

Abstract
BACKGROUND AND PURPOSE
Glutamate excitotoxicity may be involved in ischaemic injury to the CNS and some neurodegenerative diseases, such as Alzheimer's disease. Donepezil, an acetylcholinesterase (AChE) inhibitor, exerts neuroprotective effects. Here we demonstrated a novel mechanism underlying the neuroprotection induced by donepezil.
EXPERIMENTAL APPROACH
Cell damage in primary rat neuron cultures was quantified by lactate dehydrogenase release. Morphological changes associated with neuroprotective effects of nicotine and AChE inhibitors were assessed by immunostaining. Cell surface levels of the glutamate receptor sub-units, NR1 and NR2A, were analyzed using biotinylation. Immunoblot was used to measure protein levels of cleaved caspase-3, total NR1, total NR2A and phosphorylated NR1. Immunoprecipitation was used to measure association of NR1 with the post-synaptic protein, PSD-95. Intracellular Ca2+ concentrations were measured with fura 2-acetoxymethylester. Caspase 3-like activity was measured using enzyme substrate, 7-amino-4-methylcoumarin (AMC)-DEVD.
KEY RESULTS
Levels of NR1, a core subunit of the NMDA receptor, on the cell surface were significantly reduced by donepexzil. In addition, glutamate-mediated Ca2+ entry was significantly attenuated by donepezil. Methyllycaconitine, an inhibitor of α7 nicotinic acetylcholine receptors (nAChR), inhibited the donepezil-induced attenuation of glutamate-mediated Ca2+ entry. LY294002, a phosphatidyl inositol 3-kinase (PI3K) inhibitor, had no effect on attenuation of glutamate-mediated Ca2+ entry induced by donepezil.
CONCLUSIONS AND IMPLICATIONS
Decreased glutamate toxicity through down-regulation of NMDA receptors, following stimulation of α7 nAChRs, could be another mechanism underlying neuroprotection by donepezil, in addition to up-regulating the PI3K-Akt cascade or defensive system.
Keywords: donepezil, glutamate, NMDA receptor, α7 nicotinic receptor, neuroprotection
Introduction
Acetylcholinesterase (AChE) inhibitors are currently used to treat Alzheimer's disease (AD) therapy. We have previously demonstrated that AChE inhibitors, such as donepezil and galantamine, exert a protective effect via the nicotinic acetylcholine receptor (nAChR)-mediated cascade (Kihara et al., 2001; Takada et al., 2003). In addition, it has been reported that AChE inhibitors inhibit the progress of brain atrophy in AD (Hashimoto et al., 2005), indicating the attenuation of neuronal death in the brain of the patients. ACh receptors provide neuroprotection against glutamate-induced excitotoxicity by stimulating the phosphatidylinositol-3 kinase (PI3K)→Akt→Bcl-2 signalling pathway (Asomugha et al., 2010; Kihara et al., 2001). Thus, a cell survival mechanism is involved in AChE inhibitor-induced protection against neuronal death.
Recent evidence has suggested that beta amyloid (Aβ), a major component of senile plaques found in the brains of AD patients, leads to the internalization of NMDA receptors and subsequently reduces glutamatergic transmission and inhibits synaptic plasticity (Snyder et al., 2005). Aβ application produced a rapid and persistent depression of NMDA-evoked currents, which might explain the cognitive dysfunction of AD.
From a different standpoint, NMDA receptor-mediated excitotoxic signals might also be suppressed by internalization of NMDA receptors. Excessive glutamate induces neuronal death (Akaike et al., 1994), which is associated with ischaemia and numerous neurodegenerative disorders including AD. Glutamate and NMDA can cause intracellular Ca2+ influx, activation of Ca2+-dependent enzymes such as nitric oxide synthase and production of toxic oxygen radicals leading to cell death (Tamura et al., 1992). Cell death would be inhibited by the attenuation of excitotoxic signals via the reduced number of cell surface NMDA receptors. In addition, Aβ-dependent internalization of NMDA receptors required α7 nAChRs (Snyder et al., 2005). Stimulation of α7 nAChR by AChE inhibitors could also lead to the internalization of NMDA receptors.
There is, however, a possibility that the stimulation of nAChRs would attenuate the down-regulation of the surface level of glutamate receptors containing the subunits NR1/2A and NMDA receptor activity. Tyrosine dephosphorylation can down-regulate the surface level of NR1/2A receptors, which was restored by Src (Suvarna et al., 2005). The stimulation of nAChR induces phosphorylation of Akt and extracellular signal-regulated protein kinase (p42/44 MAP kinase, ERK) through Src in various cell types (Nakayama et al., 2002; Dasgupta et al., 2006; Wada et al., 2007). The stimulation of nAChR would activate Src, which in turn could phosphorylate NMDA receptors, and surface level of NR1/2A receptors would be restored.
It has been reported that AChE inhibitors have no effect on the baseline synaptic transmission or the magnitude of long-term potentiation induction (Barnes et al., 2000). In addition, α7 nAChR stimulation is reported to enhance glutamatergic transmission (Gray et al., 1996). Thus, α7 nAChRs might regulate the neuronal activity to prevent hyper-activity that leads to neuronal death without interfering with synaptic transmission. In other words, cholinergic stimulation does not appear to decrease basal transmission, even though NMDA receptors would be internalized by nAChR stimulation.
In the present study, we hypothesized that donepezil would internalize the functional NMDA receptors through α7 nAChR stimulation, which in turn would attenuate the neuronal death induced by excessive glutamate, although synaptic NMDA receptors would not be altered.
Methods
Primary neuron cultures
All animal care and experimental procedures were in accordance with the guidelines published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Primary cultures were obtained from fetal rat cerebral cortex (E18) using procedures described previously (Kihara et al., 1997). Briefly, single cells dissociated from the cerebral cortex of fetal rats were plated onto 48- or 12-well plates, 6-well plates with coverslips, 8-well chambers or 60 mm Falcon dishes. The 48-well plates were used for the evaluation of cellular damage, 12-well plates were for Ca2+ measurements, 35 mm Falcon dishes with coverslips and 8-well chambers for immunostaining, 60 mm Falcon dishes for biotinylation of cell surface experiments and 6-well plates for immunoprecipitation, caspase-3-like enzyme activity and cleaved caspase-3 immunoblot analyis. The cell density was 1.8 × 105 cells/cm2. Wells and coverslips were pre-treated by incubation with polyethyleneimine (0.1% w/v in 0.15 M borate buffer, pH 8.4) overnight at room temperature (24°C) and then rinsed three times with sterile water before seeding cells in starter medium. Cultures were incubated in the Neurobasal medium with 2% B27 supplement anti-oxidant minus (Invitrogen, Carlsbad, CA, USA), 25 µM glutamate and 0.5 mM L-glutamine. After 4 days in culture, medium was replaced with glutamate free medium (Neurobasal medium supplemented 2% B27 and 0.5 mM L-glutamine). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. Only mature cultures (8 days in vitro) were used for the experiments. All experiments were carried out in Neurobasal medium with B27 supplement and L-glutamine at 37°C.
Immunostaining
Immunostaining was performed by methods described previously (Sawada et al., 1998; Kihara et al., 2001). To summarize, the neurons were fixed by 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 30 min at 4°C. After rinsing with PBS three times, 0.2% Triton in PBS was added for 10 min and then the cells were incubated overnight at 4°C with antibody to microtubule associated protein 2a+2b (anti-MAP2). After washing with PBS three times, slides were incubated with secondary antibodies (biotinylated anti-mouse IgG (H+L) diluted 1:200 in 0.05% NaN3, 1% BSA) for 1 h at room temperature. Following a PBS wash, the fixed cells were incubated with avidin-biotin-immunoperoxidase complex (ABC) for 1 h at room temperature and then washed with Tris-buffered saline (TBS) three times. Cells were visualized by the addition of 0.7 mg mL–1 3,3′-diaminobenzidine solution with 0.018% H2O2.
