

Abstract
BACKGROUND AND PURPOSE
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) is an attractive therapeutic target of type 2 diabetes and metabolic syndrome. Emodin, a natural product and active ingredient of various Chinese herbs, has been demonstrated to possess multiple biological activities. Here, we investigated the effects of emodin on 11β-HSD1 and its ability to ameliorate metabolic disorders in diet-induced obese (DIO) mice.
EXPERIMENTAL APPROACH
Scintillation proximity assay was performed to evaluate inhibition of emodin against recombinant human and mouse 11β-HSDs. The ability of emodin to inhibit prednisone- or dexamethasone-induced insulin resistance was investigated in C57BL/6J mice and its effect on metabolic abnormalities was observed in DIO mice.
KEY RESULTS
Emodin is a potent and selective 11β-HSD1 inhibitor with the IC50 of 186 and 86 nM for human and mouse 11β-HSD1, respectively. Single oral administration of emodin inhibited 11β-HSD1 activity of liver and fat significantly in mice. Emodin reversed prednisone-induced insulin resistance in mice, whereas it did not affect dexamethasone-induced insulin resistance, which confirmed its inhibitory effect on 11β-HSD1 in vivo. In DIO mice, oral administration of emodin improved insulin sensitivity and lipid metabolism, and lowered blood glucose and hepatic PEPCK, and glucose-6-phosphatase mRNA.
CONCLUSIONS AND IMPLICATIONS
This study demonstrated a new role for emodin as a potent and selective inhibitor of 11β-HSD1 and its beneficial effects on metabolic disorders in DIO mice. This highlights the potential value of analogues of emodin as a new class of compounds for the treatment of metabolic syndrome or type 2 diabetes.
Keywords: emodin, 11β-hydroxysteroid dehydrogenase type 1, diet-induced obese mice, insulin resistance, metabolic syndrome, type 2 diabetes
Introduction
Insulin resistance plays a key role in the pathogenesis of type 2 diabetes, and is often associated with a group of metabolic disorders, such as hyperglycaemia, hypertension, dyslipidaemia, obesity and atherosclerotic vascular disease (DeFronzo and Ferrannnini, 1991). Glucocorticoids antagonize the action of insulin and induce insulin resistance when in excess (Livingstone et al., 2000; Masuzaki et al., 2001; Beauregard et al., 2002). The high circulating glucocorticoid level in Cushing's syndrome leads to visceral obesity and several clinical features associated with insulin resistance (Beauregard et al., 2002). Pharmacological blockade of glucocorticoid action by glucocorticoid receptor (GR) antagonist can ameliorate diabetes and insulin resistance, which highlights the importance of glucocorticoids in the development of type 2 diabetes and metabolic syndrome (Wang et al., 2006).
The action of glucocorticoids on target tissue is not only dependent on the circulating levels, but is regulated in a tissue-specific manner by the enzymes of 11β-hydroxysteroid dehydrogenase (11β-HSD) 1 and 2 (Draper and Stewart, 2005). 11β-HSD1 is a low-affinity, NADP(H)-dependent dehydrogenase/oxoreductase that functions predominantly as an oxoreductase in intact cells, organs and in vivo. It is highly expressed in liver, gonad, adipose tissue and brain, and amplifies local glucocorticiod action by converting cortisone into cortisol in humans, and 11-dehydrocorticosterone into corticosterone in rodents (Seckl and Walker, 2001). 11β-HSD2 is predominantly expressed in aldosterone target cells such as kidney and colon, and catalyses the opposite reaction, thereby preventing excessive activation of the mineralocorticoid receptor and sequelae including sodium retention, hypokalaemia and hypertension.
Accumulating evidence suggests that 11β-HSD1 plays an important role in the development of obesity, insulin resistance and type 2 diabetes. 11β-HSD1 mRNA expression or activity is specifically increased in liver or fat of Zucker rat, ob/ob mice and db/db mice, and may contribute to the phenotype of type 2 diabetes (Masuzaki et al., 2001). Mice with transgenic overexpression of 11β-HSD1 selectively in adipose tissue had increased intra-adipose glucocorticoid concentrations with no change in plasma levels, and exhibited visceral obesity, insulin resistance, hyperglycaemia and hyperlipidaemia (Masuzaki et al., 2001). In contrast, 11β-HSD1-deficient mice exhibited enhanced glucose tolerance, attenuated gluconeogenic responses, increased insulin sensitivity, improved lipid and lipoprotein profile and resistance to hyperglycaemia and weight gain induced by a high-fat diet (Kotelevtsev et al., 1997; Morton et al., 2001). In humans, increased 11β-HSD1 activity or mRNA has been observed in subcutaneous fat, which was positively correlated with obesity, insulin resistance and other features of metabolic disorders (Sandeep et al., 2005). The non-selective 11β-HSD1 inhibitor carbenoxolone increases hepatic insulin sensitivity in healthy men and non-obese type 2 diabetic patients, as measured by an increase in glucose infusion rate during euglycaemic hyperinsulinaemic clamp (Andrews et al., 2003). Carbenoxolone and several selective inhibitors of 11β-HSD1 have also been shown to ameliorate hyperglycaemia, and improve glucose tolerance and insulin sensitivity in rodent models of diabetes or metabolic syndrome (Alberts et al., 2003; Hermanowski-Vosatka et al., 2005). Therefore, all these findings suggest that the pharmacological inhibition of 11β-HSD1 could provide a very attractive therapy of type 2 diabetes or metabolic diseases.
Due to the unacceptable side effects caused by the inhibition of 11β-HSD2, including sodium retention and hypertension, the ideal inhibitors must be selective for 11β-HSD1 rather than 11β-HSD2. Several natural compounds and their derivatives including glycyrrhetinic acid and carbenoxolone have been investigated as 11β-HSD1 inhibitors, but in general they are non-selective. To discover new selective 11β-HSD1 inhibitors from natural products, we performed a screening of our phytocompound collection based on a scintillation proximity assay (SPA) against recombinant human and mouse 11β-HSDs. A series anthraquinone compounds had been shown to inhibit 11β-HSD1 of both humans and mice. Among those compounds, emodin (1,3,8-trihydroxy-6-methylanthraquinone), an anthraquinone derivative isolated mainly from the root and rhizome of Rheum palmatum, has been identified as the most potent selective 11β-HSD1 inhibitor. Therefore, in the present study, emodin was firstly identified as a selective 11β-HSD1 inhibitor based on both its in vitro and in vivo activities, and, secondly, its effect on metabolic abnormalities was investigated in diet-induced obese (DIO) mice with insulin resistance and dyslipidaemia.
Methods
Animals
C57BL/6J male mice were purchased from Shanghai SLAC Laboratory Animal Co. Ltd (Shanghai, China) and maintained on a 7:00 h–19:00 h light schedule with an ad libitum diet of standard lab chow, unless otherwise specified. For DIO mice study, the C57BL/6J male mice were fed with a high-fat diet (60 kcal% fat, 20 kcal% protein and 20 kcal% carbohydrate, Cat. D12492i, Research Diet, New Brunswick, NJ, USA). Animal experiments were approved by the Animal Care and Use Committee, Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Construction of stably transfected cells
The full-length cDNAs of human or murine 11β-HSD1 and 11β-HSD2 were isolated from the cDNA libraries provided by NIH Mammalian Gene Collection and cloned into pcDNA3 expression vector by PCR. HEK-293 cells were transfected with each cDNA expression construct via lipofactamine technology. Transfected cells were selected by cultivation in the presence of 700 µg·mL−1 of G418. Non-resistant cells were removed by replacing the cell culture medium every other day for 12–14 days. The single surviving colony was picked up and expanded. The protein expression of human or mouse 11β-HSD1 and 11β-HSD2 was confirmed, respectively, by Western blot. The enzymes of 11β-HSDs were purified, respectively, according to the method previously described (Mundt et al., 2005).
