

Abstract
BACKGROUND AND PURPOSE
Myocardial ischaemia is associated with perturbations of electrophysiological profile of cardiac myocytes. The persistent sodium current (INap) is one of the major contributors to ischaemic arrhythmias and appears as an attractive therapeutic target. We investigated the effects of F 15845, a new anti-anginal drug on INap and in integrative models of INap-induced arrhythmias.
EXPERIMENTAL APPROACH
Sodium current was investigated using patch clamp technique on wild-type and ΔKPQ-mutated hNav1.5 channels transfected in HEK293 cells. Effects of F 15845 on action potentials (APs) were studied by the glass microelectrode technique and its anti-arrhythmic activities were investigated in ischaemia- and aconitine-induced arrhythmias in the rat.
KEY RESULTS
We demonstrated that F 15845 is a potent blocker of INap acting from the extracellular side of the channel. Blockade of INap was voltage dependent and characterized by an almost pure tonic block. F 15845 shortened AP from rabbit Purkinje fibres, confirming its lack of pro-arrhythmic activity, and prevented AP lengthening induced by the INap activator veratridine. F 15845 did not affect APs from rabbit atria and guinea pig papillary muscle where INap is not functional, confirming its inability to affect other cardiac ionic currents. F 15845 was effective at preventing fatal ventricular fibrillation and ventricular tachycardia during coronary ligation without modifying heart rate and blood pressure, and dose dependently increased the dose threshold of aconitine required to induce ventricular arrhythmias.
CONCLUSIONS AND IMPLICATIONS
F 15845, a novel anti-anginal drug targeting INap, demonstrates new anti-arrhythmic properties which may be of therapeutic benefit against ischaemia-induced arrhythmias
Keywords: anti-arrhythmia agents; F 15845; ion channels; ischaemia, pharmacology
Introduction
Enhancement of the persistent sodium current (INap) through Na+ channels modified by ischaemic conditions is a major source of Na+ entry during cardiac ischaemia (Wu and Corr, 1994; Le Grand et al., 1995; Ju et al., 1996; Hammarstrom and Gage, 2002; Shryock and Belardinelli, 2008; Sossalla et al., 2008). For example, it has been shown that thrombin and lysophosphatidylcholine dramatically increase INap (Undrovinas et al., 1992; Tamareille et al., 2002; Pinet et al., 2008). Prolonged opening of Na+ channels generates an electric current that contributes to, at least, two major concerns of ischaemia: (i) electrophysiological alterations, and (ii) defects of intracellular Na+ and Ca2+ homeostasis. First, INap prolongs the action potential favouring the occurrence of early afterdepolarizations (EADs). Second, Na+ ions are exchanged for Ca2+, via the reverse mode of the Na+/Ca2+ exchanger (Tani and Neely, 1990), resulting in a pro-arrhythmogenic Ca2+ overload. The latter can also trigger delayed afterdepolarization (DAD) and cause a dispersion of repolarization.
On this basis, the selective inhibition of INap is expected to prevent or limit the occurrence of arrhythmias during ischaemia. Recently, inhibition of INap by ranolazine has been shown to reduce Ca2+ overload-induced arrhythmias and left ventricular dysfunction during ischaemia/reperfusion (Belardinelli et al., 2006; Hale et al., 2008; Hasenfuss and Maier, 2008). However, ranolazine potency and selectivity for INap is not optimal (Antzelevitch et al., 2004). Furthermore, it is not known whether the anti-arrhythmic activity of ranolazine is due to blockade of INap alone or to a combination of non-selective interactions with other targets. We recently described a novel, more selective and potent blocker of INap, F 15845 (Le Grand et al., 2008; Vacher et al., 2009; Vie et al., 2009), which is currently entering phase II clinical trials for the treatment of angina. However, the anti-arrhythmic properties of F 15845 during cardiac ischaemia have not yet been fully investigated.
Hence, the aims of the present study were, first, to assess the effects of F 15845 on INap in HEK293 cells expressing wild-type and mutated hNav1.5. Second, we examined the impact of INap inhibition on the action potential duration in atrial and ventricular tissues and Purkinje fibres. Finally, the anti-arrhythmic profile of F 15845 was investigated in models of INap-induced ventricular arrhythmias in rats.
Methods
Animals were housed and tested in an Association for the Assessment and Accreditation of Laboratory Animal Care-accredited facility in strict compliance with all applicable regulations and protocols were carried out in compliance with French regulations and local Ethical Committee guidelines for animal research.
Cell culture and patch clamp experiments
Nav1.5 HEK293 cells stably transfected with human SCN5A (Van Bemmelen et al., 2004) were grown under standard conditions as previously described (Van Bemmelen et al., 2004; Pignier et al., 2007; Vacher et al., 2009). ΔKPQ-SCN5A-transfected cells were obtained as described below. The HEK293 cell line was transiently transfected by calcium phosphate method with 0.8 µg pIRES-hB1-CD8, 0.8 µg ΔKPQ-SCN5A and 0.8 µg pCDNA3.1. Cells were seeded in 35 mm Petri dishes at a density of 10 000 cells per dish and cultured for 2 days.
Sodium current was recorded using the patch clamp technique in the whole cell configuration as previously reported (Pignier et al., 2007). For INa measurements in SCN5A-transfected HEK 293 cells, K+ currents were abolished by a nominally K+-free medium and by addition of CsCl. The internal solution (pipette) contained (in mM): NaCl 10, CsCl 110, CaCl2 1, HEPES 10, EGTA 10, Mg-ATP 5, D(+)-glucose 10, pH 7.3 (CsOH). The external solution had the following composition (in mM): NaCl 30, CsCl 100, MgCl2 2, CaCl2 2, HEPES 10, D(+)-glucose 5, pH 7.4 (CsOH). For the recording of INa-ΔKPQ, the electrochemical gradient for Na+ was increased by raising the external concentration of Na+ in extracellular solution of following composition (in mM): NaCl 130, CsCl 5, MgCl2 1.2, CaCl2 2, HEPES 10, D(+)-glucose 5, pH 7.4 (CsOH). Internal solution was composed of (in mM) CsCl 60, Cs-aspartate 70, CaCl2 1, MgCl2 1, HEPES 10, EGTA 11, ATPNa2 5, pH 7.2 (CsOH). Small cells were selected and cells with INa amplitude >10 nA were rejected. Because of the fast kinetic of activation of Na+ channels, all experiments were carried out at room temperature (19–22°C).
