antibody, the sections were washed 3X, once in PBST and twice in 0.05M Tris buffer, then exposed to DAB solution containing 125 μl of 5 mg/ml DAB (Sigma), 11.125 ml 50 mM Tris buffer pH 7.6, and 500 μl saturated nickel ammonium sulfate. Incubations during chromagen development were no longer than 10 min, and were followed by two quick rinses in 50 mM Tris to stop the reaction. Finally, the sections were dried, taken through graded alcohols, de-fatted in Neoclear (EMD), and mounted with Permount (Pierce). AD and ND sections were immunoreacted simultaneously using netwells in well-less plates. For fluorescence microscopy, the sections were washed 3X, blocked with either 3% normal goat serum or 3% BSA, and incubated for 2 h. The sections were then washed 2X, incubated in primary antibody in 0.25% BSA at 4°C overnight, washed again, and incubated in species–specific, fluorophore-conjugated secondary antibodies (Molecular Probes) at room temperature for 2 h. After a final wash, the sections were mounted, taken through Sudan Black to reduce autofluorescence, and coverslipped with Vectashield mounting media (Vector). Deletion of primary antibody or incubation with pre-immune serum resulted in abolition of specific immunoreactivity in all cases (data not shown). Adjacent serial sections were stained with cresyl violet for cell layer identification and verification that the island neurons of layer II were intact. For some sections, nuclei were counterstained with 4',6'-diamidino-2-phenylindole (DAPI) (Invitrogen) before mounting.
Table 1.
Antibodies
| Antibody | Host/Type | Dilution | Source/Catalogue# | Antigen/Epitope | Reference |
|---|---|---|---|---|---|
| NSE | Chicken polyclonal | 1:500 | Chemicon/AB9698 | Synthetic peptides from rat and human neuron specific enolase | www.millipore.com |
| NeuN | Mouse monoclonal/biotin conjugate | 1:1000 | Chemicon/MAB377B | Uncharacterized vertebrate neuron-specific nuclear protein | (Mullen et al., 1992) |
| GFAP | Rabbit polyclonal | 1:1000 | Chemicon/ab5804 | Bovine glial fibrillary acidic protein (holoprotein) | (Elmariah et al., 2005) |
| RAC1 | lectin | 1:1000 | Vector/B-1085 | Ricinus communis agglutinin I | (Mannoji et al., 1986) |
| pS396 | Rabbit polyclonal | 1:1000 | Invitrogen/44-752G | Phosphopeptide from serine 396 region of human tau | (Alonso et al., 2001) |
| PHF1 | Mouse monoclonal | 1:1000 | Gift of Dr. P. Davies | Human tau, phosphorylated serine 396 and 404 regions | (Chapman et al., 2003) |
| MBD3 | Mouse monoclonal | 1:400 | Abcam/ab45027 | Human MBD3/CKAFMVTDEDIRKQEE | (Boyes and Bird, 1991) |
| DOC1 | Rabbit polyclonal | 1:400 | Abcam/ab31794 | Human DOC1/TSSQYRQLLSDYGPPS | www.abcam.com |
| DNMT1 | Rabbit polyclonal | 1:400 | Abcam/ab19905 | Human DNMT1/Within residues 100-200 | www.abcam.com |
| 5-methylcytidine | Mouse monoclonal | 1:500 | Genway/20-783-71663 | 5-methylcytidine/epitope not available from manufacturer | (Espada et al., 2004) |
| RPL26 | Rabbit polyclonal | 1:500 | Abcam/ab59567 | Human RPL26/MKFNPFVTSDRSKNRKR | www.abcam.com |
| HDAC1 | Rabbit polyclonal | 1:400 | Abcam/ab19845 | Human HDAC1/Within residues 450 to the C-terminus | (Bhaskara et al., 2008) |
| P66 alpha | Rabbit polyclonal | 1:400 | Abcam/ab13714 | Human P66α/amino acids 137-150 | www.abcam.com |
| MTA2 | Rabbit polyclonal | 1:400 | Abcam/ab8106 | Human MTA2/amino acids 652-668 | (Caballero et al., 2007) |
| HDAC2 | Rabbit monoclonal | 1:400 | Abcam/ab32117 | Human HDAC2/residues within C-terminal end | (Weichert et al., 2008) |
| rRNA 5.8s | Mouse monoclonal | 1:500 | Novus/NB100-662 | 5.8s protein associated with rRNA | (Aakalu et al., 2001) |
| 5-methylcytosine | Mouse Monoclonal | 1:500 | Genway/20-003-40005 | 5-methylcytosine/epitope not available from manufacturer | (Maki et al., 2008) |
| RbAp48 | Mouse monoclonal | 1:400 | Abcam/ab488 | RbAp48/amino acids 1-425 | (Verstappen et al., 2008) |
| MBD2 | Goat polyclonal | 1:400 | Abcam/ab58241 | MBD2/RNDPLNQNKGKPDLN | www.abcam.com |
Double-label immunohistochemistry was also employed to evaluate possible associations of epigenetic factors with ribosomes and neurofibrillary tangles. Briefly, sections were washed, blocked with either 3% normal goat serum or 3% BSA, and incubated for 1-2 h. Sections were then washed 2X, incubated in primary antibodies raised in different species in 0.25% BSA/PBST at 4°C overnight. After primary incubation sections were washed 3X in PBST, and incubated in species–specific, fluorophore-conjugated secondary antibodies (Molecular Probes) at room temperature for 2 h. After a final wash, the sections were mounted, taken through Sudan Black to reduce autofluorescence, and coverslipped with Vectashield mounting media (Vector). Immunostained tissue sections were examined on Olympus IX51 and Olympus IX70 microscopes equipped with epifluorescence illumination or with confocal laser scanning using argon and krypton lasers (Olympus IX70). The findings were documented photographically with an Olympus DP-71 color digital camera or, for confocal microscopy, by Fluoview software (Olympus).