Measurement of 11β-HSD1 and -HSD2 activity in vitro
The SPA was used to screen for inhibitors of 11β-HSDs (Mundt et al., 2005), with the microsome fractions prepared from the HEK-293 cells stably transfected with either human or mouse 11β-HSD1 or 11β-HSD2 as the enzyme source. Briefly, different concentrations of compound were added to 96-well microtitre plates, followed by the addition of 80 µL of 50 mM HEPES buffer, pH 7.4 containing 25 nM [1,2-(n)3H]-cortisone and 1.25 mM NADPH (for 11β-HSD1 assay) or 12.5 nM [1,2,6,7-(n)3H]-cortisol and 0.625 mM NAD+(for 11β-HSD2 assay). Reactions were initiated by the addition of 11β-HSD1 or 11β-HSD2, enzyme preparation as microsome fractions from HEK293 cells in a final concentration of 80 µg·mL−1 for 11β-HSD1, and 160 µg·mL−1 for 11β-HSD2, respectively. After a 60 min incubation at 37°C, the reaction was stopped by the addition of 35 µL of 10 mg·mL−1 protein A-coated yttrium silicate beads suspended in SuperBlock Blocking Buffer with 3 µg·mL−1 of murine monoclonal cortisol antibody and 314 µM glycyrrhetinic acid. The plates were incubated under plastic film on an orbital shaker for 120 min at room temperature before counting. The amount of [3H]-cortisol generated in 11β-HSD1 enzyme reaction or remaining from the 11β-HSD2 enzyme reaction was captured by the beads and determined in a microplate liquid scintillation counter. The % inhibition was calculated relative to a non-inhibited control. Data were obtained from at least three independent experiments. IC50 values were calculated from concentration–response curves by a non-linear regression analysis using Prism Version 4.
Molecular modelling
The program DOCK4.0 (Kuntz, 1992; Ewing and Kuntz, 1997) was employed for the docking study. The starting structure was PDB entry 2IRW (Patel et al., 2007), and residues around the ligand in this structure at a radius of 5 Å were isolated for constructing the grids of docking. During the docking calculations, Kollman-all-atom charges (Weiner et al., 1986) were assigned to the protein, and Gasterger–Hückel charges (Gasteiger and Marsili, 1980) were assigned to the small molecules. Conformational flexibility of the small molecules was implemented in the docking search. The ligand–receptor binding energy was approximately set to be the sum of the van der Waals and electrostatic interaction energies. After an initial evaluation of the orientation and scoring, a grid-based minimization was carried out for the ligand to locate the nearest local energy minimum within the receptor binding site. Position and conformation of each docked molecule were optimized by using the single anchor search and torsion minimization method.
Acute administration in normal mice
To evaluate the activity of acute administration of emodin, C57 BL/6J mice deprived of food overnight were administered emodin (100 or 200 mg·kg−1) or vehicle (0.5% carboxymethylcellulose; CMC) p.o. Two hours later, animals were killed by cervical dislocation, and the liver and mesenteric fat were isolated immediately, washed in ice-cold PBS, frozen in liquid nitrogen and stored at −80°C. The liver and mesenteric fat were homogenized (0.1 g·mL−1) in cold homogenization buffer (20 mM Na2HPO4, 5% glycerol, 1 mM EDTA, pH 7.0), and 10 µg of liver homogenates or 30 µg mesenteric fat homogenates was used to analyse the 11β-HSD1 activity by SPA, as previously described.
Effect of emodin on prednisone- or dexamethasone-induced insulin-resistant mice
Male C57BL/6J mice (8 weeks old) were randomly assigned to six groups based on body weight. The experimental groups and respective treatment were as follows: (i) control – 0.5% CMC; (ii) prednisone acetate – 100 mg·kg−1; (iii) prednisone acetate (100 mg·kg−1) plus emodin (100 mg·kg−1); (iv) prednisone acetate (100 mg·kg−1) plus emodin (200 mg·kg−1); (v) dexamethasone (4 mg·kg−1); and (vi) dexamethasone (4 mg·kg−1) plus emodin (200 mg·kg−1). Prednisone or dexamethasone was administered by oral gavage twice daily to induce a state of glucocorticoid excess and insulin resistance in mice. Emodin was administered orally twice daily 1 day before, and then at the same time as prednisone or dexamethasone. After 14 days of treatment, insulin tolerance was determined in mice deprived of food overnight (0.5 U·kg−1 insulin administered by an i.p. injection) to investigate the effect of emodin on prednisone- or dexamethasone-induced insulin resistance.
Effect of emodin in DIO mice
C57BL/6J male mice (3–4 weeks) were fed a formulated research diet containing 60% of the calories from fat for 12 weeks before, and throughout the duration of the experiment. DIO mice were assigned to three groups and subjected to gavage treatment twice per day with vehicle (0.5% CMC), emodin 50 or 100 mg·kg−1, respectively, for 35 days. Fasting blood glucose values and initial body weights were comparable between groups. The blood glucose levels were measured via blood drops obtained by clipping the tail of the mice using a ONE TOUCH BASIC plus Glucose Monitor (Lifescan, Milpitas, CA, USA), unless otherwise specified. The food intake and body weight of the animals were recorded every 3 days. Glucose tolerance test was determined in mice deprived of food for 5 h (2 g·kg−1 glucose administered by gavage) at day 24 of the treatment. The blood samples were collected via the retro-orbital sinus, and the serum glucose and insulin concentrations were measured with an enzymatic colorimetric method and insulin elisa kit, respectively. An insulin tolerance test was performed in the 5 h-fasted mice (0.5 U·kg−1 insulin administered by an i.p. injection) at day 28 of the treatment. On the last day of treatment, 5 h-fasted mice were anaesthetized with an i.p. injection of sodium pentobarbital (40 mg·kg−1). Serum was collected for determination of insulin, triacylglycerol, cholesterols and non-esterified free fatty acid (NEFA) concentration. The liver and different fat pads including epididymal fat, mesenteric fat, perirenal fat and subcutaneous fat were dissected, weighed, immediately frozen in liquid nitrogen and stored at −80°C.
Quantification of mRNA
Real-time PCR was used to quantify mRNA levels of 11β-HSD1 in liver and fat tissues. PEPCK and G6Pase mRNA levels in liver were also determined. Total RNA was extracted from frozen livers and fat tissues using Trizol reagent. The mRNA was quantified using a BIO-RAD (Bio-rad Laboratories Inc., Hercules, CA, USA) SYBR Green Supermix and an MJ research DNA engine Opticon2. Primers were as follows: for 11β-HSD1, 11β-HSD1F (GGAGGAGATGACGGCAAT), and 11β-HSD1R (CC TTGAACTCGGAGCAGC); for PEPCK, PEPCKF (TGATGACTGTCTTGCTTTCG) and PEPCKR (GCATA ACGGTCTGGACTTCT); for G6Pase, G6PaseF (CTCGCTATCTCCAAGTGAAT) and G6PaseR (TGCTGTAGTAGTCGGTGTCC); for β-actin, β-actinF (TGCTGTCCCTGTATGCCTCTG) and β-actinR (TTGATGTCACGCACGAT TTCC). The Opticon Monitor (software version 1.11) was used for analysis. Results were normalized to endogenous control β-actin mRNA expressions.
Data analysis
The data are presented as mean with the SEM as indicated. All data were normally distributed. To determine treatment effects, statistical analysis was performed by use of a two-tailed unpaired t-test. P values of less than 0.05 were considered statistically significant.