For immunofluorescence staining, cells were fixed with 0.1 M phosphate buffer containing 4% PFA for 15 min. Fixed cells were rinsed with PBS three times and blocked with 5% goat serum in PBS with 0.005% saponin for 30 min and incubated overnight at 4°C with antibodies to the glutamate receptor NR1 subunit (1:500) or to the post-synaptic density protein (95 kDa) – (PSD-95; 1:200), diluted in PBS containing 1% goat serum and 0.005% saponin. The next day, the cells were washed three times and then incubated for 1 h at room temperature with Cy3-AffiniPure goat anti-mouse IgG (1:800) or Cy2-AffiniPure goat anti-mouse IgG (1:800). Cultures were washed with PBS, treated with FluorSave Reagent (Calbiochem, Darmstadt, Germany) and covered. Cells were visualized under a laser sharp confocal scanning microscope (Bio-Rad, Hercules, CA, USA).
Quantification of the cell damage
Neuronal cells were pre-treated by an addition of nicotine, donepezil or galantamine to the culture medium in the given concentrations for 48 h, followed by the exposure to glutamate with simultaneous administration of nicotine or AChE inhibitors for 24 h. Glutamate (dissolved in sterile water just before use) was added to the culture medium at a given concentration for 24 h. Antagonists were simultaneously added with nicotine or AChE inhibitors. Preliminary dose-response studies had indicated that 10 µM MK801 produced maximal inhibition of glutamate excitotoxicity. Methyllycaconitine (MLA; 1 µM), 10 µM LY294002 and 5 µM PP2 produced maximal inhibition of donepezil-induced protection against glutamate excitotoxicity.
The condition of the cells was determined morphologically by phase contrast microscopy and damage was assessed by the measurement of actate dehydrogenase (LDH) release into the medium, indicating loss of membrane integrity. LDH activity was spectrophotometrically measured using a MTX-LDH assay kit according to the manufacturer's instructions. Total LDH activity was defined as the sum of intracellular and extracellular LDH activity obtained by 10 mM glutamate treatment [10 mM glutamate killed all neurons in our culture (data not shown)] and released LDH was defined as the percentage of extracellular compared with total LDH activity. Absorption at 570 nm was measured by a microplate spectrophotometer (Model 680 plate reader, Bio-Rad).
Intracellular Ca2+ imaging
Intracellular Ca2+ concentrations were measured with a Ca2+-sensitive fluorescent dye, fura 2-acetoxymethylester, on a fluorescence imaging system (ARGUS/HiSCA, Hamamatsu Photonics K.K., Shizuoka, Japan) according to the methods described previously (Shirakawa et al., 2002). Cortical neurons cultured on glass coverslips were incubated in Krebs-Ringer buffer (137 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 25 mM D-(+)-glucose, 10 mM HEPES, pH 7.4) containing 5 µM fura 2- acetoxymethylester and 0.01% cremophor EL (polyoxethylated castor oil) for 30 min at 37°C and then rinsed with buffer. The cells were alternatively illuminated with light (wavelengths of 340 and 380 nm) at an interval of 5 s and the emission was measured at 500 nm. The peak or highest value of amplitude of the fluorescence ratio (340/380 nm) just after glutamate application was adopted as an index of glutamate-induced Ca2+ influx.
Preparation of cell extracts for immunoblot
After each treatment, the cell culture dishes were placed on ice and washed with ice-cold PBS, drained and then ice-cold lysis buffer consisting of PBS containing 0.1% Triton X-100, protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan) and phosphatase inhibitors cocktail 2 (Sigma, St. Louis, MO, USA) was added. The cells were scraped off using a cold plastic cell scraper, transferred into a pre-cooled microfuge tube and centrifuged at 15 700×g for 20 min at 4°C. The supernatants were used as the cell extracts for immunoblot analysis of caspase-3. The protein concentration of each sample was determined with a micro BCA Protein Assay kit (Pierce, Rockford, IL, USA).
Biotinylation of cell surface proteins
According to methods previously reported, cell surface proteins were evaluated with the biotinylation technique (Uemura et al., 2003). After treatment, neurons were washed three times with ice-cold PBS. Neurons were then incubated in PBS containing 0.5 mg/mL Sulfo-NHS-LC-biotin for 30 min on ice. Neurons were washed three times with PBS and then lysed in 300 µL PBS with 1% protease inhibitor cocktail (Nacalai Tesque), 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS) and 1% phosphatase inhibitor cocktail 1 (Sigma). To determine the total protein concentration by immunoblotting, 20 µL of the cell lysate was removed and diluted in sample buffer. To isolate biotinylated proteins, the rest of the cell lysate was incubated with immobilized streptavidin agarose for 1 h at 4°C and centrifuged at 2300×g for 1 min at 4°C. Immunoblotting was carried out subsequently.
Immunoprecipitation
Cultures were lysed with 0.2 mL lysis buffer per well (20 mM Tris-HCl pH 7, 25 mM β -glycerophosphate, 2 mM EGTA, 1% Triton-X, 1 mM vanadate, aprotinin, 1 mM phenylmethylsulfonyl fluoride, 2 mM dithiothreitol) at 4°C. After lysis, cells were scraped, transferred into 1.5 mL tubes and then centrifuged at 15 700×g for 30 min at 4°C. The supernatants were then incubated with protein G-Sepharose beads for 2 h at 4°C and were normalized by protein concentration. Immunoprecipitations were carried out by incubating the lysates with monoclonal anti-PSD-95 (1:100) overnight at 4°C. The lysates were then incubated with protein G-Sepharose beads with rotation for 2 h at 4°C. The beads were rinsed three times with lysis buffer and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Immunoblot analysis
Combined lysates were dissolved in SDS sample buffer containing 4% 2-mercaptoethanol (Daiichi Chemical, Tokyo, Japan) and heated at 95°C for 3 min. SDS-PAGE was performed on a 4–20% gradient gel (Daiichi Chemical). After electrophoresis, proteins were electrotransferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA) according to the manufacturer's instruction (Bio-Rad). The membrane was washed with 20 mM Tris-HCl, pH 7.6, 135 mM NaCl, containing 0.1% Tween 20 (TBST) and blocked with TBST containing 5% non-fat dry milk for 2 h. The membrane was incubated with rabbit anti-cleaved-caspase-3 (1:500), rabbit anti-NR1 (1:500), rabbit-anti-NMDAε1 (H-54) (1:500) or rabbit anti-phospho-NMDA receptor1 (1:500) at 4°C. After incubation with horseradish peroxidase-conjugated secondary antibody (GE Healthcare Life Sciences, Piscataway, NJ, USA) at room temperature for 1 h in TBST containing 5% non-fat dry milk, immunoreactivities of the protein bands were detected by ECL (Enhanced ChemiLuminescence) Plus Kit according to manufacture's instruction manuals (GE Healthcare Life Sciences).
Caspase3-like enzyme activity
Caspase3-like activity was measured by a spectrophotometric assay as described previously (Yamaguchi et al., 2001). Briefly, neurons were suspended in buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA and 10 mM EGTA), incubated with 10 µM digitonin (Sigma) at 37°C for 15 min, followed by centrifugation at 15 700×g for 5 min. Protein supernatant was incubated with 50 µM enzyme substrate, 7-amino-4-methylcoumarin (AMC)-DEVD, at 37°C for 1 h. Levels of released AMC were measured using an excitation wavelength of 380 nm and an emission wavelength of 460 nm with a spectrofluorometer (Wallac 1420 ARVOsx multilabel counter; PerkinElmer, Inc., Waltham, MA, USA). One unit was defined as the amount of enzyme required to release 0.22 nmol of AMC/min at 37°C.