Veratridine-induced INap
Sodium current was elicited by square depolarizing pulses of 350 ms duration from a holding potential value of −110 mV (as described in the legends of the figures) delivered at a frequency of 0.2 Hz. In order to verify the stability of voltage-clamp, every five pulses, the holding potential was shifted to –90 mV for one pulse. For the study of whole-cell current parameters, sodium currents were generated using a double-pulse protocol to obtain current–voltage (I–V) curves and steady-state inactivation and activation curves. Current density was calculated by dividing whole-cell current amplitude by whole-cell capacitance. From a holding potential of –110 mV, 350 ms depolarizing pulses to different membrane potentials (10 mV increments, i.e. the conditioning pulse, up to +40 mV) were followed by a 1 ms return to –110 mV and then by a 350 ms test pulse to –30 mV. Data used for the I–V curves and activation curves were measured from the conditioning pulse. Activation curves were estimated according to the formula: GNa=INa/(Vm– Vrev) where GNa is the conductance, INa is the amplitude of the sodium current for the test potential Vm and Vrev is the apparent reverse potential for Na+. Steady-state inactivation curves were plotted from data recorded during the test pulse. The data for activation and steady-state inactivation were fitted with a simple Boltzmann function: I/Imax = {1 + exp[Vm – V0.5)/k]} –1 where I/Imax is the relative current, V0.5 is the half-maximum voltage of activation or inactivation and k is the slope factor. Sodium current presenting an incomplete inactivation (persistent INa, INap) was induced with the alkaloid veratridine. The following two parameters of INa were measured: the amplitudes of peak of veratridine-modified INa and of the persistent INa. Amplitudes were measured as maximal amplitudes during the first 5 ms of the depolarizing pulse, and the persistent INa as the mean current amplitude of the last 10 ms of the pulse (that is the magnitude of INa at 340 to 350 ms of the depolarizing pulse) and they were expressed as current density in pA/pF.
ΔKPQ mutation-induced INap
Sodium current was elicited by square depolarizing pulses of 350 ms duration from a holding potential value of –80mV delivered at a frequency of 0.2 Hz. The following two parameters of INa were measured: the amplitudes of peak (corresponding to mean amplitude of the first 10 ms of the depolarizing pulse), and persistent INap as mean of the last 50 ms of the depolarizing pulse. Steady-state inactivation data were obtained from a double-pulse protocol. From a holding potential value of –80 mV, a conditioning pulse was applied (500 ms duration) from −135 to −25 mV (10 mV increments). This was followed by a test pulse (−10 mV, 20 ms duration) followed by a return to −80 mV. Steady-state inactivation curves were plotted from normalized current amplitude (test pulse) against voltage of conditioning pulse and fitted with a simple Boltzmann function: I/Imax = {1 + exp[Vm – V0.5)/k]} – 1.
Microelectrode experiments
Experiments on cardiac action potentials were performed as described previously from rabbit (Le Grand et al., 2000) and guinea pig (Vacher et al., 2009) heart preparations. Male rabbits (SPF, 1.8–2.0 kg, New-Zealand, ESD, France) were killed with an overdose of pentobarbital sodium and the heart was rapidly excised. Purkinje fibres were carefully dissected under a binocular microscope from the left ventricle. After a stabilization period of 30 min, the preparations were superfused with an oxygenated modified Krebs solution of the following composition (in mM): 137 NaCl, 4 KCl, 0.5 MgSO4, 1.8 CaCl2, 0.9 NaH2PO4, 20 NaHCO3, 5 glucose, 0.5 2,3-butanedione monoxime, pH 7.4 (NaOH). After a stabilization period of 30 min, the preparations were then electrically stimulated (model 6 BP, FHC, Pulsar, Brunswick, ME, USA) with rectangular pulses of 2 ms duration and 1.5 times the threshold voltage through a bipolar Ag electrode. The preparations were allowed to equilibrate for at least 1 h at a stimulation rate of 1 Hz.
Action potentials were recorded from cells on the surface of the preparations by conventional glass microelectrodes (5–20 MΩ) filled with 3M KCl, which were coupled to a high-input impedance preamplifier (VF 102 Biologic, Echirolles, France). Action potentials were displayed on a dual-beam oscilloscope (model TDS 420, Tektronix, Heerenveen, the Netherlands) and simultaneously digitized (10 KHz) and analysed by computer (Hewlett Packard, Vectra VL 800, CA, USA) using interactive software (Notocord V3.4, Croissy/Seine, France). Action potential parameters measured were maximum upstroke velocity (dV/dt), amplitude, overshoot, resting potential and action potential duration at 50 and 90% repolarization levels (APD50 and APD90, respectively).
Ischaemia-induced arrhythmias in anaesthetized rats
Male Sprague-Dawley rats weighing 220–300 g at the date of the experiments were purchased from Iffa Credo (France). Animals were anaesthetized with pentobarbital (60 mg·kg−1, i.p.). The caudal vein was cannulated for i.v. administration of compounds. Then, the animals were ventilated at 60 respirations min−1 (2.5 mL/respiration, Ventilator model 683, Harvard Apparatus, Holliston, MA, USA) under anaesthesia. The temperature of a heating pad (Homeothermic blanket control unit, Harvard Apparatus) was adjusted to 37°C. A standard limb lead II electrocardiogram was recorded via a bioelectric amplifier (ECG amplifier, Gould Instrument Systems, Valley View, OH, USA). The ECG analogue signal was digitized and simultaneously recorded by means of data acquisition software (AcqKnowledge, Biopac, Systems Inc., Goleta, CA, USA). A left thoracotomy was performed and the heart was exposed. Silk suture (4.0) was placed around the left coronary artery ∼1 mm from its origin for ligation. The compound or vehicle was administrated i.v. over 5 min using a single bolus 10 min before the initiation of the myocardial infarction. Ischaemia-induced ventricular arrhythmias were recorded during the ischaemia. Ventricular arrhythmias were evaluated according to the guidelines of the Lambeth Conventions (Walker et al., 1988).
Aconitine-induced arrhythmias in anaesthetized rats
Male Sprague-Dawley rats weighing 300–325 g at the date of the experiments were purchased from Charles River (France). Animals were anaesthetized with pentobarbital (60 mg·kg−1, i.p.). The caudal vein was cannulated for i.v. administration of compounds. Then, the animals were intubated and ventilated at 50 respirations min−1 (7.5 mL·kg−1 per respiration). A standard limb lead II electrocardiogram was recorded as described previously. The effects of F 15845 (0.63 and 2.5 mg·kg−1) on the ability of aconitine to induce ventricular arrhythmias in rats were then evaluated using an i.v. route of administration. After a stabilization period of 10 min, vehicle or F 15845 was injected (1 mL·kg−1). Perfusion of aconitine was started (20 µM at 400 µL·min−1·kg−1) 3 min later. Delays in the appearance of ventricular arrhythmias (determined in agreement with «The Lambeth Conventions»Walker et al., 1988) were converted in dose of aconitine.
Drugs
F 15845 (3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate) was synthesized by the Division of Medicinal Chemistry I, Centre de Recherche Pierre Fabre. F 15845 was dissolved in dimethyl sulphoxide (DMSO) as a 10 mM stock solution prepared freshly for each in vitro experiment. The highest final concentration of DMSO was 0.1% (F 15845 10 µM). Veratridine was purchased from Sigma Chemical (St. Louis, MO, USA) and was dissolved in distilled water. F 15845 (salt to base ratio: 1.31) was dissolved in polyethylene glycol (PEG) 300 for each in vivo experiment. After dissolution, sterile saline (0.9%) was added to obtain a final solution containing 40% PEG in sterile saline (0.9%).
Statistical analysis
All values are expressed as means ± SEM. Intra-group statistical analysis of results (drug versus baseline) was performed by paired t-test after testing for homogeneity of variance with analysis of variance (anova) with repeated measures. Intergroup statistical analyses of results (drug versus vehicle) were performed using one-way anova followed by Dunnett's test when anova was significant. Any P value less than 0.05 was considered significant (SigmaStat 2.03).