Materials
Emodin and other compounds were purchased from Nanjing Zelang Medical Technology Co. Ltd. (Qixia District, Nanjing, Jiang Su, China). The pcDNA expression vector and Trizol Reagent (Cat. no. 15596-018) were purchased from Invitrogen (Carlsbad, CA, USA). [1,2-(n)3H]- cortisone (Cat. TRK1075) was from Amersham (Buckinghamshire, UK). [1,2,6,7-(n)3H]- cortisol (Cat. NET396) was from PerkinElmer (Boston, MA, USA). SPA beads (Cat. RPN143) were from GE (Piscataway, NJ, USA). SuperBlock Blocking Buffer (Cat. 37515) was from Pierce (Rockford, IL, USA). The murine monoclonal cortisol antibody (Cat. P01-92-94M-P) was from East Coast Biologics (North Berwick, ME, USA). Glycyrrhetinic acid was from Sigma (St. Louis, MO, USA). The M-MLV reverse transcriptional enzyme (Cat. M170A) was from Promega (Madison, WI, USA). All the primers were synthesized by Sangon Corporation (Song Jiang, Shanghai, China). SYBR Green Supermix (cat.170-8880) was from Bio-Rad. The high-fat forage (Cat. D12492i) was from Research Diet (Research Diets Inc., New Brunswick, NJ, USA). Blood glucose values were measured using a One-Touch Basic Glucose Monitor (Lifescan, Milpitas, CA, USA). Serum insulin was analysed with a mice insulin elisa kit (cat. 90080, Crystal Chem, Downers Grove, IL, USA). Serum NEFA was determined with an enzymatic colorimetirc method using oleic acid as a standard (NEFA C, cat. 994-75409, Wako Chemicals, Neuss, Germany). Serum triacylglycerols and cholesterols were analysed with an enzymatic colorimetric method (Hitachi 7060 Biochemical Autoanalyser, Chlyoda-Ku, Tokyo, Japan).
Results
Emodin selectively inhibited 11β-HSD1 activity in vitro
The potency and selectivity of a series anthraquinone compounds on the inhibition of mouse or human 11β-HSD1 or 2 were determined by SPA. IC50 values are presented in Table 1. Emodin, aloe-emodin and rheochrysidin showed a strong inhibitory effect on recombinant mouse 11β-HSD1 with IC50 of 86, 98 and 81 nM, respectively. Emodin also inhibited human 11β-HSD1 with IC50 of 186 nM, whereas aloe-emodin and rheochrysidin were less potent with the IC50 of 879 and 542 nM, respectively. The other two anthraquinone compounds, rhein and 3-methylchrysazin, exhibited much weaker inhibitory effects on both mouse and human 11β-HSD1. All of the five anthraquinone compounds showed good selectivity for mouse 11β-HSD2 with an IC50 > 1 mM, and emodin did not have a significant inhibitory effect on human 11β-HSD2. Therefore, a series anthraquinone compounds were identified as selective 11β-HSD1 inhibitors, emodin being the most potent.
Table 1.
In vitro inhibitory effects of anthraquinone compounds on 11β-HSD1 and 2
| Mouse | Human | Mouse | Human | |
|---|---|---|---|---|
| Compound | 11β-HSD1 | 11β-HSD1 | 11β-HSD2 | 11β-HSD2 |
| Emodin | 86 | 186 | >1 mM | >1 mM |
| Aloe-emodin | 98 | 879 | >1 mM | ND |
| Rhein | 2840 | >10 000 | >1 mM | ND |
| Rheochrysidin | 81 | 542 | >1 mM | ND |
| 3-Methylchrysazin | 400 | 3540 | >1 mM | ND |
The values shown are IC50 (nmol·L−1). ND, not determined.
Molecular modelling of emodin and 11β-HSD1
To explain the interaction mode of emodin to human 11β-HSD1, molecular docking simulation was performed employing the program DOCK4.0 based on the X-ray crystal structure of the 11β-HSD1 complex (PDB entry 2IRW). This complex structure is composed of human 11β-HSD1, a synthetic inhibitor with high activity, and a co-substrate nicotinamide adenine dinucleotide phosphate (NADP). The emodin was docked into the binding site flexibly; meanwhile, the structure of 11β-HSD1 and NADP was fixed. The conformation with the lowest interaction energy was taken out for further analysis.
In the initial crystal structure, hydrogen bonds provide strong interactions between the ligand and the protein, as well as its co-substrate NADP. The carbonyl group of the ligand forms two hydrogen bonds with Tyr183 and Ser170. Interestingly, the docking results showed that emodin also formed strong hydrogen bonds with the receptor, as shown in Figure 1. The hydroxyl on C4 formed hydrogen bonds with Ser170, and the carbonyl group on C8 formed two hydrogen bonds with Ser170 and Tyr183 (Figure 1B). However, emodin did not form a hydrogen bond with NADP as did the ligand in the crystal structure. Instead, emodin formed hydrophobic contacts with the NADP (Figure 1B). Furthermore, residues Leu126, Val227 and Tyr177 were involved in the hydrophobic contacts with emodin (Figure 1B).
Figure 1.
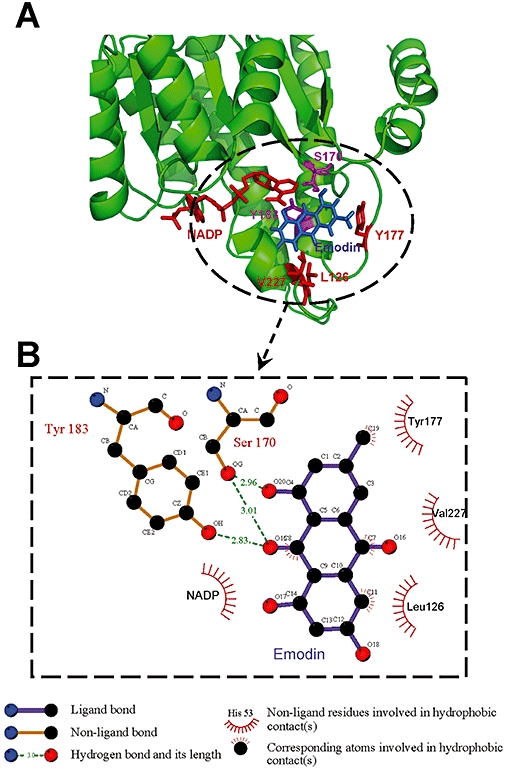
Molecular modelling of emodin and 11β-HSD1. Molecular docking simulation was performed employing the program DOCK4.0, based on the X-ray crystal structure of 11β-HSD1 complex (PDB entry 2IRW). The complex structure (A), hydrophobic contacts among 11β-HSD1, emodin and NADP (B).
Emodin inhibited 11β-HSD1 activity in vivo
The in vivo efficacy of emodin at inhibiting 11β-HSD1 activity was evaluated in C57BL/6J mice. Two hours after p.o. administration of 100 or 200 mg·kg−1 emodin, the mice were killed, and the liver and mesenteric fat were removed and assayed for 11β-HSD1 activity. As shown in Figure 2, oral administration of 100 or 200 mg·kg−1 of emodin significantly inhibited liver 11β-HSD1 enzymatic activity by 17.6 and 31.3% (Figure 2A) and mesenteric fat 11β-HSD1 enzymatic activity by 21.5 and 46.7% (Figure 2B), respectively. The results demonstrate that emodin inhibits 11β-HSD1 activity in vivo.
Figure 2.