2.3 Cell Quantification and statistical analysis
For cell quantification, number-coded sections from three AD and three ND subjects were randomly selected from the pool of 20 AD and 20 ND subjects for each of the methylation factors (i.e., samples from different or overlapping subjects were evaluated for each factor). Measurements were made by an investigator blind to the subjects’ experimental condition. At 40X magnification, the total number of neurons, which were readily discriminated from glia by their large nuclei, within each of three randomly selected entorhinal cortex layer II islands, and the total number of neurons within each of those islands that exhibited clear immunoreactivity for the antibody being tested was recorded. A conventional two-tailed t-test of the percentage of cells labeled per island was then conducted to analyze the results.
3. Results
3.1 Immunoreactivity for markers of DNA methylation
Nuclear labeling with 5-methylcytosine and 5-methylcytidine has been used to assess methylation status in many reports (e.g., Halle et al., 1995, Havlis and Trbusek, 2002). In addition, both of these markers have also been reported to be associated with ribosomal RNA (rRNA) (Dunn, 1960, Tantravahi et al., 1981, Obara et al., 1982, Negre et al., 1989).
3.1.1 Nuclear Staining
Nuclear immunoreactivity for 5-methylcytosine and 5-methylcytidine was clearly evident in entorhinal cortex layer II neurons in ND cases, but was markedly decreased in AD cases (Fig. 1). The percentage of neurons showing positive 5-methylcytosine nuclear immunoreactivity was 91.3% ± 1.3% in ND and 39.9% ± 3.4% in AD subjects (t = 514.0, P < 0.0001). Positive 5-methylcytidine nuclear immunoreactivity was observed in 90.1% ± 1.3% of ND neurons, but only 51.8% ± 6.1% of AD neurons (t = 383.1, P < 0.0001). There was no overlap in these data for ND and AD cases. By contrast, in pathologically-spared regions of brain such as cerebellum, immunoreactivity for 5-methylcytosine and 5-methylcytidine was similar in ND and AD sections (Fig. 1D and supplementary Fig. 1). Staining of entorhinal cortex glia could be discerned at high power in ND cases, but was dramatically decreased or not seen in AD cases (Fig. 2). Deletion of primary antibody or pre-absorption with primary antigen completely abolished staining with 5-methylcytosine, 5-methylcytidine, and all other antibodies employed in this study.
Fig. 1.
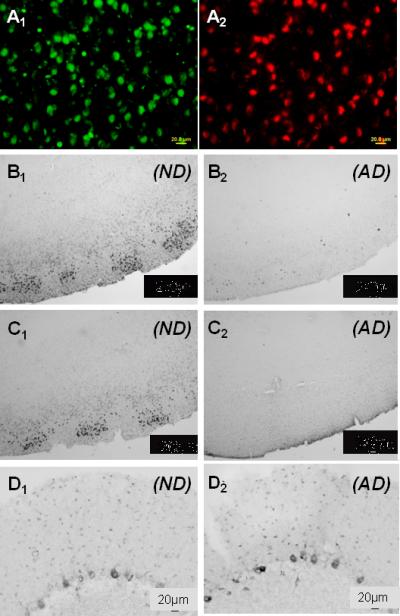
Representative micrographs of 5-methylcytosine and 5-methylcytidine immunoreactivity in entorhinal cortex layer II and cerebellar neurons from AD and ND cases. A1) High power micrograph of entorhinal cortex labeled with an antibody to 5-methylcytosine. A2) Same field labeled with an antibody to neuron-specific enolase to show co-localization (ND case). B1) Low power micrograph of 5-methylcytosine immunoreactivity in entorhinal cortex of an ND case to show general staining pattern. B2) 5-methylcytosine, AD case, entorhinal cortex. C1) 5-methylcytidine, ND case, entorhinal cortex. C2) 5-methylcytidine, AD case, entorhinal cortex. D1) 5-methylcytosine, ND case, cerebellum. D2) 5-methylcytosine, AD case, cerebellum. Staining with 5-methylcytidine in AD and ND cerebellum was virtually indistinguishable from that with 5-methylcytosine (see supplementary Fig. 1).
Fig. 2.

Representative high power micrographs of 5-methylcytosine immunoreactivity in entorhinal cortex microglia and astrocytes. A1) Immunoreactivity for the microglial marker RAC1 in an ND case. A2) Same field labeled with 5-methylcytosine. A3) Merged image to show co-localization (arrows). B1) RAC1 immunoreactivity in an AD case, where microglia were typically more abundant than in AD cases. B2) Same field labeled with 5-methylcytosine. B3) Merged image. C1) Immunoreactivity for the astrocyte marker GFAP in an ND case. C2) Same field labeled with 5-methylcytosine. C3) Merged image. D1) GFAP immunoreactivity in an AD case. D2) Same field labeled with 5 methylcytosine. D3) Merged image. (Calibration bars in all fields equal 20 μm).