Results
Effects of F 15845 on persistent sodium current
Although INap may be detected under physiological conditions, its small amplitude complicates its study. Thus, we chose two different models in which INap is amplified. First, veratridine (40 µM) increases INap by stabilizing the channel in a bursting mode (Sunami et al., 1993). Second, we used Nav1.5 channels harbouring the ΔKPQ deletion mutation linked to congenital long QT type 3 syndrome in which sodium current presents an incomplete inactivation.
Veratridine-induced persistent sodium current
INa were studied in human SCN5A-transfected HEK 293 cells using a depolarizing step to –30 mV from a holding potential of either –110 or –90 mV in order to modify the ratio of resting over inactivated channels. Cell superfusion with 40 µM veratridine induced activation of INap as illustrated in Figure 1A,B. F 15845 at 1 µM reduced INap amplitude by 10% and 25% with holding potentials values of –110 and –90 mV, respectively (Figure 1D). The current–voltage relationship of veratridine-induced INap was slightly modified by 1 µM F 15845 (Figure 1C). The sodium current reached maximal density at –55 mV (−36 ± 7 pA/pF in control and –30 ± 5 pA/pF with 1 µM F 15845, P < 0.05, n= 6). At 10 µM, F 15845 strongly reduced veratridine-induced INap at every potential, from –70 to –10 mV (Figure 1C). Maximal INap density was reduced to –17 ± 3 pA/pF (at –55 mV, P < 0.01, n= 6). Finally, F 15845 concentration-dependently reduced veratridine-induced INap (Figure 1D) with an estimated IC50 of 5.3 µM with a 95% confidence interval of [3.2; 8.1 µM] and Hill coefficient of 1.2 when the membrane HP was held at –110 mV. F 15845 was more potent at reducing veratridine-induced INap when cells were depolarized to –90 mV with an IC50 of 1.8 µM [1.1; 2.8 µM] and nH of 0.8. This observation suggests that F 15845 is more potent when the sodium channels are more likely to be in an inactivated state (i.e. depolarized state).
Figure 1.
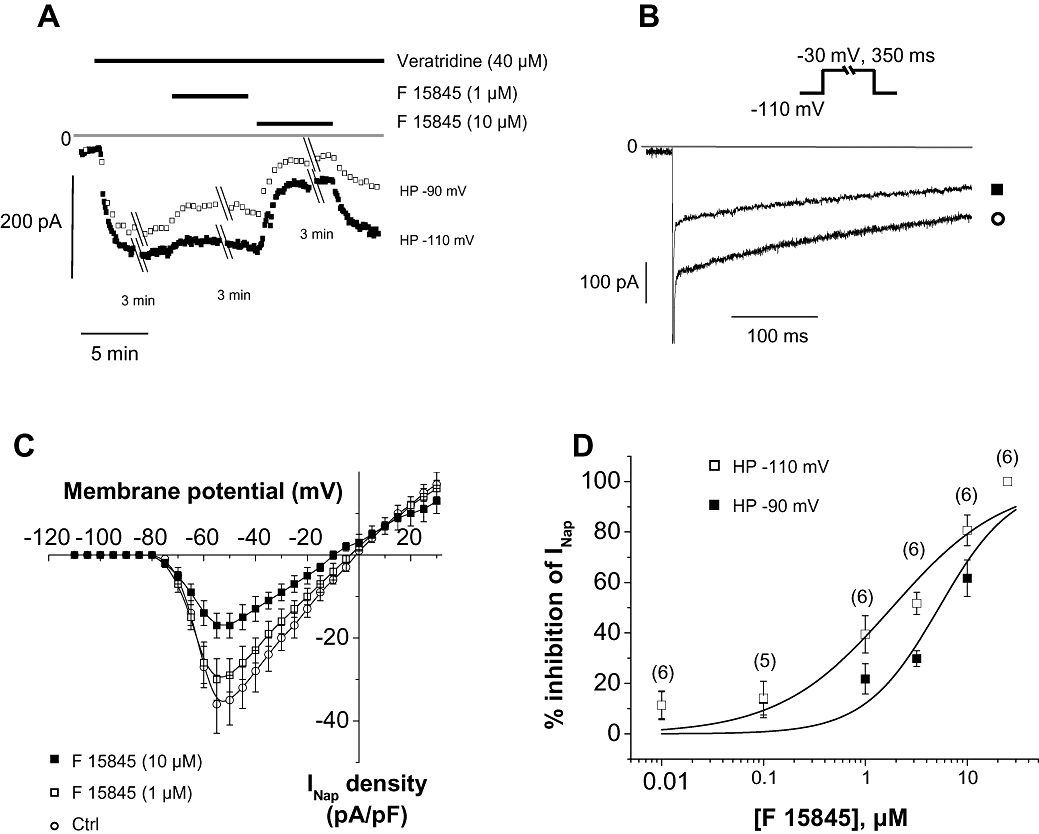
Effects of F 15845 on veratridine-induced INap. (A) Representative recordings of amplitude of INap elicited at –30 mV from a holding potential value of either –110 mV or –90 mV in presence of veratridine (40 µM) and during consecutive superfusions of 1 µM and 10 µM F 15845. (B) Veratridine-induced INap traces recorded in absence and in presence of 10 µM F 15845. (C) Mean current-voltage curves of veratridine-induced INap in absence and in presence of F 15845 1 µM and 10 µM elicited from a holding potential value of –110 mV. Data are means ± SEM of 6 cells. (D) Concentration–response curves of F 15845 on veratridine-induced INap. Sodium currents were elicited at –30 mV from a holding potential value of either –110 mV or –90 mV. Number of experiments are indicated in parentheses.
In order to investigate such a voltage-dependent or state-dependent mechanism, the macroscopic properties of veratridine-modified INa were evaluated. Steady-state activation and inactivation (Figure 2A and inset) of current were studied by means of a double-pulse protocol as described previously (Pignier et al., 2007). F 15845 at 10 µM did not affect activation parameters of veratridine-modified peak INa with mean V0.5 values of –51.8 ± 3.0 mV and –51.1 ± 2.3 mV in control and in presence of 10 µM F 15845, respectively (NS, n= 5). The steady-state inactivation curve was shifted to the left by 10 µM F 15845 with mean V0.5 values of –84.2 ± 1.2 mV and –93.4 ± 1.5 mV (P < 0.05, n= 6) in control and in presence of 10 µM F 15845, respectively (Figure 2A,B). The negative shift in the steady-state inactivation V0.5 induced with F 15845 was concentration-dependent (Figure 2B). V0.5 values were significantly shifted leftwards in the presence of F 15845 with mean V0.5 values of –3.5 ± 0.3 mV (1 µM F 15845, n= 11) and –9.1 ± 1.1 mV (10 µM F 15845, n= 6) versus –1.0 ± 0.4 mV (vehicle group, n= 8). In addition to the veratridine-induced persistent INa, a second ‘sustained INa’ called the window current is due to the overlap of activation and steady-state inactivation curves in a restricted range of potentials where sodium channels fail to inactivate completely (Figure 2A). In the presence of F 15845, this range of potentials was markedly reduced by a leftwards shift of the steady-state inactivation curve (see inset, Figure 2A) suggesting the reduced availability of non-inactivated channels. However, this observation could not be taken into account for the inhibition of veratridine-induced INap by F 15845 that was observed from −50 mV to −10 mV, a potential range far away from the one of the window current (−80 to −65 mV).
Figure 2.