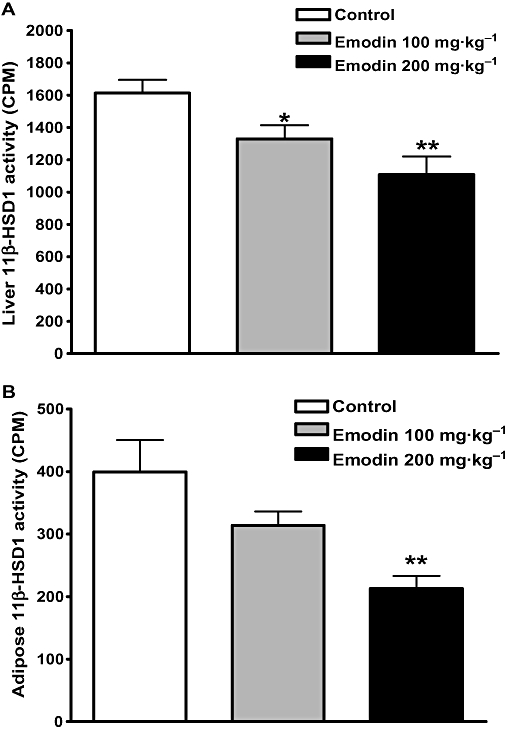
Oral administration of emodin inhibited hepatic 11β-HSD1 activity in C57BL/6J mice. Emodin was orally administered to male C57BL/6J mice, and analysis of 11β-HSD1 activity in 10 µg liver homogenates (Figure 2A) and 30 µg mesenteric fat homogenates (Figure 2B) was conducted with SPA at 2 h post-dose. Data are expressed as mean ± SEM for n= 10 mice. *P < 0.05; **P < 0.01 versus control mice.
Emodin antagonized insulin resistance induced by glucocorticoids
It is well documented that prolonged exposure to elevated glucocorticoid levels produces insulin resistance, a hallmark of diabetes mellitus. Dexamethasone is a synthetic active glucocorticoid, which has a strong affinity for the GR, whereas prednisone is a synthetic cortisone analogue, which has little affinity for the GR. However, prednisone can be catalysed by the liver 11β-HSD1 to convert it into its active metabolite, prednisolone, which has relatively high glucocorticoid activity. The insulin tolerance test showed that treatment of C57BL/6J mice with dexamethasone or prednisone for 14 days reduced the glucose-lowering effect in response to the insulin challenge, indicating the presence of insulin resistant (Figure 3). When concurrently treated with 100 or 200 mg·kg−1 emodin, the glucose-lowering effects after insulin injection were increased in prednisone-treated mice, which suggests improved insulin sensitivity. In contrast, the insulin resistance induced by dexamethasone was not improved by the concurrent treatment with 200 mg·kg−1 emodin (Figure 3). These results indicate that emodin can reverse prednisone-, but not dexamethasone-induced insulin resistance in mice, which confirms its inhibitory effect on 11β-HSD1 in vivo.
Figure 3.
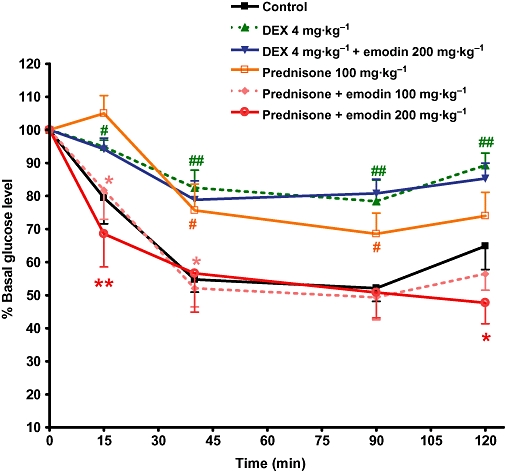
Emodin antagonized insulin resistance induced by glucocorticoids. Mice were treated as described in Methods. The insulin tolerance test was conducted in mice deprived of food overnight (0.5 U·kg−1 insulin administered by an i.p. injection) at day 14 of treatment. Values are expressed as mean ± SEM for n= 8 mice. #P < 0.05; ##P < 0.01 versus control mice. *P < 0.05; **P < 0.01 versus prednisone-treated mice.
Emodin improved metabolic abnormalities of DIO mice
C57BL/6J mice fed a high-fat diet developed moderate obesity, mild hyperglycaemia, dyslipidaemia and insulin resistance. Emodin (100 mg·kg−1) administered by oral gavage b.i.d. for 7 days reduced fasting glucose concentrations to 77.2% of the vehicle control mice, and these remained significantly lower (P < 0.01) throughout the treatment period (Figure 4A). After 24 days of treatment with emodin, the DIO mice exhibited a significant reduction in blood glucose levels at all time-points following oral glucose challenge (Figure 4B) (P < 0.05 to P < 0.01). This was accompanied by a reduction in serum insulin concentrations at 15, 30 and 60 min after glucose loading in the 100 mg·kg−1 emodin-treated mice (Figure 4C). Treatment with emodin for 28 days also evoked a significantly greater reduction in blood glucose values 40 and 90 min after insulin injection (P < 0.05 to P < 0.01), indicating an improved insulin tolerance in emodin-treated DIO mice (Figure 4D). Moreover, the serum insulin level was also significantly reduced, to 66.2% of control mice, after 35 days of treatment with 100 mg·kg−1 emodin (P < 0.05, Figure 4E).
Figure 4.
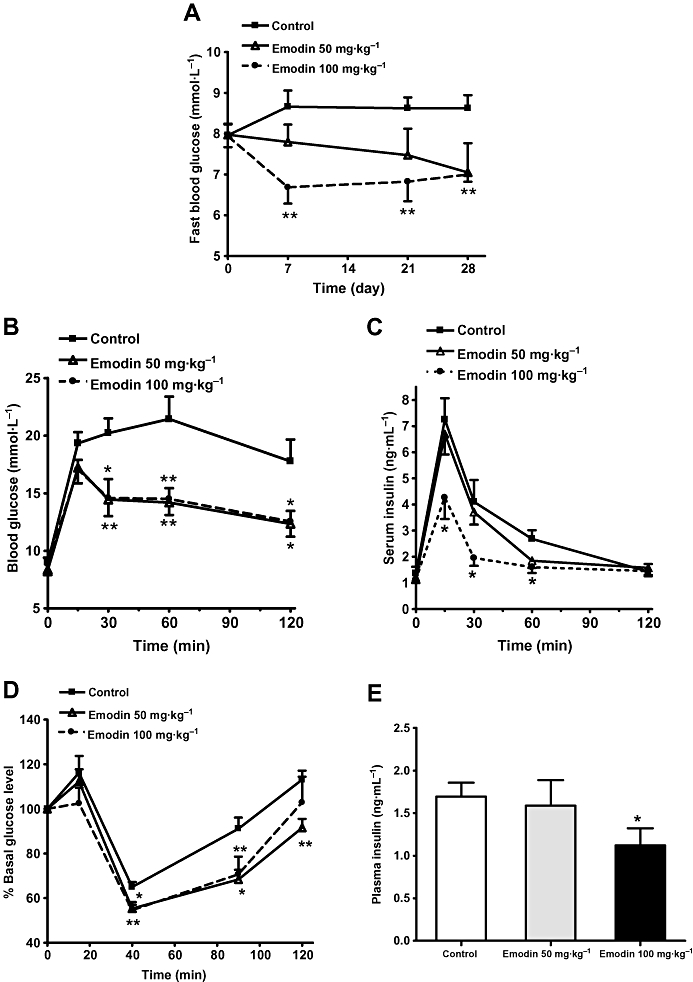
Emodin lowered blood glucose and increased insulin sensitivity of DIO mice. DIO mice were treated as described in Methods. Fasting blood glucose concentrations (A) were measured regularly during the treatment period. Glucose tolerance (B) and the corresponding insulin levels (C) were determined at day 24 of the treatment. Insulin tolerance (D) was determined at day 28 of the treatment. Serum insulin concentration (E) was measured in mice deprived of food for 5 h (5 h fasted mice) at the end of the treatment period. Values are expressed as mean ± SEM for n= 8 mice. *P < 0.05; **P < 0.01 versus control mice.