3.1.2 Cytoplasmic Staining
In ND neurons, cytoplasmic 5-methylcytidine and 5-methylcytosine immunoreactivity was evident at the light level (Fig. 3A1, 3C1). With confocal microscopy, the immunoreactivity had a punctate morphology (Fig. 3A2, 3C2), and was highly co-localizated with immunoreactivity for the ribosomal protein RPL26 (Fig. 3A3, 3C3). By contrast, 5-methylcytosine and 5-methylcytidine staining was faint to absent in the cytoplasm of AD neurons (Fig. 3B1, 3D1), and where it could be discerned, there was no co-localization with RPL26 (Fig. 3B2, 3B3, 3D2, 3D3).
Fig. 3.
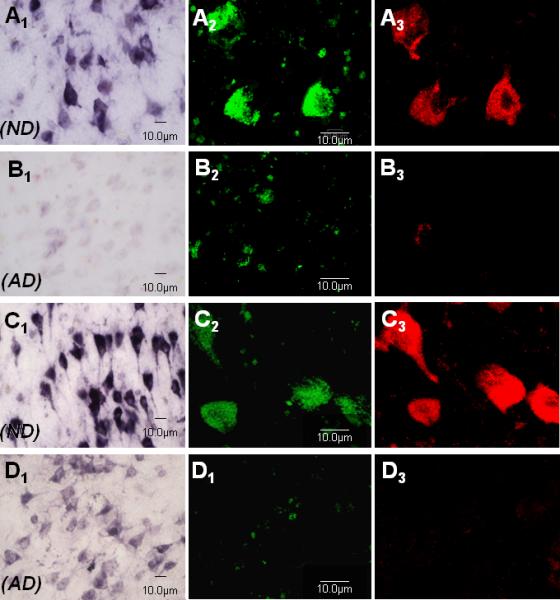
Representative micrographs of cytoplasmic immunoreactivity for 5-methylcytosine and 5-methylcytidine (green fluorophore in confocal micrographs) and their co-localization with immunoreactivity for the ribosomal protein RPL26 (red fluorophore in confocal micrographs) in AD and ND entorhinal cortex layer II neurons. Confocal planes were chosen that emphasized cytoplasmic rather than nuclear staining. A1) 5-methylcytosine, ND case, bright field. A2) 5-methylcytosine, ND case, confocal microscopy. A3) Same field as A2 for RPL26. B1) 5-methylcytosine, AD case, bright field. B2) 5-methylcytosine, AD case, confocal microscopy. B3) Same field as B2 for RPL26. C1) 5-methylcytidine, ND case, bright field. C2) 5-methylcytidine, ND case, confocal microscopy. C3) Same field as C2 for RPL26. D1) 5-methylcytidine, AD case, bright field. D2) 5-methylcytidine, AD case, confocal microscopy. D3) Same field as D2 for RPL26.
3.2 Immunoreactivity for DNA methylation stabilizing factors
To investigate mechanisms that might underlie the changes observed in neuronal DNA methylation, we next examined a wide range of proteins that methylate CpG sites or help maintain normal methylation status.
3.2.1 Nuclear Staining
Nuclear immunoreactivity for DNMT1, a major methyltransferase (Mortusewicz et al., 2005), and six different components of the MeCP1/MBD2 methylation complex, including MTA2, HDAC1, HDAC2, p66α, RbAp48, and MBD2/3, was significantly diminished in entorhinal cortex layer II neurons of AD compared to ND subjects (Fig. 4) (see also Supplementary Fig. 2). Mean percent of immunopositive neurons (± SEM) and comparisons by t-test were: DNMT1—ND (91.9% ± 1.0%), AD (7.6% ± 2.4%) (t = 33.0, P < 0.0001); HDAC1—ND (65.1% ± 4.2%), AD (17.0% ± 4.2%) (t = 8.1, P < 0.0001); HDAC2—ND (91.7% ± 2.7%), AD (12.7% ± 3.3%) (t = 18.6, P < 0.0001); p66α—ND (79.8% ± 4.6%), AD (12.7% ± 3.0%) (t = 12.3, P < 0.0001); MTA2—ND (94.7% ± 1.5%), AD (11.1% ± 2.4%) (t = 29.8, P < 0.0001); RbAp48—ND (93.7% ± 0.4%), AD (21.4% ± 2.8%) (t = 25.1, P < 0.0001); and MBD2/3—ND (98.3% ± 1.4%), AD (9.1% ± 2.0%), (t = 37.1, P < 0.0001). By contrast, when these factors were evaluated in cerebellum, a region that exhibits little to no AD pathology, immunoreactivity in ND and AD sections was again equivalent (Supplementary Fig. 1).
Fig. 4.

Representative micrographs of immunoreactivity for selected components of the MeCP1/MBD2 complex in entorhinal cortex layer II neurons. A1) MTA2, ND case. A2) MTA2, AD case. B1) HDAC2, ND case. B2) HDAC2, AD case. C1) RBAP, ND case. C2) RBAP, AD case. Similar results were obtained for all eight of the factors evaluated (see Supplementary Fig. 3).