Statistical analysis
Statistically significant differences between groups were determined by Kruskal–Wallis non-parametric analysis of variance followed by Dunn's post test or Student's t-test. The level of statistical significance was taken at P < 0.05.
Materials
The sources of materials were as follows: Neurobasal medium and B27 supplement were obtained from Invitrogen; (-)-nicotine di-d-tartrate, MK801, MLA, phosphatase inhibitor cocktail 2, Ac-DEVD-AMC, monoclonal anti-post synaptic density (95 kDa) protein (PSD-95), clone 7E3-IB8 and monoclonal anti-microtubule associated protein 2 (2a+2b) clone AP-20 were from Sigma; Donepezil was provided by Eisai Co., Ltd. (Tokyo, Japan); PI3K inhibitor LY294002 and 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo [3,4-d] pyrimidine (PP2) was from Calbiochem; EGTA, boric acid and protease inhibitor cocktail were from Nacalai Tesque; Sulfo-NHS-LC-biotin and streptavidin agarose were from Pierce; anti-phospho-NMDA receptor 1 (Ser890) antibody and anti-cleaved caspase-3 (Asp175) antibody were from Cell Signaling (Danvers, MA, USA); anti-NR1 antibody was from Upstate Biotechnology, Inc. (Lake Placid, NY, USA); anti-NMDAε1 (H-54) antibody was from SantaCruz Biotechnology, Inc. (Santa Cruz, CA, USA); Cy2-AffiniPure goat anti-mouse IgG and Cy3-AffiniPure goat anti-rabbit IgG were from Jackson ImmunoResearch (West Grove, PA, USA); LDH assay kit from Kyokuto (Tokyo, Japan); Protein G sepharose 4 Fast Flow was from GE Healthcare Life Sciences; biotinylated anti-mouse IgG (H+L) and Vectastain ABC kit were from Vector Laboratories (Burlingame, CA, USA).
The nomenclature used for the receptors, agonists and antagonists follows Alexander et al. (2009).
Results
Glutamate cytotoxicity was mediated via Ca2+ influx
Cortical neurons that were incubated with glutamate for 24 h showed a significant increase in LDH release in a concentration-dependent manner, which indicates a reduction of the neuronal cell viability (Figure 1A). Total LDH activity was defined as the sum of intracellular and extracellular LDH activity obtained by 10 mM glutamate treatment [in preliminary experiments, 24 h-treatment with 10 mM glutamate left no MAP-2 positive cells, that is, no neuronal cells, in culture (data not shown)] and released LDH was defined as the percentage of extracellular activity compared with total LDH activity. Glutamate (30 µM) was used in the following studies of excitotoxicity as it was the EC50 for LDH release. We chose this concentration as it would enable us to detect either inhibition (protection) or potentiation of glutamate-induced cell toxicity.
Figure 1.
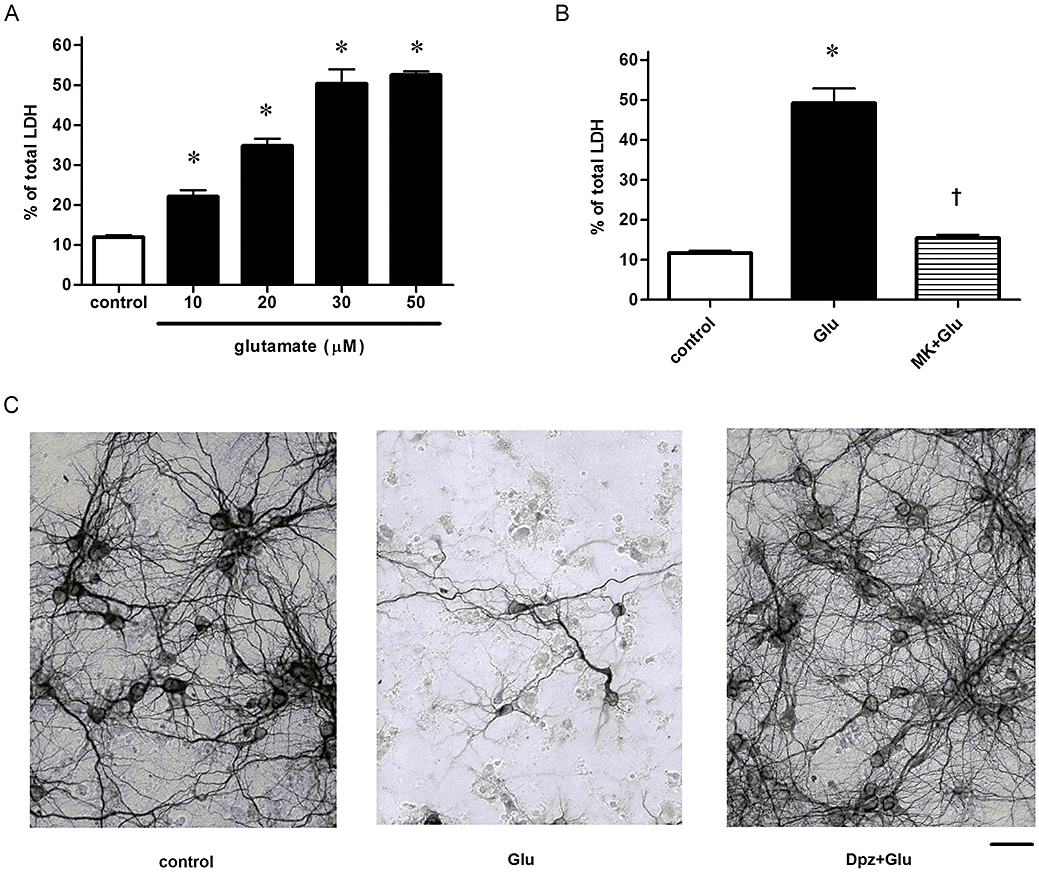
Glutamate (Glu)-induced neuronal death is mediated via NMDA receptor. (A) Glu excitotoxicity was concentration-dependent. Cultures were exposed to 24 h incubation. n = 32. *P < 0.05 compared with control (vehicle alone). Statistically significant differences between groups were determined by Kruskal–Wallis analysis of variance (anova) with post hoc multiple comparisons. (B) Glu excitotoxicity was inhibited by the NMDA receptor antagonist MK801 (10 µM; MK). n = 32. *P < 0.05 compared with control (vehicle alone), †P < 0.05 compared with Glu alone. Statistically significant differences between groups were determined by Kruskal–Wallis anova followed by Dunn's post-test. (C) Immunostained images showing neuroprotective effects of donepezil (Dpz) against Glu excitotoxicity. Bar = 1 × 10−4 m.
Glutamate-induced neuronal death (30 µM, 24 h) was attenuated by the NMDA receptor antagonist, MK801 (10 µM), as previously demonstrated (Figure 1B). We have previously reported that simultaneous administration of EGTA reduced glutamate-induced excitotoxicity, and glutamate-induced caspase-3-like activity was also inhibited by EGTA, indicating the involvement of Ca2+ (Yazawa et al., 2006).