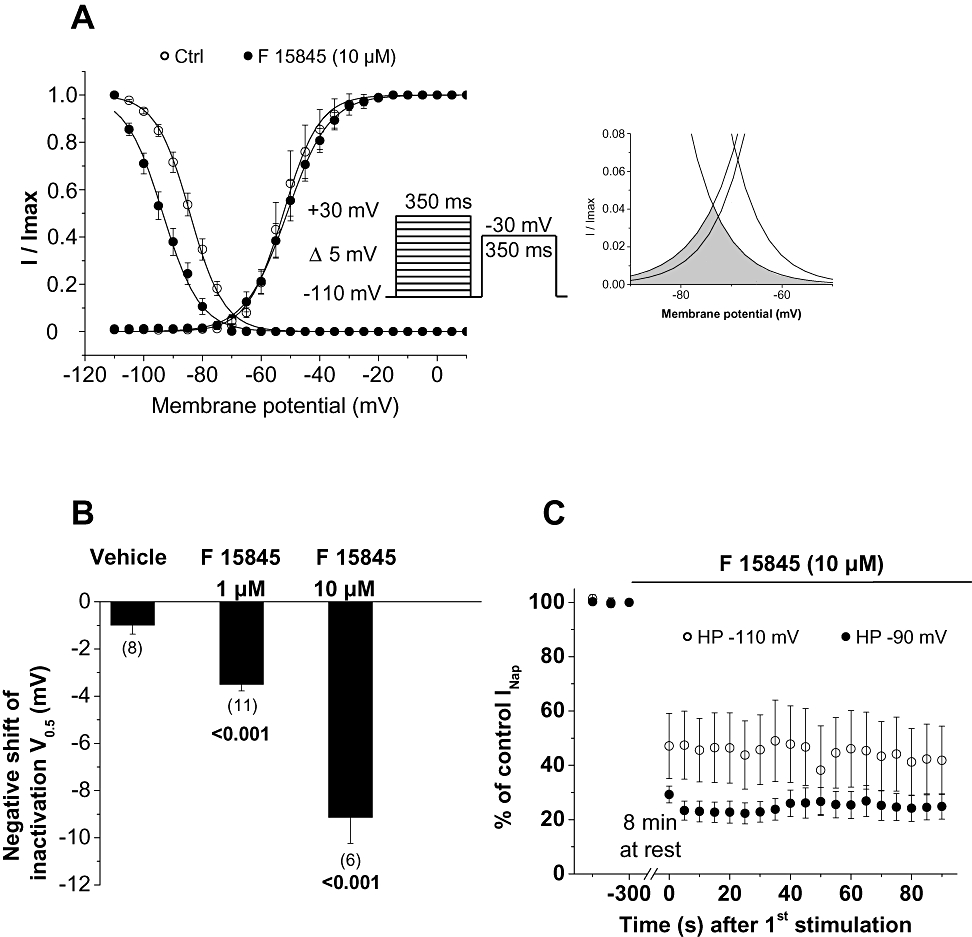
Effects of F 15845 on veratridine-modified INa availability. (A) Steady-state availability of veratridine-modified peak sodium current in the absence and presence of 10 µM F 15845. Curves were obtained as described in Methods section. Availability of window current is shown in expanded scale in inset. (B) Histogram representing the effect of vehicle (dimethyl sulphoxide 0.1%), F 15845 at 1 µM and 10 µM on the shift of steady-state inactivation V0.5. Data are means ± SEM, and numbers of cells are indicated in parentheses. Statistical significance is given versus vehicle group. (C) F 15845: Tonic versus phasic block of veratridine-induced INap. After a stabilization period in the presence of 40 µM veratridine, F 15845 was applied for 8 min without any stimulation (cells were maintained at a holding potential value of either –110 mV or –90 mV), after which, INap was elicited at –30 mV (0.2 Hz) for 90 s. INap amplitudes obtained were normalized to the last INap amplitude of the stabilization period. Tonic block was determined as block obtained with the 1st stimulation, and phasic block as the block observed with the last stimulation. Data are means ± SEM of 4–7 cells.
Tonic block of INap was investigated by measuring the first INap amplitude after an 8 min superfusion with 10 µM F 15845 without any stimulation either at holding potential of –110 and –90 mV (Figure 2C). The remaining INap amplitude at T0 was 47.1 ± 10.4% of control (n= 7, HP-110 mV) compared to 41.8 ± 10.6% (n= 6, HP-110 mV) at T90 with a holding potential of –110 mV and 29.3 ± 3.1% (T0, n= 4) and 24.9 ± 4.7% (T90, n= 4) with a holding potential of –90 mV. These results indicate that F 15845 was devoid of phasic block in these experimental conditions and confirm that tonic block was more potent at a depolarized membrane potential.
To determine the site of action of the compound, i.e. intra-cytoplasmic versus extra-cytoplasmic, F 15845 was applied through the patch clamp pipette solution. INap density was measured 10 min after patch rupture and compared to normal intra-pipette solution (vehicle group, 0.1% DMSO). The INap densities measured were similar in the absence or in presence of F 15845 in the pipette with mean current densities at –30 mV (from a HP of –110 mV) of –11.9 ± 2.8 pA/pF (vehicle, n= 6) and –9.2 ± 1.9 pA/pF (F 15845, n= 7) suggesting a lack of effect of F 15845 applied from the cytoplasmic side of the channel. Given that the effects of F 15845 were voltage dependent (Figure 2), these experiments were repeated at a holding potential of –90 mV. Likewise, the intracellular application of F 15845 had no effect on INap elicited at –30 mV with mean INap densities of −9.5 ± 2.1 pA/pF (vehicle, n= 6) and −6.5 ± 1.3 pA/pF (F 15845, n= 7).
ΔKPQ mutation -induced persistent sodium current
Transfected cells with ΔKPQ mutated channels exhibited sodium current with an incomplete inactivation as shown in Figure 3A. This current was sensitive to a high concentration of tetrodotoxin (TTX) (30 µM), which ascertains the nature of channels at the origin of INap (Figure 3A). F 15845 concentration-dependently reduced INap (Figure 3B). As previously observed for veratridine-induced INap, F 15845 10 µM significantly shifted the steady-state inactivation curve leftward with a mean V0.5 values of –76.6 ± 0.3 mV and –80.4 ± 0.3 mV (P < 0.001, n= 6) in control and in the presence of F 15845, respectively (Figure 3C).
Figure 3.