3.2.2 Cytoplasmic Staining
Methylation of pre-rRNA is necessary for correct maturation of rRNA, and three of the methylation factors that were evaluated, DNMT1, HDAC1, and MBD2, have previously been reported to interact with rRNA as important components of ribosome synthesis (Ghoshal et al., 2004, Zhou and Grummt, 2005). Consistent with these findings in peripheral cells, DNMT1, MBD2, as well as p66α, another member of the MeCP1/MBD2 complex, exhibited punctate cytoplasmic immunoreactivity and co-localization with the ribosome markers RPL26 and 5.8s rRNA in ND neurons (Fig. 5). HDAC1, however, showed only diffuse cytoplasmic reactivity without a punctate appearance and without RPL26 or 5.8s rRNA co-localization (not shown). In AD neurons, by contrast, ribosome-associated cytoplasmic immunoreactivity for DNMT1, MBD2, and p66α was weak or not observed (Fig. 5). Moreover, consistent with the important role of these methylation factors in the synthesis of ribosomes, there was a noticeable decrease in immunoreactivity for the ribosome markers themselves in AD neurons (Fig. 5). Compromised expression of rRNA was also suggested by the absence in AD neurons of punctate nuclear immunoreactivity for SNF2H, a component of NoRC, which is integral to rRNA synthesis, whereas punctate nuclear SNF2H immunoreactivity was clearly evident in ND neurons (see supplementary Fig. 4).
Fig. 5.

Representative micrographs of cytoplasmic immunoreactivity for p66α (green fluorophore), its co-localization with 5.8s rRNA (red fluorophore), and AD decrements in 5.8s rRNA and the ribosomal protein RPL26 in entorhinal cortex layer II neurons. A1) 5.8s rRNA, ND case, confocal microscopy. A2) Same field as A1 for p66α. B1) 5.8s rRNA, AD case, confocal microscopy. B2) Same field as B1 for p66α. Similar results were observed for MBD2 and DNMT1. C1) 5.8s rRNA, ND case, bright field. C2) 5.8s rRNA, AD case, bright field. D1) RPL26, ND case, bright field. D2) RPL26, AD case, bright field.
3.3 Relationship of methylation markers to neurofibrillary tangles
Even in many ND cases, entorhinal cortex layer II is characterized by high densities of neurons with neurofibrillary tangles. Interestingly, there appeared to be an inverse relationship of entorhinal cortex layer II nuclear immunoreactivity for DNA methylation markers and markers for late-stage tangles (Fig. 6). This was especially true for 5-methylcytosine methylation in AD cases, where it was extremely difficult to find any PHF1 or PS396 immunopositive tangle-bearing neurons that also exhibited discernable nuclear 5-methylcytosine immunoreactivity (Fig 6A). Similar but less extreme results were seen for MeCP1 complex components (Fig. 6C, 6D). Even in ND cases, the majority of layer II PHF1 and PS396 immunopositive neurons showed little to no co-localized MeCP1 component immunoreactivity, and where such co-localization was seen the MeCP1 component staining appeared to be cytoplasmic, not nuclear (Fig. 6D, arrow).
Fig. 6.

Decreased 5-methylcytosine and p66α nuclear immunoreactivity in entorhinal cortex layer II island neurons that were immunoreactive for the late-stage tangle markers PS396 and PHF1. A1) 5-methylcytosine immunoreactivity, AD case. A2) Same field, PS396 immunoreactivity. A3) Merged image. B1) 5-methylcytosine immunoreactivity, ND case. B2) Same field, PS396 immunoreactivity. B3) Merged image. C1) p66α immunoreactivity, AD case. C2) Same field, PHF1 immunoreactivity. C3) Merged image. D1) p66α immunoreactivity, ND case. D2) Same field, PHF1 immunoreactivity. D3) Merged image.
4. Discussion
Perhaps because epigenetics is itself a relatively new field and has been primarily investigated in the context of oncology, epigenetic changes in AD are just beginning to be explored (Scarpa et al., 2003, Siegmund et al., 2007, Silva et al., 2008, Spremo-Potparavic et al., 2008, Wang, Oelze, and Schumacher, 2008, Wu et al., 2008). However, the changes in manifold individual genes in multiple cellular pathways that are emerging from large-scale gene expression array studies of AD (Robinson et al., 1994, Loring et al., 2001, Dunckley et al., 2006, Liang et al., 2007, Weeraratna et al., 2007, Liang et al., 2008a, Liang et al., 2008b, Liang et al., 2008c) suggest that epigenetic mechanisms could play a role in the disorder because of the ability of such mechanisms to modulate the coordinate expression of large numbers of genes. Here we provide, for the first time in human brain, immunohistochemical evidence that a wide range of epigenetic markers and regulators are expressed in neurons from control entorhinal cortex layer II and are dramatically decreased in AD patients, consistent with the high vulnerability of this brain region to AD pathology. In addition, we show co-localization of methylation stabilizing factors with rRNA (5.8s rRNA) and the ribosomal protein RPL26, a decrement in AD samples, and an apparent decrease in AD ribosomes themselves. We also show loss of SNF2H, a core component of the NoRC that is directly involved in rRNA expression.
The present findings suggest that global DNA and RNA methylation status in entorhinal cortex layer II neurons is significantly diminished in AD. A complete loss of methylation is, of course, unlikely in any cell, and, indeed, extensive digital enhancement of superficially blank fields in AD cases often revealed faint nuclear and cytoplasmic immunoreactivity for most of the methylation markers.