Donepezil protects neurons from glutamate cytotoxicity via α7 nAChR and a Src family tyrosine kinase
Cultures were exposed to glutamate (30 µM) for 24 h to induce neuronal death. Donepezil, an AChE inhibitor, was used as a pre-treatment for 48 h and then continued with the glutamate stumulus for 24 h. Under these conditions, donepezil exerted a neuroprotective effect against glutamate excitotoxicity in a concentration-dependent manner (Figure 2A). In the following studies, 10 µM donepezil was used because it produced a maximal response. We selected the concentration as the inhibitory effects of MLA, LY294002 or PP2 could be observed clearly.
Figure 2.
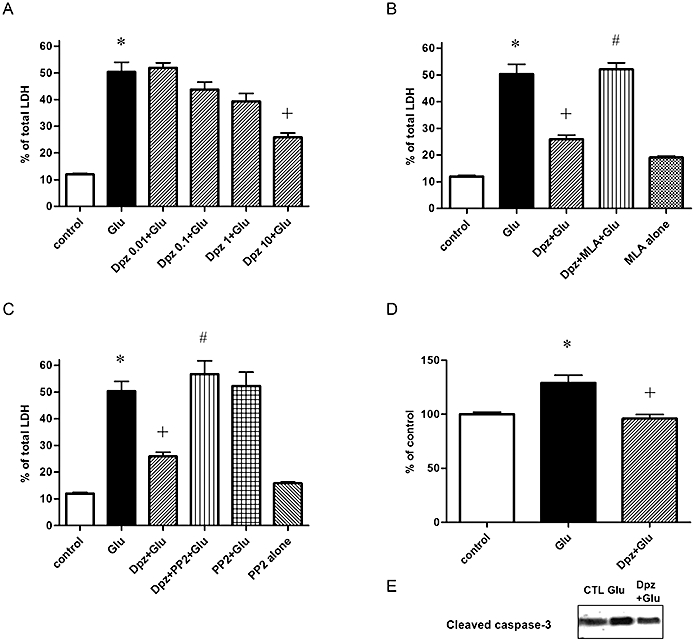
Neuroprotective effect of donepezil (Dpz) against glutamate (Glu)-induced caspase-3 activation and excitotoxicity was mediated via α7 nAChR and a Src family tyrosine kinase. (A) Dpz-induced neuroprotection (10 µM) against glutamate (Glu) excitotoxicity was concentration-dependent. Pre-treatment with Dpz for 48 h followed by 24-h Glu (30 µM) with Dpz incubation. n = 32. *P < 0.05 compared with control (vehicle alone), †P < 0.05 compared with Glu alone. Statistically significant differences between groups were determined by Kruskal–Wallis analysis of variance (anova) followed by Dunn's post-test. (B) Dpz (10 µM)-induced neuroprotection against Glu (30 µM, 24 h) excitotoxicity was mediated via α7 nAChR. Simultaneous administration of MLA (1 µM), α7 nAChR antagonist, attenuated Dpz-induced neuroprotection. n = 32. *P < 0.05 compared with control (vehicle alone), †P < 0.05 compared with Glu alone, #P < 0.05 compared with Dpz + Glu. Statistically significant differences between groups were determined by Kruskal–Wallis anova followed by Dunn's post-test. (C) Dpz-induced neuroprotection (10 µM) against Glu (30 µM, 24 h) excitotoxicity was mediated via a Src family tyrosine kinase. PP2 (5 µM), a Src family tyrosine kinase inhibitor, reduced Dpz-induced neuroprotection. n = 32. *P < 0.05 compared with control (vehicle alone), †P < 0.05 compared with Glu alone, #P < 0.05 compared with Dpz + Glu. Statistically significant differences between groups were determined by Kruskal–Wallis anova followed by Dunn's post-test. (D) Glu-induced caspase-3-like activity (30 µM) was reduced by Dpz. Glu increased caspase-3-like activity and reached its peak at 3 h treatment. Pre-treatment of Dpz (10 µM, 48 h) attenuated the Glu-induced caspase-3 like activity. n = 6. *P < 0.05 compared with Glu alone. Statistically significant differences between groups were determined by Kruskal-Wallis anova followed by Dunn's post-test. (E) Cleaved caspase-3, an activated form of caspase-3, was also increased by Glu and inhibited by Dpz pre-treatment (10 µM, 48 h).
The neuroprotection against glutamate excitotoxicity was significantly attenuated by simultaneous incubation with the α7 nAChR antagonist MLA (1 µM) (Figure 2B) or PP2 (5 µM), a Src family tyrosine kinase inhibitor (Figure 2C). These results support the hypothesis that α7 nAChR and Src family tyrosine kinases were involved in the donepezil-induced protection.
Donepezil reduces glutamate-induced caspase-3 activation
We examined the effect of donepezil on glutamate-induced activation of caspase-3 using caspase-3-like enzyme activity spectrophotometric assay. Glutamate (30 µM) activated caspase-3-like activities and this activation reached its peak around 3 h after treatment. Pre-treatment with donepezil for 48 h prevented glutamate-induced caspase-3 activation significantly after 3 h treatment with glutamate (Figure 2D). Immunoblot assays demonstrated that cleaved caspase-3, an activated form of caspase-3, was also increased by glutamate and inhibited by donepezil pre-treatment (Figure 2E).
Donepezil attenuates glutamate-induced Ca2+ influx; α7 nAChR and a Src family tyrosine kinase, but not PI3 kinase, are involved
NMDA receptor stimulation causes intracellular overload of Ca2+ and this raised intracellular Ca2+ subsequently activates caspase-3. Glutamate-induced Ca2+ influx was therefore measured. To determine the cellular mechanisms responsible for the protective effect of donepezil, fura-2 was used to measure glutamate-mediated Ca2+ influx in neurons. Neurons were pre-incubated for 48 h with either control media or donepezil (10 µM), and then neurons were stimulated with a toxic dose of glutamate (30 µM). Glutamate stimulation induced a significant increase of intracellular Ca2+ in a concentration-dependent manner (Figure S1). Pre-treatment with donepezil significantly reduced the glutamate-induced increase of intracellular Ca2+ (Figure 3). This attenuation of the intracellular Ca2+ overload contributes at least in part to the reduction of the glutamate-induced neuronal death. Reduction of the glutamate-induced Ca2+ influx was inhibited by the α7 nAChR antagonist MLA (Figure 3A). Pre-treatment with nicotine also attenuated the glutamate-induced increase of intracellular Ca2+, which was also inhibited by MLA (Figure S2).
Figure 3.