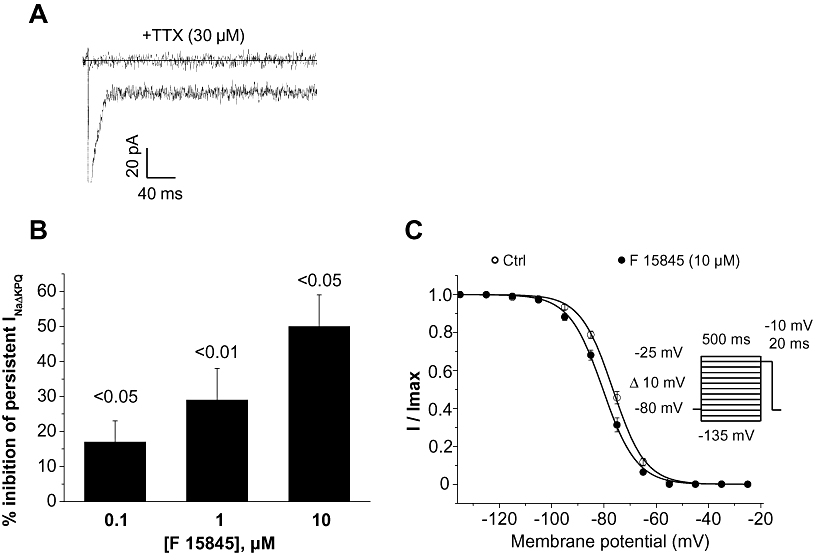
Effects of F 15845 Δ-KPQ -induced INap. (A) Experimental recordings of ΔKPQ-Nav1.5-induced INap in HEK293 cells in the absence and presence of 30 µM tetrodotoxin (TTX). INa-ΔKPQ was elicited at –10 mV from a holding potential value of –80 mV. (B) Effects of F 15845 at different concentrations on INa-ΔKPQ. (C) Steady-state inactivation curves of peak INa-ΔKPQ in the absence and presence of F 15845. Curves were obtained as described in Methods section. Data are means ± SEM of six cells.
Effects of F 15845 on rabbit Purkinje fibres and left atria action potentials
Figure 4A–C shows the effects of F 15845 on action potentials obtained in rabbit-isolated Purkinje fibres and left atria. Purkinje fibres are known to spontaneously exhibit an active INap during the plateau phase of the action potential (Wasserstrom and Salata, 1988). F 15845 from 0.1 to 10 µM shortened the action potential duration (Figure 4B,D). Thus, F 15845 0.1 to 10 µM reduced APD50 and APD90; mean maximal changes: −133.8 ± 11.4 ms, n= 6, P < 0.001 and –97.3 ± 10.8 ms, n= 6, P < 0.001, respectively. Importantly, F 15845 did not affect the resting potential, overshoot, action potential amplitude nor dV/dtmax even at the highest concentration, demonstrating its selectivity towards INap.
Figure 4.
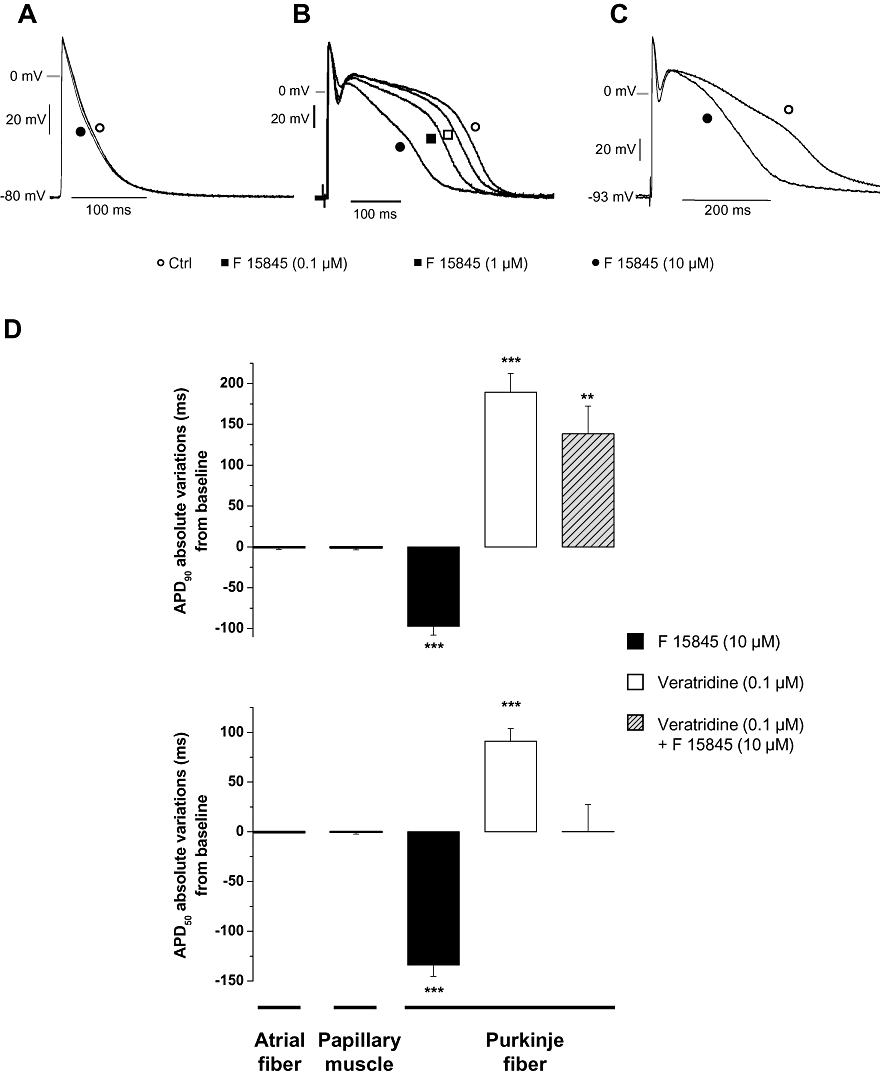
Effects of F 15845 on action potentials from rabbit Purkinje fibres and left atria. (A) Experimental action potential recordings of rabbit left atria (elicited at 1 Hz) in the absence and presence of 10 µM F 15845. (B) Experimental action potential recordings from rabbit Purkinje fibres (elicited at 1 Hz) in the absence and presence of various concentrations of F 15845 (0.1 µM; 1 µM and 10 µM). (C) Experimental recordings of veratridine-modified action potential (elicited at 1 Hz) in the absence and presence of 10 µM F 15845. Veratridine was superfused at 0.1 µM to induce the INap exhibited with the apparent increase in the duration of plateau phase. (D) Graphs representing the effects of F 15845 on APD50 (lower graph) and APD90 (upper graph) in left atria, Purkinje fibres from rabbit hearts and in papillary muscles from guinea pig hearts. Data are means ± SEM of absolute variation (ms). ** < 0.01 and ***P < 0.001 vs. baseline. Values of guinea pig papillary muscle were obtained from a previous study.
The effects of F 15845 on veratridine-intoxicated Purkinje fibres from rabbit were then evaluated (Figure 4C,D). Superfusion of veratridine 0.1 µM led to a marked lengthening of action potential that, in turn, can lead to EADs (data not shown). In the presence of 0.1 µM veratridine, APD50 was increased by 91.1 ± 12.6 ms (P < 0.001 vs. baseline, n= 6), and APD90 by 189.3 ± 23.2 ms (P < 0.001 vs. baseline, n= 6), whereas resting membrane potential, maximal amplitude and maximal dV/dtmax remained unchanged. Application of 10 µM F 15845 counteracted the veratridine-induced lengthening of APD with mean changes relative to control values APD50+0.4 ± 27.1 ms (n= 6) and +138.3 ± 34.1 ms (P < 0.01, n= 6) for APD50 and APD90, respectively (Figure 4C,D).
The effects of F 15845 on the action potential were also studied in rabbit left atria from where INap is not functional in normal conditions. In this model, F 15845 (10−5 M) did not affect any of the action potential parameters (Figure 4A,D): APD50 and APD90, maximal upstroke velocity nor conduction time. These findings emphasized once again the selectivity of this compound and are in complete agreement with those reported previously in guinea pig papillary muscles (Vacher et al., 2009).