Emodin also improved the lipid profiles in DIO mice. After 35 days of treatment with 100 mg·kg−1 emodin, the serum triglyceride and total cholesterol levels were significantly reduced by 19.3 and 12.5% (P < 0.05), respectively, compared with vehicle control mice (Figure 5A,B). Emodin (100 mg·kg−1) also caused a 22.7% reduction of NEFA level, although this did not reach statistical significance (P= 0.077) (Figure 5C).
Figure 5.
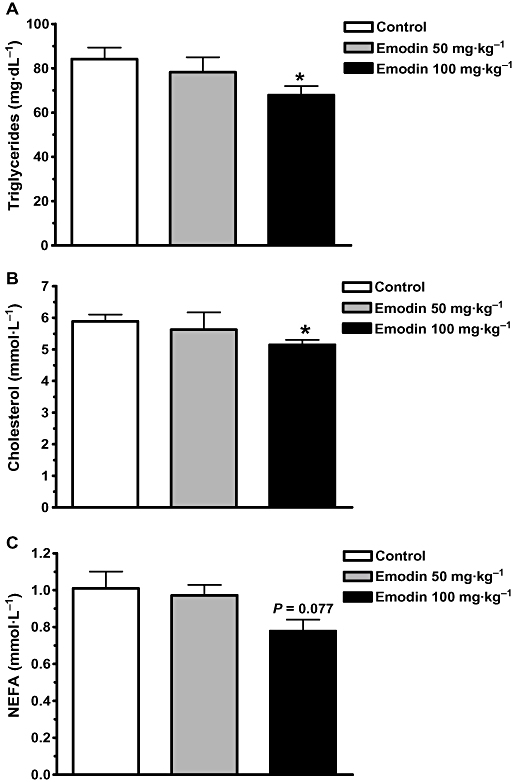
Emodin decreased serum lipids of DIO mice. DIO mice were treated as described in Methods. Serum triacylglycerol (A), cholesterols (B) and NEFA (C) concentrations were evaluated at the end of the treatment period. Data are mean ± SEM for n= 8 mice. *P < 0.05 versus control mice.
Chronic treatment with emodin lowered body weight and appetite in DIO mice. DIO mice treated with 100 mg·kg−1 emodin showed a steady decline in body weight that was significantly different from vehicle-treated animals from day 18 of the treatment; their body weights were reduced by 13.9% (P < 0.01) at the end of treatment (Figure 6A). Emodin also affected the animals' feeding behaviour, resulting in a 17% reduction in food intake compared with the vehicle-treated animals (P < 0.05; Figure 6B). Furthermore, it caused a preferential reduction in mesenteric fat pad and perirenal fat pad weights by 29 and 47%, respectively. The subcutaneous fat weight in emodin-treated DIO mice was reduced compared with vehicle-treated control mice (Figure 6C), but it essentially had no effect on epididymal fat weight (data not shown).
Figure 6.
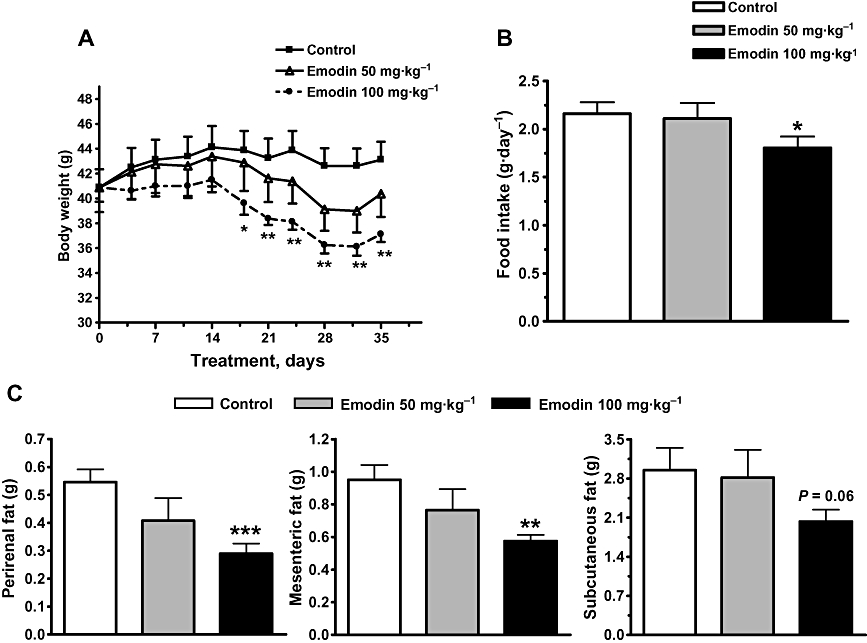
Emodin decreased body weight, food intake and fat pad weight of DIO mice. DIO mice were treated as described in Methods. Body weight (A) and food intake (B) were recorded regularly during the treatment period. Different fat pads (C) were also weighed at the end of the experiment. Values are mean ± SEM for n= 8 mice. *P < 0.05; **P < 0.01; ***P < 0.001 versus control mice.
Emodin suppressed 11β-HSD1 activity and reduced the mRNA levels of gluconeogenic genes in DIO mice
The enzymatic activity of 11β-HSD1 in liver and adipose tissues was measured 35 days after the treatment of DIO mice with 100 mg·kg−1 emodin. A significant decrease in 11β-HSD1 activity was observed in both the liver and mesenteric adipose tissues of emodin-treated DIO mice (P < 0.01). The 11β-HSD1 activity in liver and mesenteric adipose tissues was decreased by 53.5 and 41.2% (Figure 7A), respectively, whereas no significant change in 11β-HSD1 mRNA expression was observed (Figure 7B). Treatment of DIO mice with 100 mg·kg−1 emodin for 35 days significantly reduced hepatic PEPCK and G6Pase mRNA to levels 25.4 and 36.5% less than that of vehicle control mice (P < 0.05; Figure 7C).
Figure 7.
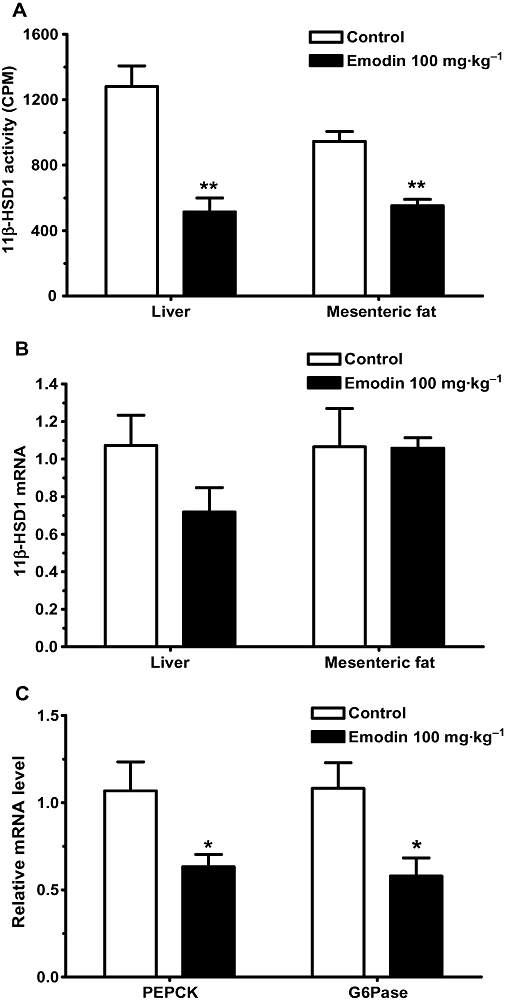
Emodin suppressed 11β-HSD1 activity and reduced the mRNA levels of gluconeogenic genes in DIO mice. DIO mice were treated as described in Methods. 11β-HSD1 activity (A) in liver and mesenteric adipose tissues was measured by SPA at the end of the treatment period. The expression of 11β-HSD1 mRNA (B), PEPCK and G6Pase mRNA (C) were also determined by real-time PCR at the end of the treatment period. Values are mean ± SEM for n= 4–8 mice. *P < 0.05; **P < 0.01 versus control mice.