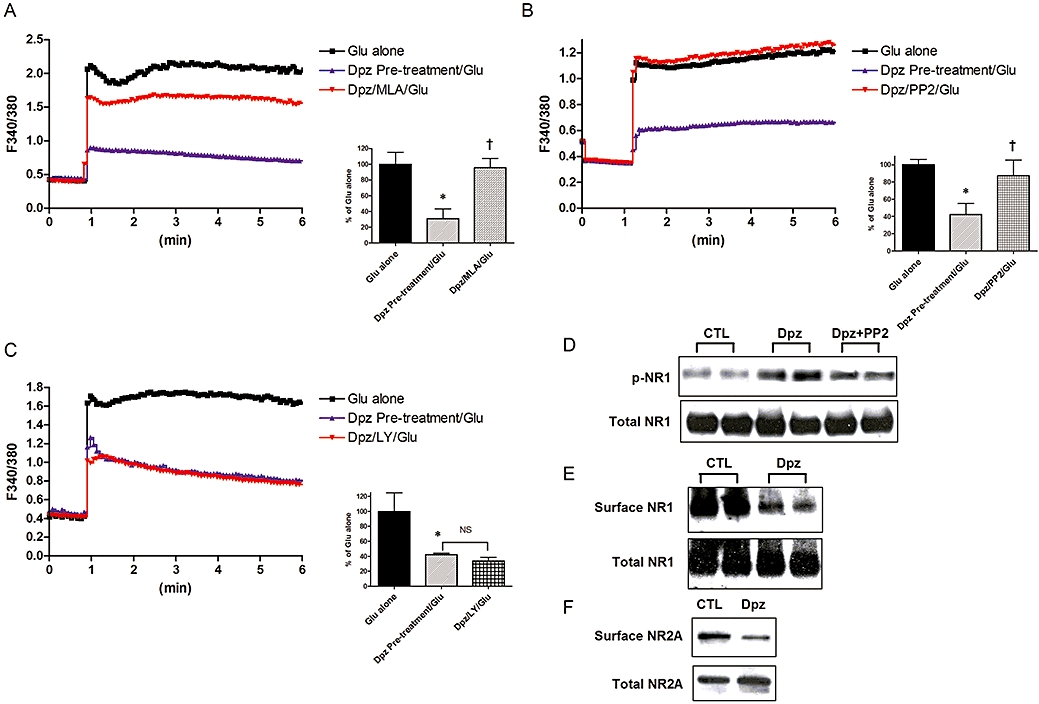
Donepezil (Dpz) abolished glutamate (Glu)-induced Ca2+ influx, which was mediated, at least in part, via Src family kinases. Dpz also induced NMDA receptor internalization and phosphorylation. Representative data of NMDA receptor subunit level altered by AChE inhibitors are shown. (A) Dpz (10 µM, 48 h) pre-treatment attenuated Glu-induced Ca2+ influx. Dpz was washed out during Glu administration. The effect of Dpz was also attenuated by MLA (1 µM). n = 20. *P < 0.05 compared with Glu alone, †P < 0.05 compared with Dpz pre-treatment (10 µM, 48 h). Statistically significant differences between groups were determined by Kruskal–Wallis analysis of variance (anova) followed by Dunn's post-test. (B) PP2 (5 µM), a Src family tyrosine kinase inhibitor, attenuated Dpz-induced reduction of Glu receptor-mediated Ca2+ influx. n = 20. *P < 0.05 compared with Glu alone, †P < 0.05 compared with Dpz pre-treatment (10 µM, 48 h). Statistically significant differences between groups were determined by Kruskal–Wallis anova followed by Dunn's post-test. (C) Simultaneous administration of the PI3K inhibitor LY294002 (10 µM) had no effect on Dpz-induced reduction of Glu receptor-mediated Ca2+ influx. n = 20. *P < 0.05 compared with Glu alone; NS, not significant. Statistically significant differences between groups were determined by Kruskal–Wallis anova followed by Dunn's post-test. (D) NR1 was phosphorylated by Dpz (10 µM, 48 h), although the total level of NR1 was not altered. n= 6. *P < 0.05 compared with control (vehicle alone), †P < 0.05 compared with Glu alone. Statistically significant differences between groups were determined by Kruskal–Wallis ANOVA followed by Dunn's post-test. (E) Surface level of NR1 was also reduced by Dpz (10 µM, 48 h), although the total level of NR1 was not altered. (F) Surface level of NR2A was also reduced by Dpz (10 µM, 48 h), although total NR2A was not significantly altered.
The effect of donepezil was also inhibited by PP2 (5 µM) (Figure 3B). PP2 alone did not affect the glutamate-induced Ca2+ influx (data not shown). LY294002 (10 µM), a PI3K inhibitor, had no influence on the reducing effect of donepezil on glutamate-induced Ca2+ influx (Figure 3C).
Reduced level of surface NMDA receptors was mediated via α7 nAChRs
The reduced level of glutamate-induced Ca2+ influx might be mediated by the modulation of NMDA receptor function. It was previously shown that phosphorylated NR1 subunits enhance glutamate-induced Ca2+ influx. Therefore, the phosphorylation state of NR1 was investigated using a phospho-specific anti-NR1 antibody.
Tyrosine dephosphorylation can down-regulate the NR1/2A-containing receptors at the cell surface (Suvarna et al., 2005), and we hypothesized that NR1 would be dephosphorylated by donepezil. NR1 phosphorylation was, however, significantly higher after treatment with donepezil (10 µM, 48 h). NR1 phosphorylation by donepezil was significantly attenuated by PP2 (5 µM) (Figure 3D).
To measure reduced cell surface expression of NR1, cell surface proteins were biotinylated and immunoprecipitated by anti-NR1 antibody (1:500). Surface level of NR1 was significantly reduced by donepezil (10 µM, 48 h) (Figure 3E) but the total level of NR1 was not significantly affected.
Aβ-induced stimulation of α7 nAChRs activated tyrosine phosphatase, leading to the dephosphorylation of NR2B, which results in the internalization of NR2B (Snyder et al., 2005). However, surface NR2B was not detected in our culture (data not shown). As shown in other studies, NR2A also forms a complex with NR1 as an NMDA receptor and the expression is delayed compared with NR2B (Zhong et al., 1994; Liu et al., 2003). NR2A was detected in our culture, and the surface NR2A level was also decreased by donepezil (Figure 3F).
Non-toxic Ca2+ entry was not affected by donepezil pre-treatment
Low concentrations of glutamate also induced glutamate receptor-mediated Ca2+ entry. Glutamate (3 µM) caused Ca2+ influx which was below the level inducing toxicity (Figure 1A). The peak of the Ca2+ entry was not significantly inhibited by MK801, although the amplitude was rapidly decreased, as previously reported (Kume et al., 2002). Pre-treatment with donepezil had no influence upon the Ca2+ influx induced by low concentrations of glutamate (Figure 4A). High potassium concentrations also increased intracellular Ca2+ (Figure 4B and C). Donepezil slightly enhanced this increase of the intracellular Ca2+. High potassium in the presence of MK801 also increased intracellular Ca2+ (Figure 4B). Donepezil could not attenuate this increase of the intracellular Ca2+, indicating that almost all the donepezil-induced attenuation is mediated by NMDA receptors.
Figure 4.