Several previous studies have examined methylation at specific gene loci in AD cortex. One report examined methylation status of 12 specific genes that have been implicated in AD pathology. Significant “drifts” in status were observed in some of the loci, but the manner in which the data were analyzed makes it difficult to determine whether methylation was increased or decreased in AD (Wang et al., 2008). A second paper found AD cytosine methylation changes in two genes, one of which, S100A2, exhibited a significant decrease in methylation. Cytidine methylation was not examined (Siegmund et al., 2007). Scarpa, Fuso, and colleagues (2003) have reported loss of methylation control of the BACE and presenilin 1 genes and its potential to cause overexpression of Aß in AD. A very early, more global analysis found no significant difference in percent CCGG methylation of DNA in AD cortex, but a number of caveats were given (Schwob et al., 1990). CCGG methylation, for example, would only cover approximately 20-30% of CpG sites. Because only DNA extracts were assayed, alterations in cytoplasmic ribosomal methylation would also have been missed. Data on postmortem delay for the samples, which could have a substantial effect on methylation integrity, were unavailable. By contrast, postmortem intervals for the present study averaged 2 hours 40 min, and were well matched among AD and ND cases. Moreover, as the authors of the previous research themselves noted, “Degenerating neurons, the cells most likely to reflect the molecular derangements of AD, probably contributed little to the bulk-extracted DNA from brain slices from patients with AD.” Our analysis, on the other hand, included all cells on the entorhinal cortex sections. Finally, in lymphocyte preparations, Silva and colleagues (2008) did not observe AD methylation changes in the SIRT3, SMARCA5, or CDH1 genes, but did report significant hypermethylation of the HTERT gene. The lymphocyte HTERT gene, however, is unusual in that its hypermethylation is believed to result in increased expression rather than the more usual silencing (Silva et al., 2008).
AD decrements in methylation status, as assayed by 5-methylcytosine and 5-methylcytidine immunoreactivity, were accompanied by decrements in all eight of the methylation stabilizing factors examined. These factors are critical components of MeCp1, NoRC, and NuRd, which, in turn, are important to maintenance of normal methylation state. The loss of many of these factors may be interlinked. For example, DNMT1 is recruited by HDAC1, and both are recruited by NoRC (Zhou et al., 2002, Zhou and Grummt, 2005). Functionally, improper maintenance of methylation in somatic cells leads to nuclear disorganization, de-differentiation, upregulation of tumor suppressor proteins, and an increase in histone acetylation (Chen et al., 1998, Santos et al., 2002, Zhu and Otterson, 2003, Hoffmann and Schulz, 2005). That methylation deficits in the present study were functionally associated with neurodegeneration is also suggested by their co-occurence with PHF1 and PS396 immunoreactivity, which are considered markers for the late stages of neurofibrillary tangle formation.
Methylation is involved in many aspects of cellular regulation, including ribosomal control and function. In addition to nuclear changes, we also report, for the first time, loss of cytoplasmic immunoreactivity for 5-methylcytidine and 5-methylcytosine, markers of methylation, and three methylation stabilizing factors, DNMT1, MBD2, and p66α, in AD entorhinal cortex island neurons. Genes for rRNA are heavily methylated in both transcribed and non-transcribed regions of the repeat unit (Bird, 1978), leading to the suggestion that changes in epigenetic factors may disrupt processing and function of rRNA (Espada et al., 2007). Our findings are wholly consistent with this hypothesis. 1) Loss of rRNA methylation alters rRNA post-transcriptional modification (Swaminathan et al., 2007), and AD neurons showed a near absence of the two key methylation markers 5-methylcytosine and 5-methylcytidine. 2) DNMT1 deficits, as reported here for AD neurons, have been previously demonstrated to disrupt DNA methylation and increase acetylation levels of lysine 16 histone H4 on rRNA genes (Espada et al., 2007). 3) The normal association of methylation stabilizing factors such as DNMT1, MBD2, and p66α with rRNA was nearly absent in entorhinal cortex layer II AD neurons. 4) SNF2H, a component of NoRC that is integral to rRNA synthesis, appeared to be lost in AD neurons. Indeed, we observed qualitative decreases in immunoreactivity for two markers of ribosomes themselves, RPL26 and 5.8s rRNA, in these neurons. It will therefore be of interest in future studies to test the hypothesis that loss of methylation markers and methylation stabilizers in AD neurons may cause deficits in rRNA gene expression, rRNAs, ribosomes, and ribosomal protein synthesis.
A final point that may warrant further research follows from the ability of epigenetic shifts to orchestrate entry or re-entry into the cell cycle (reviewed in (Golubnitschaja, 2007), a process that would presumably lead to apoptosis in postmitotic neurons (Nagy et al., 1998, Arendt, 2000, Nagy, 2000, Copani et al., 2001, Bowser and Smith, 2002, Herrup and Arendt, 2002). Many reports have suggested a derangement of cell cycle events in AD, including the observation that premature centromere division of the X chromosome is increased in AD (Spremo-Potparavic et al., 2008). Treatment of terminally differentiated PC12 neurons with the demethylating agent 5-azacytodine, for example, causes the cells to retract their processes, express cell cycle proteins, and apoptose (Hossain et al., 1997). Similarly, loss of methylation and DNMT1 in cultured neurons has been shown to lead to increased expression of cell cycle-related genes in vitro (Mattson, 2003). Thus, the decrements in methylation and methylation stabilization observed in the present study could provide an explanation for the many previous reports that have suggested an aberrant re-entry of AD neurons into the cell cycle and/or their apoptosis.