Effects of F 15845 on ischaemia-induced arrhythmias in anaesthetized rats
The effects of F 15845 on the incidence of ischaemia-induced ventricular arrhythmias are shown in Figure 5. F 15845 (0.63 mg·kg−1 i.v.) did not affect the incidence of ventricular premature beat (VPB), salvos, VT or fibrillation. However, F 15845 (from 2.5 mg·kg−1 onwards) dose-dependently reduced the incidence of VT and fibrillation (Figure 5C,D) but not that of VPB and salvos. F 15845 reduced the VT duration in a dose-related manner (Figure 5C). Similarly, F 15845 exerted a protective effect against the number of fibrillations (Figure 5D).
Figure 5.
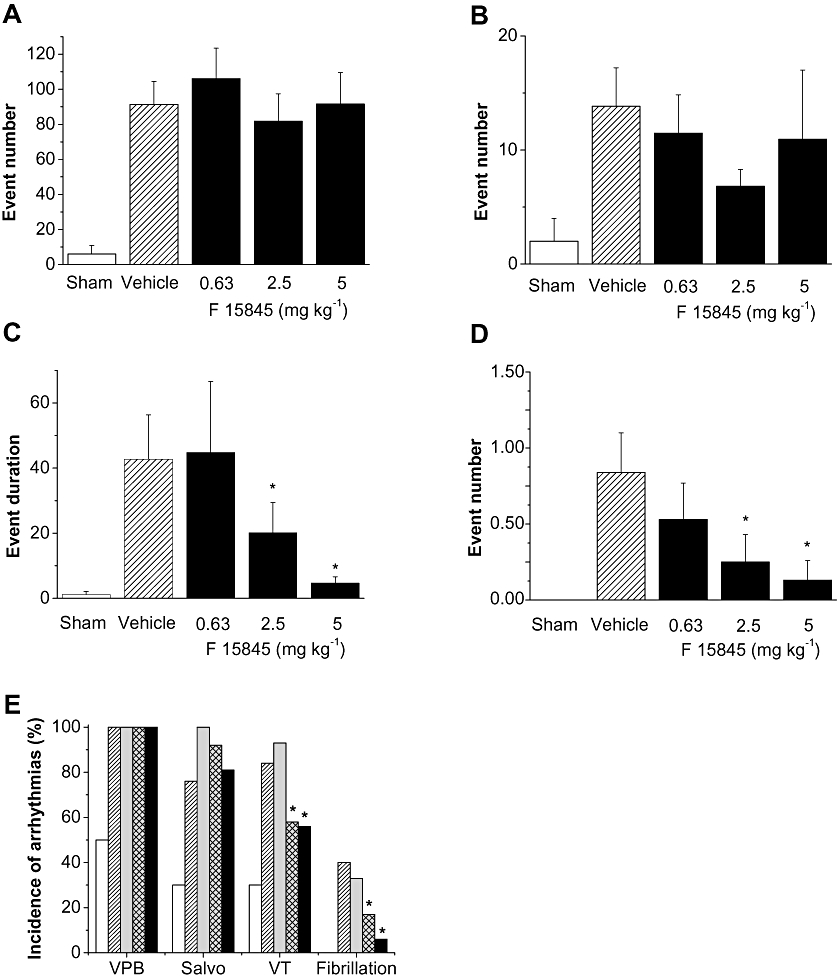
Effects of F 15845 on ischaemia-induced ventricular arrhythmias in anaesthetized rats. Histograms showing the number events of ischaemia-induced ventricular arrhythmias in sham-operated animals (n= 13) and in the presence of vehicle [40% polyethylene glycol (PEG) in sterile saline, n= 25] and F 15845 0.63 mg·kg−1 (n= 15), 2.5 mg·kg−1 (n= 12) and 5 mg·kg−1 (n= 16). (A) Ventricular premature beat, (B) salvos (C) ventricular tachycardia and (D) fibrillations. Data are means ± SEM. *P < 0.05 versus vehicle group. (E) The incidence of ischaemia-induced ventricular arrhythmias in the presence of vehicle (40% PEG in sterile saline, n= 25) and F 15845 0.63 mg·kg−1 (n= 15), 2.5 mg·kg−1 (n= 12) and 5 mg·kg−1 (n= 16). Open columns show the incidence of arrhythmias in sham operated animals (n= 13). VPB, ventricular premature beat; VT, ventricular tachycardia. P < 0.01, *P < 0.05 vs. vehicle group.
Effects of F 15845 on aconitine-induced arrhythmias in anaesthetized rats
Aconitine is a potent sodium current activator known to cause ventricular arrhythmias. In anaesthetized rats, the threshold of the i.v. dose of aconitine necessary to initiate VPB, VT and VF was measured in the absence and the presence of F 15845. As shown in Figures 6, F 15845 0.63 mg·kg−1 did not increase the threshold dose of aconitine needed to induce VPB, VT and VF. However, in the presence of 2.5 mg·kg−1 F 15845, an increased dose of aconitine was needed to trigger VPB, VT and VF when compared to the vehicle group. These results demonstrate that F 15845 exerts an anti-arrhythmic effect in vivo and that this effect is mediated through INap blockade.
Figure 6.
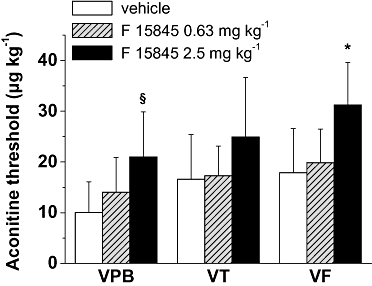
Effects F 15845 on aconitine-induced ventricular arrhythmias in rats. Effects of F 15845 (0.63 and 2.5 mg·kg−1) on the ability of aconitine to induce ventricular arrhythmias in rat. After a stabilization period of 10 min, vehicle or F 15845 were injected (1 mL·kg−1). After 3 min, perfusion of aconitine was initiated (20 µg·mL−1 at 400 µL·min−1·kg−1).VPB, ventricular premature beat; VT, ventricular tachycardia, VF, ventricular fibrillation. *P < 0.05 versus vehicle group (n= 5 animals per group).
Discussion and conclusion
The present results establish that F 15845, a highly effective blocker of INap, can reduce the current activated either by veratridine or the ΔKPQ mutation of the channel. The interaction of F 15845 with the channel is dependent upon state of the channel (and more potent when the cell is depolarized) and free from phasic block. The compound prevents the INap-induced prolongation of AP depolarization without affecting any other AP parameters. In addition, F 15845 diminished the incidence of ventricular arrhythmias in vivo in both a coronary ligation and an aconitine model of arrhythmias.
F 15845 selectively inhibited the persistent component of the Na+ current and the activity of F 15845 on that component is enhanced under depolarizing conditions (Vacher et al., 2009). This finding is important because in pathological situations such as cardiac ischaemia, cardiomyocytes are depolarized. In addition, F 15845 produced a large tonic block and almost no phasic block. Ideally, the tonic block achieved at very negative membrane potentials corresponds to that of closed/rested channels. Moreover, it is known that voltage-dependent inhibition related to inactivated state of channels can participate in tonic block. F 15845 at 1 and 10 µM induced a loss of channel availability, as suggested by the negative shift of steady-state half-inactivation voltage. These results indicate that F 15845 interacts with the rested/closed and or rested/inactivated states of channels, leading to a decrease in the number of available sodium channels functioning in the persistent mode.