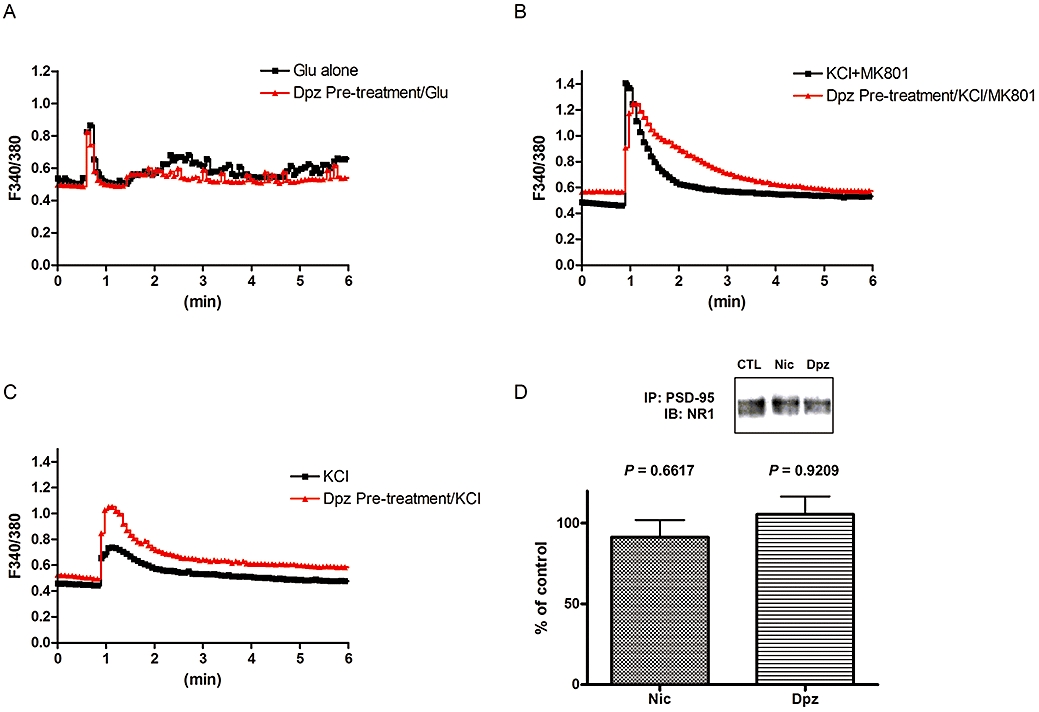
Donepezil (Dpz) or nicotine (Nic) had no influence upon Ca2+ influx induced by low concentrations of glutamate (Glu) or non-NMDA receptor-induced Ca2+ entry. (A) Ca2+ influx induced by a low concentration (3 µM), or non-toxic dose, of Glu was not attenuated by Dpz pre-treatment (10 µM, 48 h). (B) High potassium (KCl, 25 mM) with MK801 induced Ca2+ entry, which was not attenuated, but slightly enhanced by Dpz pre-treatment (10 µM, 48 h). (C) High potassium (KCl, 25 mM) induced Ca2+ entry, which was not attenuated by Dpz pre-treatment (10 µM, 48 h). (D) Synapse-related NR1 was not internalized by Dpz. Immunoprecipitation of the NR1 subunit protein with PSD-95 did not demonstrate any difference in membrane-bound NR1 subunits between Dpz (10 µM, 48 h) or Nic (10 µM, 48 h) treated or vehicle-treated cells. Statistically significant differences between groups were determined by Student's t-test compared with control (CTL).
However, the NMDA receptors involved in normal neuro-transmission might also be up-regulated by donepezil. To test this possibility, we measured PSD-95, a post-synaptic protein involved in clustering NMDA receptors (Kennedy, 1997). We investigated the localization of PDS-95 and NMDA receptor proteins. Immunoprecipitation of the NR1 subunit protein with PSD-95 did not demonstrate any difference in membrane-bound NR1 subunits between donepezil or nicotine treated, and vehicle-treated, cells (Figure 4D). Moreover, the co-localization of NR1 and PSD-95 was maintained even in the donepezil or nicotine-treated groups (Figure S3). From these data, we concluded that the synaptic NMDA receptors were not changed by donepezil.
Discussion
In the present study, we demonstrated that α7 nAChR stimulation by donepezil would lead to the internalization of NMDA receptors, by following the core receptor subunit, NR1. Such internalization might contribute to the attenuation of Ca2+ influx induced by excessive glutamate, the subsequent activation of caspase-3 and neuronal death.
There are at least two mechanisms of neuroprotection: one is the up-regulation of a defensive system and the other is the reduction of the toxic system. Nicotine and AChE inhibitors have been shown to attenuate neuronal death induced by glutamate or Aβ (Akaike et al., 1994; Shimohama et al., 1996; Kihara et al., 1997; 1998; 2001; 2004; Takada et al., 2003). For the former mechanism, we have previously demonstrated that α7 nAChR stimulation would activate PI3K, which phosphorylates the cell-survival pathway mediated by Akt/PKB and subsequently enhances the expression of the anti-apoptotic protein Bcl-2 (Kihara et al., 2001). AChE inhibitors such as donepezil or galantamine also exert a neuroprotective effect via this nAChR-PI3K system (Takada et al., 2003; Kihara et al., 2004; Takada-Takatori et al., 2006).
For the latter mechanism, reduction of NMDA receptor-mediated Ca2+ influx is likely to be one of the steps involved, as neuronal death induced by NMDA receptors has been shown to be mediated by increased Ca2+ influx, which subsequently activates caspase-3 (Yazawa et al., 2006).
Donepezil attenuated glutamate-induced neuronal death, which was inhibited by MLA and PP2 as previously reported (Takada et al., 2003). In addition, donepezil attenuated the glutamate-induced caspase-3 activation in our neuronal cultures. Glutamate-induced Ca2+ influx was also attenuated by donepezil, indicating that reduced influx of Ca2+ might be the cause of the attenuation of caspase-3 activation. Attenuation of the glutamate-induced Ca2+ influx was significantly inhibited by MLA, used in the concentration previously reported (Dajas-Bailador et al., 2000; Snyder et al., 2005), indicating the involvement of α7 nAChRs.
Attenuation of the glutamate-induced Ca2+ influx was not inhibited by LY294002. This pathway of NMDA receptor modulation by α7 nAChR stimulation is therefore independent of the up-regulation of the defensive system, which is mediated via the PI3K cascade.
Reduction of the NMDA receptor-mediated Ca2+ influx might inhibit normal, basal, neurotransmission or signal transduction. However, we propose that the internalization of NMDA receptors occurs at extrasynaptic sites rather than synaptic sites. PSD-95 co-localizes with NMDA receptors predominantly at synaptic sites and the NMDA receptor-PSD-95 complex blocks the internalization of NMDA receptors (Roche et al., 2001). This might be the reason why the Ca2+ influx sufficient to cause excitotoxicity, induced by high concentrations of glutamate, was inhibited by donepezil, but donepezil had no influence on the influx induced by low concentrations of glutamate or slightly increased the high potassium-induced increase of intracellular Ca2+.
We observed co-localization of NR1 and PSD-95. Immunoprecipitation data indicated that PSD-95-bound NR1 levels were not significantly reduced by donepezil. It was reported that NR1 is not directly associated with PSD-95, though another subunit such as NR2A or 2B might link those molecules (Huang et al., 2000). In the present study, the cell surface level of NR1 was decreased, although levels of synaptic NR1 may be maintained, and the low concentration of glutamate- or high potassium-induced Ca2+ influx was not affected, or conversely enhanced by donepezil. Thus, it is the toxic Ca2+ influx mediated by NMDA receptors that would chiefly be attenuated by nAChR stimulation.
It has been shown that most NR2A subunits are incorporated in synapses in mature neurons, whereas NR2B subunits predominate at extrasynaptic sites (Tovar and Westbrook, 1999). However, recent reports suggested that NR2A subunit-containing NMDA receptors contribute also to extrasynaptic NMDA receptors during in vitro differentiation (Mohrmann et al., 2000; Thomas et al., 2006). Hardingham et al. (2002) have proposed that NMDA supports neuronal survival, if it is acting at a synapse but that it triggers excitotoxicity, if acting at extrasynaptic sites. Extrasynaptic NR2A subunits might also contribute to the excitotoxicity and the reduced level of NR2A subunits, following exposure to AChE inhibitors, could contribute to the attenuation of this excitotoxicity.
It has been reported that the expression of NR2A mRNA increases after day 7 in culture, and that of NR2B increases between days 1 and 7, with little further change after day 7 (Zhong et al., 1994; Liu et al., 2003). In our cultures, experiments were performed at day 10 and NR2A was detected by immunoblot analysis. We cannot completely exclude the possibility that nAChR stimulation decreased the trafficking of NR2A to the cell surface, but the total level of NR2A was slightly increased, in contrast to the decrease of the surface level.
It has been demonstrated that a tyrosine-based motif is essential for internalization of NR1 or NR2 (Roche et al., 2001; Scott et al., 2004). A tyrosine residue of the NMDA receptor might be phosphorylated by a Src family tyrosine kinase, such as Fyn. Akt phosphorylation induced by α7 nAChR is mediated via a Src family tyrosine kinase (Kihara et al., 2001). In the present study, α7 nAChR-Src signalling induced phosphorylation and internalization of NR1. PP2 alone did not influence the glutamate-induced Ca2+ entry in our culture, indicating that NMDA receptors might be significantly phosphorylated only when nAChRs were stimulated.