Development and aging are processes that have been consistently linked to epigenetic orchestration (reviewed in (Wu et al., 2008), and aging remains one of the most salient risk factors for AD. Epigenetic shifts have also been considered as major causative factors in disease states, particularly cancer (reviewed in (Esteller, 2008, Gonzalo et al., 2008, Vucic et al., 2008). Thousands of genes have been reported to increase or decrease their expression in AD neurons (Liang et al., 2008a), suggesting that there may be some over-arching, but presently unknown principle underlying these massive genomic changes. It will therefore be of great interest to explore, in prospective studies, interactions of AD pathology with the epigenetic mechanisms and factors that are highlighted by the present descriptive findings.
Supplementary Material
Acknowledgements
This research was supported by NIA AGO-7367 (JR), NIA 030429 (PC), and by the Arizona Alzheimer's Research Consortium (JR/PC). We thank Dr. Thomas Beach, Lucia Sue, and their staff for provision of tissue samples and postmortem evaluations, and Dr. Marwan Sabbagh and his staff for antemortem neurologic evaluations and diagnoses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement. To the best of their knowledge, the authors do not have an actual or potential conflict of interest with regard to the present research.
References
- Aakalu G, Smith WB, Nguyen N, Jiang C, Schuman EM. Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron. 2001;30:489–502. doi: 10.1016/s0896-6273(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Adcock IM, Tsaprouni L, Bhavsar P, Ito K. Epigenetic regulation of airway inflammation. Curr Opin Immunol. 2007;19:694–700. doi: 10.1016/j.coi.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Alonso AD, Zaidi T, Novak M, Barra HS, Grundke-Iqbal I, Iqbal K. Interaction of tau isoforms with Alzheimer's disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J Biol Chem. 2001;276:37967–73. doi: 10.1074/jbc.M105365200. [DOI] [PubMed] [Google Scholar]
- Arendt T. Alzheimer's disease as a loss of differentiation control in a subset of neurons that retain immature features in the adult brain. Neurobiol Aging. 2000;21:783–96. doi: 10.1016/s0197-4580(00)00216-5. [DOI] [PubMed] [Google Scholar]
- Ashraf SI, Ip YT. Transcriptional control: repression by local chromatin modification. Curr Biol. 1998;8:R683–6. doi: 10.1016/s0960-9822(98)70435-x. [DOI] [PubMed] [Google Scholar]
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. The essentials of DNA methylation. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- Bird AP. Use of restriction enzymes to study eukaryotic DNA methylation: II. The symmetry of methylated sites supports semi-conservative copying of the methylation pattern. J Mol Biol. 1978;118:49–60. doi: 10.1016/0022-2836(78)90243-7. [DOI] [PubMed] [Google Scholar]
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–13. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Bowser R, Smith MA. Cell cycle proteins in Alzheimer's disease: plenty of wheels but no cycle. J Alzheimers Dis. 2002;4:249–54. doi: 10.3233/jad-2002-4316. [DOI] [PubMed] [Google Scholar]
- Boyes J, Bird A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell. 1991;64:1123–34. doi: 10.1016/0092-8674(91)90267-3. [DOI] [PubMed] [Google Scholar]
- Caballero R, Setien F, Lopez-Serra L, Boix-Chornet M, Fraga MF, Ropero S, Megias D, Alaminos M, Sanchez-Tapia EM, Montoya MC, Esteller M, Gonzalez-Sarmiento R, Ballestar E. Combinatorial effects of splice variants modulate function of Aiolos. J Cell Sci. 2007;120:2619–30. doi: 10.1242/jcs.007344. [DOI] [PubMed] [Google Scholar]
- Chan MF, Van Amerongen R, Nijjar T, Cuppen E, Jones PA, Laird PW. Reduced rates of gene loss, gene silencing, and gene mutation in Dnmt1-deficient embryonic stem cells. Mol Cell Biol. 2001;21:7587–600. doi: 10.1128/MCB.21.22.7587-7600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman G, Beaman BL, Loeffler DA, Camp DM, Domino EF, Dickson DW, Ellis WG, Chen I, Bachus SE, Lewitt PA. In situ hybridization for detection of nocardial 16S rRNA: reactivity within intracellular inclusions in experimentally infected cynomolgus monkeys--and in Lewy body-containing human brain specimens. Exp Neurol. 2003;184:715–25. doi: 10.1016/S0014-4886(03)00337-6. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- Copani A, Condorelli F, Canonico PL, Nicoletti F, Sortino MA. Cell cycle progression towards Alzheimer's disease. Funct Neurol. 2001;16:11–5. [PubMed] [Google Scholar]
- Dunckley T, Beach TG, Ramsey KE, Grover A, Mastroeni D, Walker DG, Lafleur BJ, Coon KD, Brown KM, Caselli R, Kukull W, Higdon R, Mckeel D, Morris JC, Hulette C, Schmechel D, Reiman EM, Rogers J, Stephan DA. Gene expression correlates of neurofibrillary tangles in Alzheimer's disease. Neurobiol Aging. 2006;27:1359–71. doi: 10.1016/j.neurobiolaging.2005.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn DB. The isolation of 5-methylcytidine from RNA. Biochim Biophys Acta. 1960;38:176–8. doi: 10.1016/0006-3002(60)91219-1. [DOI] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–50. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espada J, Ballestar E, Fraga MF, Villar-Garea A, Juarranz A, Stockert JC, Robertson KD, Fuks F, Esteller M. Human DNA methyltransferase 1 is required for maintenance of the histone H3 modification pattern. J Biol Chem. 2004;279:37175–84. doi: 10.1074/jbc.M404842200. [DOI] [PubMed] [Google Scholar]