The ΔKPQ mutation (responsible for LQT3 syndrome, Bennett et al., 1995; Wang et al., 1996) induces changes in Na+ channel gating, which also enhance Na+ current. This current resembles that activated by veratridine (Fredj et al., 2006). F 15845 concentration-dependently blocked the current mediated by the ΔKPQ mutation with a potency close to that which inhibited veratridine-induced INap. Because the ΔKPQ mutation can delay cellular repolarization and induce Ca2+ spontaneous diastolic activities (Fredj et al., 2006), F 15845 should exhibit anti-arrhythmic properties at least equivalent to that of ranolazine (Wu et al., 2004).
Arrhythmias result from abnormal impulse initiation or conduction (or a combination of both). Among the possible causes for impulse initiation disturbances are those triggered by afterdepolarizations. It is well known that both EADs and DADs are calcium-dependent events sensitive to changes in the action potential waveform. We suggest that activation of INap is a major player in these arrhythmogenic events and, consequently, that a selective blocker of this current will be endowed with anti-arrhythmic properties.
Sodium channel activity contributes to the upstroke, but also to the plateau of the action potential and repolarization. This sustained activity derives from two separate mechanisms: one is channel bursting in which the channel fails to inactivate (Gintant et al., 1984; Carmeliet, 1987; Wasserstrom and Salata, 1988); the other results from the overlap of the steady-state activation and inactivation curves and is known as the ‘window current’ (Coulombe et al., 1985). Each of these leads to a prolongation of the ventricular action potential and promotes EADs in ventricular myocytes (Coulombe et al., 1985; Boutjdir et al., 1994; Song et al., 2004; Maltsev and Undrovinas, 2008). Athough INap is of weak intensity, its long duration results in a build-up of Na+ (Noble and Noble, 2006; Maltsev and Undrovinas, 2008; Zaza et al., 2008), which drives the intracellular Ca2+ overload (Haigney et al., 1994; Noble and Noble, 2006). In addition, the prolongation of the action potential duration can also reactivate inactivated Ca2+ channels. In line with this, blockade of INap has been found to attenuate the calcium loading-induced EADs (Haigney et al., 1994; Song et al., 2006; Zaza et al., 2008). F 15845 concentration-dependently shortened APD in rabbit Purkinje fibres under normal conditions (Wasserstrom and Salata, 1988) but did so more extensively following full activation of INap. This result demonstrates that the anti-arrhythmic properties of F 15845 are mediated by inhibition of INap-induced prolongation of the plateau phase, and by doing so, it prevents the genesis of EADs. Finally, F 15845 was devoid of significant effects on the action potential shape of atrial and ventricular myocytes in which INap has no functional role in normal situations.
Excessive Ca2+ loading also triggers oscillatory Ca2+ release from the sarcoplasmic reticulum during diastole which, in turn, activates a transient inward current. This current is responsible for DADs (Lederer and Tsien, 1976). Recently, several lines of evidence indicate that an increase in INap is associated with an increase in transient inward current, DADs, and sustained triggered activity via a mechanism that probably involves Ca2+ overload (Undrovinas et al., 2006). This interpretation is consistent with earlier findings showing that low concentrations of TTX and ranolazine reduced DADs induced by Ca2+ overload (Undrovinas et al., 2006; Song et al., 2008). Therefore, F 15845 should present a similar anti-arrhythmic profile against DADs.
In vivo, F 15845 is effective in preventing fatal ventricular fibrillation and ventricular tachycardia (VT) during coronary ligation without modifying heart rate and blood pressure. The anti-arrhythmic effects of F 15845 during ischaemia are therefore directly related to inhibition of INap, given its high selectivity for this target (Vacher et al., 2009). We are convinced that the underlying mechanism of ischaemia-induced damages are mediated by enhanced INap activity (Tani and Neely, 1990; Zaza et al., 2008). The anti-arrhythmic component of the activity of INap blockers during ischaemia, as reported here and elsewhere (Wu et al., 2004; Sossalla et al., 2008), illustrates the role of INap in the early process of ischaemia-induced Na+ accumulation. Indeed, it also reinforces the role played by INaP in the intracellular increase in Ca2+ at the origin of arrhythmias either through activation of the transient inward current Iti, or electrical uncoupling of cardiac myocytes (see below). The favourable properties of INap inhibitors in cardiac ischaemia thus derive from attenuation of Na+ and Ca2+ overload (Noble and Noble, 2006) of which VTs and fibrillation are direct manifestations.
To further support its selectivity, the effects of F 15845 were evaluated in a model of arrhythmias induced by aconitine, the mechanism of which involves cardiac sodium channels (Honerjäger and Meissner, 1983). Aconitine binds with high affinity to the open state of the voltage-sensitive sodium channels causing a persistent activation of the channels, which become refractory to excitation. The electrophysiological mechanism of aconitine-induced arrhythmias is due to DAD and EAD. The present results show that F 15845 dose-dependently increases the dose threshold of aconitine required to induce ventricular arrhythmias. This result again demonstrates that the anti-arrhythmic property of F 15845 in vivo is linked to INap blockade.
In conclusion, the results of our in vitro and in vivo studies show that F 15845 can reduce arrhythmias induced by Na+ and Ca2+ overload. The anti-arrhythmic activity of F 15845 is mediated by its interaction with ischaemic (inactivated or closed) state of the sodium channel. Finally, in addition to its anti-anginal activity and its cardioprotective activity (Vie et al., 2009), F 15845 constitutes a new generation of INap blockers that prevents the ischaemia-induced arrhythmias and ventricular dysfunction. Such properties of F 15845 may be of therapeutic benefit and warrant further investigations in pathological situations that are associated with enhanced INap.
Acknowledgments
We are indebted to our colleagues in the Division of Cardiovascular Diseases II, Medicinal Chemistry I and General Pharmacology Department for performing the experimentations described in the present manuscript.
Glossary
Abbreviations
- AP
action potential
- APD50, APD90
action potential duration at 50% and 90% repolarization
- DAD
delayed afterdepolarization
- EAD
early afterdepolarization
- INap
persistent sodium current
Conflicts of interest
All the authors with exception of H. Abriel and J-S Rougier are employees of Pierre Fabre Laboratory.