As LTP requires NMDA receptor activation and Ca2+ currents, Src family-induced phosphorylation of NMDA receptors would be involved. Nicotine enhanced NMDA receptor-mediated currents in the hippocampus and this was accompanied by increased tyrosine phosphorylation of NR2B subunits (Yamazaki et al., 2006). It is not clear, however, whether the phosphorylated NR2B is synaptic or extrasynaptic in their report.
Aβ-induced α7 nAChR stimulation was shown to activate calcineurin, which then promoted the dephosphorylation and activation of the tyrosine phosphatase, striatal enriched phosphatase (STEP46). STEP46 promotes the dephosphorylation of Tyr1472 of NR2B. In regard to long-term depression (LTD), however, a calcineurin inhibitor was shown to have no effect on LTD of NMDA receptor-mediated excitatory postsynaptic potentials (Morishita et al., 2005).
NR2 subunits might also be modified by the α7 nAChR-STEP46 cascade, which might lead to the internalization of NR2, although dephosphorylation would occur at extrasynaptic sites, and synaptic NR2 might be maintained by Src-mediated phosphorylation. Under these conditions, synaptic transmission would be maintained and excess glutamate-induced excitotoxicity might be blocked.
The regulation of NMDA receptor-mediated Ca2+ current is likely to be complex and finely controlled. The balance between Src-mediated phosphorylation and calcineurin-dependent dephosphorylation would determine the phosphorylation state and the surface level of NMDA receptors and subsequently the level of glutamate-induced Ca2+ influx and cell response.
In AD brains, it was revealed that the protein levels of NMDA receptors are reduced (Nordberg, 1992; Hynd et al., 2004). This might be because Aβ is increased in an AD brain, which causes α7 nAChR-mediated internalization of NMDA receptors. From another point of view, however, the residual neurons exhibit down-regulated NMDA receptors. That is, internalization of NMDA receptors might be a biological response to counter excitotoxicity. Phosphorylated NR2A and NR2B are reported to be reduced in AD when compared with controls (Sze et al., 2001). In this report, it was demonstrated that losses of NMDA receptor subunit proteins correlated with changes in synaptobrevin levels (a presynaptic protein). NMDA receptors in synaptic regions would be responsible for cognitive functions and would be reduced in AD brains.
In conclusion, phosphorylation of NR1 by Src family tyrosine kinase was induced by α7 nAChR stimulation and promoted NMDA receptor internalization. This would protect against glutamate excitotoxicity by reducing Ca2+ influx through these receptors.
Acknowledgments
We thank Dr Kengo Uemura (Department of Neurology, Alzheimer Disease Research Unit, Massachusetts General Hospital, Boston, MA, USA) for kindly providing support for the biotinylation technique and for helpful suggestions. We are grateful to Eisai Co., Ltd. for providing donepezil.
Glossary
Abbreviations
- AD
Alzheimer's disease
- AChE
acetylcholinesterase
- Aβ
beta amyloid
- DMXBA
3-(2,4-dimethoxybenzylidene)-anabaseine
- LDH
lactate dehydrogenase
- LTP
long-term potentiation
- LTD
long term depression
- MAP2
microtubule associated protein 2
- MLA
methyllycaconitine
- nAChR
nicotinic acetylcholine receptor
- PI3K
phosphatidylinositol 3-kinase
- PP2
[4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine]
- PSD-95
post synaptic density protein (95 kDa)
- SP
senile plaques
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Glutamate-induced Ca2+ influx, which was mediated via, at least in part, NMDAR. A: Glutamate-induced Ca2+ entry was increased in a concentration dependent manner. B, C: Effects of MK801 on glutamate-induced Ca2+ entry. High concentration of glutamate induced high Ca2+ entry, and MK801 attenuated the peak of the amplitude. When low concentration of glutamate was applied, peak was not suppressed by MK801, although rapidly the amplitude was decreased.
Figure S2 Nicotinic acetylcholine receptor stimulation abolished glutamate-induced Ca2+ influx, which was mediated via, at least in part, α7 nAChR. NMDAR internalization and phosphorylation were induced by nicotine. Representative data of NMDA receptor subunit levels altered by nicotine were shown. A: Nicotine (Nic, 10 µM 48 h) pre-treatment abolished glutamate (Glu, 30 µM)-induced Ca2+ entry. Nicotine was washed out during glutamate administration. Effect of nicotine was attenuated by MLA (1 µM), indicating the involvement of α7 nAChR. Average F340/F380 for each treatment at 5 s was statistically analyzed. n = 20. *P < 0.05 compared with glutamate alone, †P < 0.05 compared with nicotine pre-treatment (Nic, 10 µM 48 h). Statistically significant differences between groups were determined by Kruskal–Wallis non-parametric analysis of variance with post hoc multiple comparisons. B: NR1 was phosphorylated by nicotine (Nic, 10 µM 48 h) treatment, although total level of NR1 was not altered. Surface level of NR1 was reduced by nicotine (Nic, 10 µM 48 h). Phosphorylation of NR1 induced by nicotine was inhibited by PP2 (5 µM) or MLA (1µM). Reduction of the surface level of NR1 induced by nicotine was also inhibited by PP2 or MLA. C: Surface level of NR1 was reduced by DMXBA (10 µM 48 h), although total NR1 was not significantly altered. D: Surface level of NR2A was reduced by nicotine (Nic, 10 µM 48 h), although total NR2A was not significantly altered.