Discussion
Emodin, a natural product and active ingredient of various Chinese herbs, has been demonstrated to possess multiple biological activities, including antitumour, antibacterial (Wang and Chung, 1997), anti-inflammatory (Chang et al., 1996) and immunosuppressive effects(Huang et al., 1992). Recent studies have shown that emodin could be a potential drug for the therapy of several proliferative diseases, such as liver cirrhosis (Woo et al., 2002), diabetic nephropathy (Wang et al., 2006), atherosclerosis (Heo et al., 2008) and tumours (Huang et al., 2007). Although a hypoglycaemic and hypolipidaemic effect of emodin had been reported in STZ-induced dyslipidaemic–diabetic rats (Zhao et al., 2008), the effects of emodin on metabolic abnormalities, especially insulin resistance and the molecular mechanisms involved, have not been thoroughly studied. Our study shows for the first time that emodin is a potent selective 11β-HSD1 inhibitor and can ameliorate metabolic disorders in DIO mice.
11β-HSD1 is highly expressed in liver and adipose tissue, where it plays key role in the regulation of the local generation of active glucocorticoids and is closely associated with the development of a cluster of metabolic abnormalities including insulin resistance, central obesity, hyperglycaemia and dyslipidaemia (Masuzaki et al., 2001; Lindsay et al., 2003; Lundgren et al., 2004; Valsamakis et al., 2004; Abdallah et al., 2005). Thus, there is a great interest in the discovery of potent selective 11β-HSD1 inhibitors for the development of therapeutic interventions in metabolic syndrome. In the present study, a screening of our compound collection provided us with an astonishing discovery that of a series anthraquinone compounds showed inhibitory activities against mouse and human 11β-HSD1. The SPA showed that emodin inhibited mouse and human 11β-HSD1 activity with IC50 values of 86 and 186 nM, respectively. As only 79% amino acids of the mouse and human 11β-HSD1 enzymes are identical, we did not expect emodin to inhibit 11β-HSD1 from both species to a similar degree. More importantly, emodin exhibited low inhibitory activity against mouse and human 11β-HSD2, with an IC50 higher than 1 mM, indicating that emodin is more than 5000-fold selective for the human and mouse 11β-HSD1 enzymes over the type 2 isoenzyme. A SPA for 11-HSD1 activity was also performed with the liver homogenates, and emodin displayed a comparable IC50 value against 11β-HSD1 in cell lysate with the recombinant enzyme (data not shown). Moreover, the in vivo inhibitory effect of emodin on 11β-HSD1 was confirmed in C57 BL/6J mice; a significant reduction of 11β-HSD1 activity in liver and mesenteric fat occurred at 2 h post-dose, which is around the half-life time of oral administration of emodin (Li et al., 2009). Therefore, emodin is a potent selective inhibitor of both the in vitro and in vivo activities of 11β-HSD1.
Chronic exposure to high circulating glucocorticoid levels causes insulin resistance (Pagano et al., 1983). In the present study, chronic treatment of C57BL/6J mice with dexamethasone or prednisone resulted in an impaired insulin tolerance, which indicated the development of insulin resistance. Concurrent treatment with emodin had no effect on dexamethasone-induced insulin resistance, whereas prednisone-induced insulin resistance could be fully reversed by emodin. Dexamethasone is a synthetic cortisol analogue, whereas prednisone is a synthetic cortisone analogue and needs to be catalysed by 11β-HSD1 in the liver to convert it into its active metabolite, prednisolone. Therefore, the finding that emodin prevented prednisone-induced insulin resistance confirmed that chronic administration of emodin can inhibit hepatic 11β-HSD1 activity in vivo.
The DIO mice showed moderate obesity, mild hyperglycaemia, dyslipidaemia and insulin resistance after being fed a high-fat diet for 12–15 weeks, which is closely similar to the obesity seen in humans consuming high-fat and energy-rich diets (Surwit et al., 1988). So, this model of obesity has been extensively used to evaluate the pharmacodynamic effects of numerous therapeutic compounds on metabolic syndrome or type 2 diabetes (Hu et al., 2007; Gounarides et al., 2008). Glucocorticoid excess antagonizes the effects of insulin, which decreases glucose uptake in peripheral tissues, increases hepatic glucose production and leads to elevated circulating levels of glucose and insulin resistance (Lambillotte et al., 1997). Selective inhibition of 11β-HSD1 could provide the means to block local activation of glucocorticoids and ameliorate the metabolic disorders (Hermanowski-Vosatka et al., 2005; Taylor et al., 2008). In the present study, emodin administration decreased blood glucose levels in DIO mice, with a parallel decrease in insulin levels. The OGTT results showed that treatment with emodin 100 mg·kg−1 resulted in a significant reduction in blood glucose levels, accompanied by a decrease in serum insulin concentrations, which indicates an increase of insulin sensitivity. This was further confirmed by the ITT results. Inhibition of 11β-HSD1 was expected to have a lipid-lowering effect, based on the ability of glucocorticoids to induce lipolysis and produce hepatic lipoprotein (Slavin et al., 1994). Emodin administration significantly reduced serum triglycerides and cholesterol levels in DIO mice, and tended to reduce the NEFA level, although this did not reach statistical significance. This modest decrease in NEFA level may be explained by the 41% inhibition of 11β-HSD1 activity in adipose tissue of emodin-treated mice, which might lead to only a slight suppression of the lipolytic activity induced by active glucocorticoids. Our results are consistent with previous reports on the effects of selective 11β-HSD1 inhibitors and on observations obtained in 11β-HSD1 KO mice (Morton et al., 2001), which suggested that emodin ameliorates metabolic disorder in DIO mice by selective inhibition of 11β-HSD1 in liver and adipose tissues.
Glucocorticoids are orexigenic (Cavagnini et al., 2000), and overexpression of 11β-HSD1 selectively in adipose tissue causes hyperphagia (Masuzaki et al., 2001). A previous study showed that the 11β-HSD1 inhibitor, BVT.2733 reduced food intake and body weight gain, but maintained energy expenditure in DIO mice, although the impared feeding caused a decrease of body weight as great as the inhibitor treatment (Wang et al., 2006). Therefore, we speculated that the decreased body weight caused by 100 mg·kg−1 emodin could be partly due to the reduced food intake, and the energy expenditure is likely to be maintained in emodin-treated mice as previously reported (Wang et al., 2006). Excess glucocorticoids enhance hypertrophy and differentiation of adipocytes, leading to central obesity and a redistribution of adipose tissue away from subcutaneous depots and into the visceral compartment (Marin et al., 1992; Vegiopoulos et al., 2007; Morton, 2010). Therefore, it is reasonable to assume administration of emodin, via inhibition of 11β-HSD1 activity, lowers the activity of GCs and this decreases the visceral fat mass, as shown here for the DIO mice.