References
- Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl. IV):iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Boutjdir M, Restivo M, Wei Y, Stergiopoulos K, El-Sherif N. Early afterdepolarization formation in cardiac myocytes: analysis of phase plane patterns, action potential, and membrane currents. J Cardiovasc Electrophysiol. 1994;7:609–620. doi: 10.1111/j.1540-8167.1994.tb01302.x. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Slow inactivation of the sodium current in rabbit cardiac Purkinje fibres. Pflugers Arch. 1987;408:18–26. doi: 10.1007/BF00581835. [DOI] [PubMed] [Google Scholar]
- Coulombe A, Coraboeuf E, Malecot C, Deroubaix E. Role of the ‘Na window’ current and other ionic currents in triggering early after-depolarizations and resulting re-excitation in Purkinje fibers. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology and Arrhythmias. New York: Grune and Stratton; 1985. pp. 43–49. [Google Scholar]
- Fredj S, Lindegger N, Sampson KJ, Carmeliet P, Kass RS. Altered Na+ channels promote pause-induced spontaneous diastolic activity in long QT syndrome Type 3 myocytes. Circ Res. 2006;99:1225–1232. doi: 10.1161/01.RES.0000251305.25604.b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gintant GA, Datyner NB, Cohen IS. Slow inactivation of a tetrodotoxin-sensitive current in canine cardiac Purkinje fibers. Biophys J. 1984;45:509–512. doi: 10.1016/S0006-3495(84)84187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigney MC, Lakatta EG, Stern MD, Silverman HS. Sodium channel blockade reduces hypoxic sodium loading and sodium-dependent calcium loading. Circulation. 1994;90:391–399. doi: 10.1161/01.cir.90.1.391. [DOI] [PubMed] [Google Scholar]
- Hale SL, Shryock JC, Belardinelli L, Sweeney M, Kloner RA. Late sodium current inhibition as a new cardioprotective approach. J Mol Cell Cardiol. 2008;44:954–967. doi: 10.1016/j.yjmcc.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Hammarstrom AK, Gage PW. Hypoxia and persistent sodium current. Eur Biophys J. 2002;31:323–330. doi: 10.1007/s00249-002-0218-2. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G, Maier LS. Mechanism of action of the new anti-ischaemia drug ranolazine. Clin Res Cardiol. 2008;97:222–226. doi: 10.1007/s00392-007-0612-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honerjäger P, Meissner A. The positive inotropic effect of aconitine. Naunyn Schmiedebergs Arch Pharmacol. 1983;322:49–58. doi: 10.1007/BF00649352. [DOI] [PubMed] [Google Scholar]
- Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–341. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand B, Vié B, Talmant JM, Coraboeuf E, John GW. Alleviation of contractile dysfunction in ischaemic hearts by slowly inactivating Na+ current blockers. Am J Physiol. 1995;269:H533–H540. doi: 10.1152/ajpheart.1995.269.2.H533. [DOI] [PubMed] [Google Scholar]
- Le Grand B, Dordain-Maffre M, John GW. Lubeluzole-induced prolongation of cardiac action potential in rabbit Purkinje Fibres. Fundam Clin Pharmacol. 2000;14:159–162. doi: 10.1111/j.1472-8206.2000.tb00405.x. [DOI] [PubMed] [Google Scholar]
- Le Grand B, Pignier C, Létienne R, Cuisiat F, Rolland F, Mas A, et al. Sodium late current blockers in ischaemia reperfusion: is the bullet magic? J Med Chem. 2008;51:3856–3866. doi: 10.1021/jm800100z. [DOI] [PubMed] [Google Scholar]
- Lederer WJ, Tsien RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibers. J Physiol. 1976;263:73–100. doi: 10.1113/jphysiol.1976.sp011622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Undrovinas A. Late sodium current in failing heart: friend or foe? Prog Biophys Mol Biol. 2008;96:421–451. doi: 10.1016/j.pbiomolbio.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92:iv1–iv5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignier C, Revenaz C, Rauly-Lestienne I, Cussac D, Delhon A, Gardette J, et al. Direct protective effects of poly-unsaturated fatty acids, DHA and EPA, against activation of cardiac late sodium current: a mechanism for ischaemia selectivity. Basic Res Cardiol. 2007;102:553–564. doi: 10.1007/s00395-007-0676-x. [DOI] [PubMed] [Google Scholar]
- Pinet C, Algalarrondo V, Sablayrolles S, Le Grand B, Pignier C, Cussac D, et al. Protease-activated receptor-1 mediates thrombin-induced persistent sodium current in human cardiomyocytes. Mol Pharmacol. 2008;73:1622–1631. doi: 10.1124/mol.107.043182. [DOI] [PubMed] [Google Scholar]
- Shryock JC, Belardinelli L. Inhibition of late sodium current to reduce electrical and mechanical dysfunction of ischaemic myocardium. Br J Pharmacol. 2008;153:1128–1132. doi: 10.1038/sj.bjp.0707522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effect of increase late Ina in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol. 2008;294:H2031–H2039. doi: 10.1152/ajpheart.01357.2007. [DOI] [PubMed] [Google Scholar]
- Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schöndube FA, et al. Ranolazine improves dysfunction in isolated myocardium from failing human hearts: role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43. doi: 10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Sunami A, Sasano T, Matsunaga A, Fan Z, Sawanobori T, Hiraoka M. Properties of veratridine-modified single Na+ channels in guinea-pig ventricular myocytes. Am J Physiol. 1993;264:H454–H463. doi: 10.1152/ajpheart.1993.264.2.H454. [DOI] [PubMed] [Google Scholar]
- Tamareille S, Le Grand B, John GW, Feuvray D, Coulombe A. Anti-ischaemic compound KC 12291 prevents diastolic contracture in isolated atria by blockade of voltage-gated sodium channels. J Cardiovasc Pharmacol. 2002;40:346–355. doi: 10.1097/00005344-200209000-00003. [DOI] [PubMed] [Google Scholar]
- Tani M, Neely JR. Na+ accumulation increases Ca2+ overload and impairs function in anoxic rat heart. J Mol Cell Cardiol. 1990;22:57–72. doi: 10.1016/0022-2828(90)90972-5. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischaemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231–1241. doi: 10.1161/01.res.71.5.1231. [DOI] [PubMed] [Google Scholar]
- Undrovinas AL, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher B, Pignier C, Létienne R, Verscheure Y, Le Grand B. F 15845 inhibits cardiac persistent sodium current and prevents angina. Br J Pharmacol. 2009;156:214–225. doi: 10.1111/j.1476-5381.2008.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bemmelen MX, Rougier JS, Gavillet B, Apothéloz F, Daidié D, Tateyama M, et al. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res. 2004;95:284–291. doi: 10.1161/01.RES.0000136816.05109.89. [DOI] [PubMed] [Google Scholar]
- Vie B, Sablayrolles S, Létienne R, Vacher B, Darmellah A, Bernard M, et al. 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5-benzoxathiepine bromhydrate (F 15845) prevents ischemia-induced heart remodelling by reduction of the intracellular Na+ overload. J Pharmacol Exp Ther. 2009;330:696–703. doi: 10.1124/jpet.109.153122. [DOI] [PubMed] [Google Scholar]
- Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction and reperfusion. Cardiovasc Res. 1988;22:447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- Wang DW, Yazawa K, George AL, Jr, Bennett PB. Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc Natl Acad Sci U S A. 1996;93:13200–13205. doi: 10.1073/pnas.93.23.13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstrom JA, Salata JJ. Basis for tetrodotoxin and lidocaine effects on action potentials in dog ventricular myocytes. Am J Physiol. 1988;254:H1157–H1166. doi: 10.1152/ajpheart.1988.254.6.H1157. [DOI] [PubMed] [Google Scholar]
- Wu J, Corr PB. Palmitoyl carnitine modifies sodium currents and induces transient inward current in ventricular myocytes. Am J Physiol. 1994;266:H1034–H1046. doi: 10.1152/ajpheart.1994.266.3.H1034. [DOI] [PubMed] [Google Scholar]
- Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea-pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605. doi: 10.1124/jpet.104.066100. [DOI] [PubMed] [Google Scholar]
- Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac ‘late sodium current’. Pharmacol Ther. 2008;119:326–339. doi: 10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]