Figure S3 Confocal images of double immunostaining. Co-localizations of NR1 and PSD-95 were found even after donepezil (Dpz, 10 µM 48 h) or nicotine (Nic, 10 µM 48 h) treatment. In contrast, some NR1 and PSD-95 were not clearly co-localized in control cells (ctl). Neuronal cells were probed with antibodies directed against NR1 and PSD-95. Cy2 and Cy3 images were acquired simultaneously and showed that NR1-positive (red) neurons were also PSD-95 positive (green), even after nicotine (Nic, 10 µM 48 h) or donepezil (Dpz, 10 µM 48 h) treatment.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Akaike A, Tamura Y, Yokota T, Shimohama S, Kimura J. Nicotine-induced protection of cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1994;644:181–187. doi: 10.1016/0006-8993(94)91678-0. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asomugha C, Linn D, Linn C. ACh receptors link two signaling pathways to neuroprotection against glutamate-induced excitotoxicity in isolated RGCs. J Neurochem. 2010;112:214–226. doi: 10.1111/j.1471-4159.2009.06447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C, Meltzer J, Houston F, Orr G, McGann K, Wenk G. Chronic treatment of old rats with donepezil or galantamine: effects on memory, hippocampal plasticity and nicotinic receptors. Neuroscience. 2000;99:17–23. doi: 10.1016/s0306-4522(00)00180-9. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Lima P, Wonnacott S. The alpha7 nicotinic acetylcholine receptor subtype mediates nicotine protection against NMDA excitotoxicity in primary hippocampal cultures through a Ca(2+) dependent mechanism. Neuropharmacology. 2000;39:2799–2807. doi: 10.1016/s0028-3908(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Dasgupta P, Rastogi S, Pillai S, Ordonez-Ercan D, Morris M, Haura E, et al. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest. 2006;116:2208–2217. doi: 10.1172/JCI28164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Rajan A, Radcliffe K, Yakehiro M, Dani J. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Hardingham G, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Kazui H, Matsumoto K, Nakano Y, Yasuda M, Mori E. Does donepezil treatment slow the progression of hippocampal atrophy in patients with Alzheimer's disease? Am J Psychiatry. 2005;162:676–682. doi: 10.1176/appi.ajp.162.4.676. [DOI] [PubMed] [Google Scholar]
- Huang Y, Won S, Ali D, Wang Q, Tanowitz M, Du Q, et al. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26:443–455. doi: 10.1016/s0896-6273(00)81176-9. [DOI] [PubMed] [Google Scholar]
- Hynd M, Scott H, Dodd P. Differential expression of N-methyl-D-aspartate receptor NR2 isoforms in Alzheimer's disease. J Neurochem. 2004;90:913–919. doi: 10.1111/j.1471-4159.2004.02548.x. [DOI] [PubMed] [Google Scholar]
- Kennedy M. The postsynaptic density at glutamatergic synapses. Trends Neurosci. 1997;20:264–268. doi: 10.1016/s0166-2236(96)01033-8. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, et al. Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann Neurol. 1997;42:159–163. doi: 10.1002/ana.410420205. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Urushitani M, Sawada H, Kimura J, Kume T, et al. Stimulation of alpha4beta2 nicotinic acetylcholine receptors inhibits beta-amyloid toxicity. Brain Res. 1998;792:331–334. doi: 10.1016/s0006-8993(98)00138-3. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, et al. alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- Kihara T, Sawada H, Nakamizo T, Kanki R, Yamashita H, Maelicke A, et al. Galantamine modulates nicotinic receptor and blocks Abeta-enhanced glutamate toxicity. Biochem Biophys Res Commun. 2004;325:976–982. doi: 10.1016/j.bbrc.2004.10.132. [DOI] [PubMed] [Google Scholar]
- Kume T, Nishikawa H, Taguchi R, Hashino A, Katsuki H, Kaneko S, et al. Antagonism of NMDA receptors by sigma receptor ligands attenuates chemical ischemia-induced neuronal death in vitro. Eur J Pharmacol. 2002;455:91–100. doi: 10.1016/s0014-2999(02)02582-7. [DOI] [PubMed] [Google Scholar]
- Liu A, Zhuang Z, Hoffman P, Bai G. Functional analysis of the rat N-methyl-D-aspartate receptor 2A promoter: multiple transcription starts points, positive regulation by Sp factors, and translational regulation. J Biol Chem. 2003;278:26423–26434. doi: 10.1074/jbc.M211165200. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, Hatt H, Gottmann K. Developmental regulation of subunit composition of extrasynaptic NMDA receptors in neocortical neurones. Neuroreport. 2000;11:1203–1208. doi: 10.1097/00001756-200004270-00012. [DOI] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka R. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T. Nicotine-induced phosphorylation of Akt through epidermal growth factor receptor and Src in PC12h cells. J Neurochem. 2002;83:1372–1379. doi: 10.1046/j.1471-4159.2002.01248.x. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Neuroreceptor changes in Alzheimer disease. Cerebrovasc Brain Metab Rev. 1992;4:303–328. [PubMed] [Google Scholar]
- Roche K, Standley S, McCallum J, Dune Ly C, Ehlers M, Wenthold R. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Sawada H, Ibi M, Kihara T, Urushitani M, Akaike A, Kimura J, et al. Dopamine D2-type agonists protect mesencephalic neurons from glutamate neurotoxicity: mechanisms of neuroprotective treatment against oxidative stress. Ann Neurol. 1998;44:110–119. doi: 10.1002/ana.410440117. [DOI] [PubMed] [Google Scholar]
- Scott D, Michailidis I, Mu Y, Logothetis D, Ehlers M. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S, Akaike A, Kimura J. Nicotine-induced protection against glutamate cytotoxicity. Nicotinic cholinergic receptor-mediated inhibition of nitric oxide formation. Ann N Y Acad Sci. 1996;777:356–361. doi: 10.1111/j.1749-6632.1996.tb34445.x. [DOI] [PubMed] [Google Scholar]
- Shirakawa H, Katsuki H, Kume T, Kaneko S, Ito J, Akaike A. Regulation of N-methyl-D-aspartate cytotoxicity by neuroactive steroids in rat cortical neurons. Eur J Pharmacol. 2002;454:165–175. doi: 10.1016/s0014-2999(02)02493-7. [DOI] [PubMed] [Google Scholar]
- Snyder E, Nong Y, Almeida C, Paul S, Moran T, Choi E, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Suvarna N, Borgland S, Wang J, Phamluong K, Auberson Y, Bonci A, et al. Ethanol alters trafficking and functional N-methyl-D-aspartate receptor NR2 subunit ratio via H-Ras. J Biol Chem. 2005;280:31450–31459. doi: 10.1074/jbc.M504120200. [DOI] [PubMed] [Google Scholar]
- Sze C, Bi H, Kleinschmidt-DeMasters B, Filley C, Martin L. N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. J Neurol Sci. 2001;182:151–159. doi: 10.1016/s0022-510x(00)00467-6. [DOI] [PubMed] [Google Scholar]
- Takada Y, Yonezawa A, Kume T, Katsuki H, Kaneko S, Sugimoto H, et al. Nicotinic acetylcholine receptor-mediated neuroprotection by donepezil against glutamate neurotoxicity in rat cortical neurons. J Pharmacol Exp Ther. 2003;306:772–777. doi: 10.1124/jpet.103.050104. [DOI] [PubMed] [Google Scholar]
- Takada-Takatori Y, Kume T, Sugimoto M, Katsuki H, Sugimoto H, Akaike A. Acetylcholinesterase inhibitors used in treatment of Alzheimer's disease prevent glutamate neurotoxicity via nicotinic acetylcholine receptors and phosphatidylinositol 3-kinase cascade. Neuropharmacology. 2006;51:474–486. doi: 10.1016/j.neuropharm.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Tamura Y, Sato Y, Akaike A, Shiomi H. Mechanisms of cholecystokinin-induced protection of cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1992;592:317–325. doi: 10.1016/0006-8993(92)91691-7. [DOI] [PubMed] [Google Scholar]
- Thomas C, Miller A, Westbrook G. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Tovar K, Westbrook G. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura K, Kitagawa N, Kohno R, Kuzuya A, Kageyama T, Chonabayashi K, et al. Presenilin 1 is involved in maturation and trafficking of N-cadherin to the plasma membrane. J Neurosci Res. 2003;74:184–191. doi: 10.1002/jnr.10753. [DOI] [PubMed] [Google Scholar]
- Wada T, Naito M, Kenmochi H, Tsuneki H, Sasaoka T. Chronic nicotine exposure enhances insulin-induced mitogenic signaling via up-regulation of alpha7 nicotinic receptors in isolated rat aortic smooth muscle cells. Endocrinology. 2007;148:790–799. doi: 10.1210/en.2006-0907. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek M, et al. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Jia Y, Niu R, Sumikawa K. Nicotine exposure in vivo induces long-lasting enhancement of NMDA receptor-mediated currents in the hippocampus. Eur J Neurosci. 2006;23:1819–1828. doi: 10.1111/j.1460-9568.2006.04714.x. [DOI] [PubMed] [Google Scholar]
- Yazawa K, Kihara T, Shen H, Shimmyo Y, Niidome T, Sugimoto H. Distinct mechanisms underlie distinct polyphenol-induced neuroprotection. FEBS Lett. 2006;580:6623–6628. doi: 10.1016/j.febslet.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Zhong J, Russell S, Pritchett D, Molinoff P, Williams K. Expression of mRNAs encoding subunits of the N-methyl-D-aspartate receptor in cultured cortical neurons. Mol Pharmacol. 1994;45:846–853. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.