- Espada J, Ballestar E, Santoro R, Fraga MF, Villar-Garea A, Nemeth A, Lopez-Serra L, Ropero S, Aranda A, Orozco H, Moreno V, Juarranz A, Stockert JC, Langst G, Grummt I, Bickmore W, Esteller M. Epigenetic disruption of ribosomal RNA genes and nucleolar architecture in DNA methyltransferase 1 (Dnmt1) deficient cells. Nucleic Acids Res. 2007;35:2191–8. doi: 10.1093/nar/gkm118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–97. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Zhang Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. 2001;15:827–32. doi: 10.1101/gad.876201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Zhang Y. The NuRD complex: linking histone modification to nucleosome remodeling. Curr Top Microbiol Immunol. 2003;274:269–90. doi: 10.1007/978-3-642-55747-7_10. [DOI] [PubMed] [Google Scholar]
- Fujita N, Takebayashi S, Okumura K, Kudo S, Chiba T, Saya H, Nakao M. Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol Cell Biol. 1999;19:6415–26. doi: 10.1128/mcb.19.9.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal K, Majumder S, Datta J, Motiwala T, Bai S, Sharma SM, Frankel W, Jacob ST. Role of human ribosomal RNA (rRNA) promoter methylation and of methyl-CpG-binding protein MBD2 in the suppression of rRNA gene expression. J Biol Chem. 2004;279:6783–93. doi: 10.1074/jbc.M309393200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubnitschaja O. Cell cycle checkpoints: the role and evaluation for early diagnosis of senescence, cardiovascular, cancer, and neurodegenerative diseases. Amino Acids. 2007;32:359–71. doi: 10.1007/s00726-006-0473-0. [DOI] [PubMed] [Google Scholar]
- Gonzalo V, Castellvi-Bel S, Balaguer F, Pellise M, Ocana T, Castells A. Epigenetics of cancer. Gastroenterol Hepatol. 2008;31:37–45. doi: 10.1157/13114573. [DOI] [PubMed] [Google Scholar]
- Halle JP, Schmidt C, Adam G. Changes of the methylation pattern of the c-myc gene during in vitro aging of IMR90 human embryonic fibroblasts. Mutat Res. 1995;316:157–71. doi: 10.1016/0921-8734(95)90002-0. [DOI] [PubMed] [Google Scholar]
- Havlis J, Trbusek M. 5-Methylcytosine as a marker for the monitoring of DNA methylation. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;781:373–92. doi: 10.1016/s1570-0232(02)00499-3. [DOI] [PubMed] [Google Scholar]
- Herrup K, Arendt T. Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer's disease. J Alzheimers Dis. 2002;4:243–7. doi: 10.3233/jad-2002-4315. [DOI] [PubMed] [Google Scholar]
- Hoffmann MJ, Schulz WA. Causes and consequences of DNA hypomethylation in human cancer. Biochem Cell Biol. 2005;83:296–321. doi: 10.1139/o05-036. [DOI] [PubMed] [Google Scholar]
- Hossain MM, Takashima A, Nakayama H, Doi K. 5-Azacytidine induces toxicity in PC12 cells by apoptosis. Exp Toxicol Pathol. 1997;49:201–6. doi: 10.1016/S0940-2993(97)80008-5. [DOI] [PubMed] [Google Scholar]
- Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, Ramsahoye B. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004;24:8862–71. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Stebbins GT, Dekosky ST, Cochran EJ, Bennett D, Mufson EJ. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49:202–13. [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, Mckeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA. Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiol Genomics. 2008a;33:240–56. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, Mckeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA. Neuronal gene expression in non-demented individuals with intermediate Alzheimer's Disease neuropathology. Neurobiol Aging. 2008b doi: 10.1016/j.neurobiolaging.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Walker DG, Caselli RJ, Kukull WA, Mckeel D, Morris JC, Hulette C, Schmechel D, Alexander GE, Reiman EM, Rogers J, Stephan DA. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol Genomics. 2007;28:311–22. doi: 10.1152/physiolgenomics.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R, Kukull W, Morris JC, Hulette CM, Schmechel D, Rogers J, Stephan DA. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A. 2008c;105:4441–6. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loring JF, Wen X, Lee JM, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer's disease. DNA Cell Biol. 2001;20:683–95. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- Maki WC, Mishra NN, Cameron EG, Filanoski B, Rastogi SK, Maki GK. Nanowire-transistor based ultra-sensitive DNA methylation detection. Biosens Bioelectron. 2008;23:780–7. doi: 10.1016/j.bios.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Mannoji H, Yeger H, Becker LE. A specific histochemical marker (lectin Ricinus communis agglutinin-1) for normal human microglia, and application to routine histopathology. Acta Neuropathol. 1986;71:341–3. doi: 10.1007/BF00688060. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Methylation and acetylation in nervous system development and neurodegenerative disorders. Ageing Res Rev. 2003;2:329–42. doi: 10.1016/s1568-1637(03)00013-8. [DOI] [PubMed] [Google Scholar]