Glucocorticoids stimulate transcription of hepatic gluconeogenic enzymes and thus play a major role in the enhancement of liver glucose output during starvation or stress (Pilkis and Granner, 1992). Thus, inhibition of 11β-HSD1 offers an effective pharmacological intervention that is likely to yield a sustained reduction of glucocorticoid-inducible hepatic gluconeogenic enzymes. PEPCK and G6Pase catalyse the rate-limiting steps of gluconeogenesis. Transcription of genes encoding both enzymes is regulated by classical glucocorticoid-inducible promoters (Imai et al., 1990; Lange et al., 1994), and is markedly attenuated in GR-deficient mice (Cole et al., 1995). Administration of emodin significantly reduced hepatic concentrations of mRNA encoding PEPCK and G6Pase, which is consistent with observations in 11β-HSD1 knock-out mice and with the selective inhibitor BVT.2733 (Alberts et al., 2003). These results support the hypothesis that emodin is a potent 11β-HSD1 inhibitor, which can reduce GR-activated hepatic gluconeogenesis; this might account for the decreased fasting blood glucose level and the improvement of the glucose tolerance seen after emodin treatment.
Glycyrrhetinic acid, a natural compound, and its hemisuccinyl derivative carbenoxolone have been well documented as 11β-HSD1 inhibitors (Walker et al., 1995). However, these two compounds display poor selectivity between the two isoforms of 11β-HSDs (Nuotio-Antar et al., 2007). Although, in a clinical study, carbenoxolone has been reported to improve hepatic insulin sensitivity and decrease glucose production in euglycaemic hyperinsulinaemic clamp, it only inhibited 11β-HSD1 in liver but had no effect in adipose tissue in vivo (Taylor et al., 2008). In our study, chronic treatment with emodin caused significant inhibition of 11β-HSD1 activity both in liver and mesenteric adipose tissue of DIO mice, whereas the 11β-HSD1 mRNA levels did not tend to change significantly.
Accumulating studies have indicated that a more effective targeting of 11β-HSD1 on adipose tissue is needed (Engeli et al., 2004; Morton et al., 2004; Tomlinson et al., 2007), our data suggest that of all the natural products showing 11β-HSD1 inhibitory activity, emodin is the most selective inhibitor of 11β-HSD1. Moreover, although the affinity of emodin for other enzymes and receptors has not been investigated, no evidence was found that emodin has any significant affinity for a panel of essential and ubiquitous enzymes and receptors, including the oestrogen, glucocorticoid, progesterone and androgen receptors.
In conclusion, our studies demonstrate a new role for emodin as a potent selective inhibitor of 11β-HSD1. Administration of emodin decreased blood glucose and serum insulin, improved insulin resistance and dyslipidaemia and decreased body weight and central fat mass in DIO mice. These results highlight the potential value of analogues of emodin as a new class of compound for the treatment of metabolic syndrome or type 2 diabetes.
Acknowledgments
This study was supported by the National Basic Research Program of China (973 Program, 2009CB522300), National Nature Science Foundation of China (grant 30873106), Shanghai Rising-Star Foundation (grant 08QH14028), National Science & Technology Major Project ‘Key New Drug Creation and Manufacturing Program’, China (Number 2009ZX09103-061) and grant 08DZ1980200 of Science and Technology Commission of Shanghai Municipality.
Glossary
Abbreviations
- 11β-HSD1
11β-hydroxysteroid dehydrogenase type 1
- 11β-HSD2
11β-hydroxysteroid dehydrogenase type 2
- DIO
diet-induced obese
- G6Pase
glucose-6-phosphatase
- GC
glucocorticoid
- GR
glucocorticoid receptor
- NEFA
non-esterified free fatty acid
- OGTT
oral glucose tolerance test
- PEPCK
phosphoenolpyruvate carboxykinase
- SPA
scintillation proximity assay
Conflict of interest
None of the authors have any conflict of interest.
References
- Abdallah BM, Beck-Nielsen H, Gaster M. Increased expression of 11beta-hydroxysteroid dehydrogenase type 1 in type 2 diabetic myotubes. Eur J Clin Invest. 2005;35:627–634. doi: 10.1111/j.1365-2362.2005.01552.x. [DOI] [PubMed] [Google Scholar]
- Alberts P, Nilsson C, Selen G, Engblom LO, Edling NH, Norling S, et al. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology. 2003;144:4755–4762. doi: 10.1210/en.2003-0344. [DOI] [PubMed] [Google Scholar]
- Andrews RC, Rooyackers O, Walker BR. Effects of the 11 beta-hydroxysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity in men with type 2 diabetes. J Clin Endocrinol Metab. 2003;88:285–291. doi: 10.1210/jc.2002-021194. [DOI] [PubMed] [Google Scholar]
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing's syndrome: diagnosis and therapy. Treat Endocrinol. 2002;1:79–94. doi: 10.2165/00024677-200201020-00002. [DOI] [PubMed] [Google Scholar]
- Cavagnini F, Croci M, Putignano P, Petroni ML, Invitti C. Glucocorticoids and neuroendocrine function. Int J Obes Relat Metab Disord. 2000;24:S77–S79. doi: 10.1038/sj.ijo.0801284. [DOI] [PubMed] [Google Scholar]
- Chang CH, Lin CC, Yang JJ, Namba T, Hattori M. Anti-inflammatory effects of emodin from ventilago leiocarpa. Am J Chin Med. 1996;24:139–142. doi: 10.1142/S0192415X96000189. [DOI] [PubMed] [Google Scholar]
- Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9:1608–1621. doi: 10.1101/gad.9.13.1608. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Ferrannnini E. Insulin resistance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–194. doi: 10.2337/diacare.14.3.173. [DOI] [PubMed] [Google Scholar]
- Draper N, Stewart PM. 11β-Hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol. 2005;86:251–271. doi: 10.1677/joe.1.06019. [DOI] [PubMed] [Google Scholar]
- Engeli S, Böhnke J, Feldpausch M, Gorzelniak K, Heintze U, Janke J, et al. Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res. 2004;12:9–17. doi: 10.1038/oby.2004.3. [DOI] [PubMed] [Google Scholar]
- Ewing T, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem. 1997;18:1175–1189. [Google Scholar]
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- Gounarides JS, Korach-André M, Killary K, Argentieri G, Turner O, Laurent D. Effect of dexamethasome on glucose tolerance and fat metabolism in a diet-induced obesity mouse model. Endocrinology. 2008;149:758–766. doi: 10.1210/en.2007-1214. [DOI] [PubMed] [Google Scholar]
- Heo SK, Yun HJ, Park WH, Park SD. Emodin inhibits TNF-alpha-induced human aortic smooth muscle cell proliferation via caspase and mitochondrial-dependent apoptosis. J Cell Biochem. 2008;105:70–80. doi: 10.1002/jcb.21805. [DOI] [PubMed] [Google Scholar]
- Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, et al. 11Beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med. 2005;202:517–527. doi: 10.1084/jem.20050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Feng Y, Liu X, Zhao XF, Yu JH, Yang YS, et al. Effect of a novel non-thiazolidinedione peroxisome proliferators-activated receptor α/γ agonist on glucose uptake. Diabetologia. 2007;50:1048–1057. doi: 10.1007/s00125-007-0622-3. [DOI] [PubMed] [Google Scholar]
- Huang HC, Chang JH, Tung SF, Wu RT, Foegh ML, Chu SH. Immunosuppressive effect of emodin, a free radical generator. Eur J Pharmacol. 1992;211:359–364. doi: 10.1016/0014-2999(92)90393-i. [DOI] [PubMed] [Google Scholar]
- Huang Q, Lu G, Shen HM, Chung MC, Ong CN. Anti-cancer properties of anthraquinones from rhubarb. Med Res Rev. 2007;27:609–630. doi: 10.1002/med.20094. [DOI] [PubMed] [Google Scholar]