- Milutinovic S, Brown SE, Zhuang Q, Szyf M. DNA methyltransferase 1 knock down induces gene expression by a mechanism independent of DNA methylation and histone deacetylation. J Biol Chem. 2004;279:27915–27. doi: 10.1074/jbc.M312823200. [DOI] [PubMed] [Google Scholar]
- Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci U S A. 2005;102:8905–9. doi: 10.1073/pnas.0501034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–11. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nagy Z. Cell cycle regulatory failure in neurones: causes and consequences. Neurobiol Aging. 2000;21:761–9. doi: 10.1016/s0197-4580(00)00223-2. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Smith AD. The cell division cycle and the pathophysiology of Alzheimer's disease. Neuroscience. 1998;87:731–9. doi: 10.1016/s0306-4522(98)00293-0. [DOI] [PubMed] [Google Scholar]
- Nan X, Cross S, Bird A. Gene silencing by methyl-CpG-binding proteins. Novartis Found Symp. 1998;214:6–16. doi: 10.1002/9780470515501.ch2. discussion 16-21, 46-50. [DOI] [PubMed] [Google Scholar]
- Negre D, Weitzmann C, Ofengand J. In vitro methylation of Escherichia coli 16S ribosomal RNA and 30S ribosomes. Proc Natl Acad Sci U S A. 1989;86:4902–6. doi: 10.1073/pnas.86.13.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, Tempst P, Reinberg D, Bird A. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23:58–61. doi: 10.1038/12659. [DOI] [PubMed] [Google Scholar]
- Obara M, Higashi K, Kuchino Y. Isolation of nucleolar methylase producing only 5-methylcytidine in ribosomal RNA. Biochem Biophys Res Commun. 1982;104:241–6. doi: 10.1016/0006-291x(82)91965-9. [DOI] [PubMed] [Google Scholar]
- Robinson CA, Clark AW, Parhad IM, Fung TS, Bou SS. Gene expression in Alzheimer neocortex as a function of age and pathologic severity. Neurobiol Aging. 1994;15:681–90. doi: 10.1016/0197-4580(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Santos AP, Abranches R, Stoger E, Beven A, Viegas W, Shaw PJ. The architecture of interphase chromosomes and gene positioning are altered by changes in DNA methylation and histone acetylation. J Cell Sci. 2002;115:4597–605. doi: 10.1242/jcs.00160. [DOI] [PubMed] [Google Scholar]
- Schwob NG, Nalbantoglu J, Hastings KE, Mikkelsen T, Cashman NR. DNA cytosine methylation in brain of patients with Alzheimer's disease. Ann Neurol. 1990;28:91–4. doi: 10.1002/ana.410280117. [DOI] [PubMed] [Google Scholar]
- Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, Jaenisch R, Laird PW, Akbarian S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Swaminathan V, Reddy BA, Ruthrotha Selvi B, Sukanya MS, Kundu TK. Small molecule modulators in epigenetics: implications in gene expression and therapeutics. Subcell Biochem. 2007;41:397–428. [PubMed] [Google Scholar]
- Tantravahi U, Breg WR, Wertelecki V, Erlanger BF, Miller OJ. Evidence for methylation of inactive human rRNA genes in amplified regions. Hum Genet. 1981;56:315–20. doi: 10.1007/BF00274686. [DOI] [PubMed] [Google Scholar]
- Verstappen G, Van Grunsven LA, Michiels C, Van De Putte T, Souopgui J, Van Damme J, Bellefroid E, Vandekerckhove J, Huylebroeck D. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum Mol Genet. 2008;17:1175–83. doi: 10.1093/hmg/ddn007. [DOI] [PubMed] [Google Scholar]
- Vucic EA, Brown CJ, Lam WL. Epigenetics of cancer progression. Pharmacogenomics. 2008;9:215–34. doi: 10.2217/14622416.9.2.215. [DOI] [PubMed] [Google Scholar]
- Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer's disease. PLoS ONE. 2008;3:e2698. doi: 10.1371/journal.pone.0002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeraratna AT, Kalehua A, Deleon I, Bertak D, Maher G, Wade MS, Lustig A, Becker KG, Wood W, 3rd, Walker DG, Beach TG, Taub DD. Alterations in immunological and neurological gene expression patterns in Alzheimer's disease tissues. Exp Cell Res. 2007;313:450–61. doi: 10.1016/j.yexcr.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichert W, Roske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T, Denkert C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14:1669–77. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- Wu J, Basha MR, Zawia NH. The environment, epigenetics and amyloidogenesis. J Mol Neurosci. 2008;34:1–7. doi: 10.1007/s12031-007-0009-4. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Grummt I. The PHD finger/bromodomain of NoRC interacts with acetylated histone H4K16 and is sufficient for rDNA silencing. Curr Biol. 2005;15:1434–8. doi: 10.1016/j.cub.2005.06.057. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Santoro R, Grummt I. The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription. Embo J. 2002;21:4632–40. doi: 10.1093/emboj/cdf460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WG, Otterson GA. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr Med Chem Anticancer Agents. 2003;3:187–99. doi: 10.2174/1568011033482440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.