- Imai E, Stromstedt PE, Quinn PG, Carlstedt-Duke J, Gustafsson JA, Granner DK. Characterization of a complex glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol. 1990;10:4712–4719. doi: 10.1128/mcb.10.9.4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest. 1997;99:414–423. doi: 10.1172/JCI119175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange AJ, Argaud D, el-Maghrabi MR, Pan W, Maitra SR, Pilkis SJ. Isolation of a cDNA for the catalytic subunit of rat liver glucose-6-phosphatase: regulation of gene expression in FAO hepatoma cells by insulin, dexamethasone and cAMP. Biochem Biophys Res Commun. 1994;201:302–309. doi: 10.1006/bbrc.1994.1702. [DOI] [PubMed] [Google Scholar]
- Li Y, Duan J, Guo T, Xie W, Yan S, Li B, et al. In vivo pharmacokinetics comparisons of icariin, emodin and psoralen from Gan-kang granules and extracts of Herba epimedii, Nepal dock root, Ficus hirta yahl. J Ethnopharmacol. 2009;124:522–529. doi: 10.1016/j.jep.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Lindsay RS, Wake DJ, Nair S, Bunt J, Livingstone DE, Permana PA, et al. Subcutaneous adipose 11 beta-hydroxysteroid dehydrogenase type 1 activity and messenger ribonucleic acid levels are associated with adiposity and insulinemia in Pima Indians and Caucasians. J Clin Endocrinol Metab. 2003;88:2738–2744. doi: 10.1210/jc.2002-030017. [DOI] [PubMed] [Google Scholar]
- Livingstone DE, Jones GC, Smith K, Jamieson PM, Andrew R, Kenyon CJ, et al. Understanding the role of glucocorticoids in obesity: tissue-specific alternations of corticosterone metabolism in obese Zucker rats. Endocrinology. 2000;141:560–563. doi: 10.1210/endo.141.2.7297. [DOI] [PubMed] [Google Scholar]
- Lundgren M, Buren J, Ruge T, Myrnas T, Eriksson JW. Glucocorticoids down-regulate glucose uptake capacity and insulin-signaling proteins in omental but not subcutaneous human adipocytes. J Clin Endocrinol Metab. 2004;89:2989–2997. doi: 10.1210/jc.2003-031157. [DOI] [PubMed] [Google Scholar]
- Marin P, Drain N, Amemiya T, Andersson B, Jern S, Bjorntorp P. Cortisol secretion in relation to body fat distribution in obese premenopausal women. Metabolism. 1992;41:882–886. doi: 10.1016/0026-0495(92)90171-6. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- Morton NM. Obesity and corticosteroids: 11β-hydroxysteriod type 1 as a cause and therapeutic target in metabolic disease. Mol Cellular Endocrinol. 2010;316:154–164. doi: 10.1016/j.mce.2009.09.024. [DOI] [PubMed] [Google Scholar]
- Morton NM, Holmes MC, Fiévet C, Staels B, Tailleux A, Mullins JJ, et al. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem. 2001;276:41293–41300. doi: 10.1074/jbc.M103676200. [DOI] [PubMed] [Google Scholar]
- Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, et al. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes. 2004;53:931–938. doi: 10.2337/diabetes.53.4.931. [DOI] [PubMed] [Google Scholar]
- Mundt S, Solly K, Thieringer R, Hermanowski-Vosatka A. Development and application of a scintillation proximity assay (SPA) for identification of selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1. Assay Drug Dev Technol. 2005;3:367–375. doi: 10.1089/adt.2005.3.367. [DOI] [PubMed] [Google Scholar]
- Nuotio-Antar AM, Hachey DL, Hasty AH. Carbenoxolone treatment attenuates symptoms of metabolic syndrome and atherogenesis in obese, hyperlipidemic mice. Am J Physiol Endocrinol Metab. 2007;293:E1517–E1528. doi: 10.1152/ajpendo.00522.2007. [DOI] [PubMed] [Google Scholar]
- Pagano G, Cavallo-Perin P, Cassader M, Bruno A, Ozzello A, Masciola P, et al. An in vivo and in vitro study of the mechanism of prednisone-induced insulin resistance in healthy subjects. J Clin Invest. 1983;72:1814–1820. doi: 10.1172/JCI111141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JR, Shuai Q, Dinges J, Winn M, Pliushchev M, Fung S, et al. Discovery of adamantane ethers as inhibitors of 11β-HSD-1: synthesis and biological evaluation. Bioorg Med Chem Lett. 2007;17:50–755. doi: 10.1016/j.bmcl.2006.10.074. [DOI] [PubMed] [Google Scholar]
- Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- Sandeep TC, Andrew R, Homer NZ, Andrews RC, Smith K, Walker BR. Increased in vivo regeneration of cortisol in adipose tissue in human obesity and effects of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor carbenoxolone. Diabetes. 2005;54:872–879. doi: 10.2337/diabetes.54.3.872. [DOI] [PubMed] [Google Scholar]
- Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1 – a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- Slavin BG, Ong JM, Kern PA. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J Lipid Res. 1994;35:1535–1541. [PubMed] [Google Scholar]
- Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- Taylor A, Irwin N, McKillop AM, Flatt PR, Gault VA. Sub-chronic administration of 11β-HSD1 inhibitor, carbenoxolone, improves glucose tolerance and insulin sensitivity in mice with diet-induced obesity. Biol Chem. 2008;389:441–445. doi: 10.1515/BC.2008.049. [DOI] [PubMed] [Google Scholar]
- Tomlinson JW, Sherlock M, Hughes B, Hughes SV, Kilvington F, Bartlett W, et al. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 activity in vivo limits glucocorticoid exposure to human adipose tissue and decreases lipolysis. J Clin Endocrinol Metab. 2007;92:857–864. doi: 10.1210/jc.2006-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsamakis G, Anwar A, Tomlinson JW, Shackleton CH, McTernan PG, Chetty R, et al. 11Beta-hydroxysteroid dehydrogenase type 1 activity in lean and obese males with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2004;89:4755–4761. doi: 10.1210/jc.2003-032240. [DOI] [PubMed] [Google Scholar]
- Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275:43–61. doi: 10.1016/j.mce.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Walker BR, Connacher AA, Lindsay RM, Webb DJ, Edwards CR. Carbenoxolone increases hepatic insulin sensitivity in man: a novel role for 11-oxosteroid reductase in enhancing glucocorticoid receptor activation. J Clin Endocrinol Metab. 1995;80:3155–3159. doi: 10.1210/jcem.80.11.7593419. [DOI] [PubMed] [Google Scholar]
- Wang HH, Chung JG. Emodin-induced inhibition of growth and DNA damage in the Helicobacter pylori. Curr Microbiol. 1997;35:262–266. doi: 10.1007/s002849900250. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Birtles S, de Schoolmeester J, Swales J, Moody G, Hislop D, et al. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 reduces food intake and weight gain but maintains energy expenditure in diet-induced obese mice. Diabetologia. 2006;49:1333–1337. doi: 10.1007/s00125-006-0239-y. [DOI] [PubMed] [Google Scholar]
- Weiner SJ, Kollman PA, Nguyen DT, Case DA. An allatom force field for simulations of proteins and nucleic acids. J Comput Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- Woo SW, Nan JX, Lee SH, Park EJ, Zhao YZ, Sohn DH. Aloe emodin suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Pharmacol Toxicol. 2002;90:193–198. doi: 10.1034/j.1600-0773.2002.900404.x. [DOI] [PubMed] [Google Scholar]
- Zhao XY, Qiao GF, Li BX, Chai LM, Li Z, Lu YJ, et al. Hypoglycaemic and hypolipidaemic effects of emodin and its effect on L-type calcium channels in dyslipidaemic-diabetic rats. Clin Exp Pharmacol Physiol. 2008;36:29–34. doi: 10.1111/j.1440-1681.2008.05051.x. [DOI] [PubMed] [Google Scholar]