

Abstract
BACKGROUND AND PURPOSE
The angiotensin II type 1 (AT1) receptor belongs to family A of 7 transmembrane (7TM) receptors. The receptor has important roles in the cardiovascular system and is commonly used as a drug target in cardiovascular diseases. Interaction of 7TM receptors with G proteins or β-arrestins often induces higher binding affinity for agonists. Here, we examined interactions between AT1A receptors and β-arrestins to look for differences between the AT1A receptor interaction with β-arrestin1 and β-arrestin2.
EXPERIMENTAL APPROACH
Ligand-induced interaction between AT1A receptors and β-arrestins was measured by Bioluminescence Resonance Energy Transfer 2. AT1A-β-arrestin1 and AT1A-β-arrestin2 fusion proteins were cloned and tested for differences using immunocytochemistry, inositol phosphate hydrolysis and competition radioligand binding.
KEY RESULTS
Bioluminescence Resonance Energy Transfer 2 analysis showed that β-arrestin1 and 2 were recruited to AT1A receptors with similar ligand potencies and efficacies. The AT1A-β-arrestin fusion proteins showed attenuated G protein signalling and increased agonist binding affinity, while antagonist affinity was unchanged. Importantly, larger agonist affinity shifts were observed for AT1A-β-arrestin2 than for AT1A-β-arrestin1.
CONCLUSION AND IMPLICATIONS
β-Arrestin1 and 2 are recruited to AT1A receptors with similar ligand pharmacology and stabilize AT1A receptors in distinct high-affinity conformations. However, β-arrestin2 induces a receptor conformation with a higher agonist-binding affinity than β-arrestin1. Thus, this study demonstrates that β-arrestins interact with AT1A receptors in different ways and suggest that AT1 receptor biased agonists with the ability to recruit either of the β-arrestins selectively, would be possible to design.
Keywords: AT1 receptor, β-arrestins, fusion proteins, ternary complex, high-affinity conformation
Introduction
The angiotensin II type 1 (AT1) receptor (nomenclature follows Alexander et al., 2009) belongs to family A of 7 transmembrane (7TM) receptors and plays a central role in blood pressure regulation and fluid homeostasis (Hunyady and Catt, 2006). The pathophysiological importance of AT1 receptors is emphasized by the use of AT1 receptor blockers and angiotensin converting enzyme inhibitors in treatment of cardiovascular diseases such hypertension and cardiac failure (Burnier and Brunner, 2000; Weir, 2007).
The AT1 receptor signals through two major pathways, namely the Gq protein-dependent signalling pathway, leading to inositol phosphate (IP) accumulation and calcium release and the G protein-independent signalling, which involves recruitment of the scaffolding proteins, β-arrestin1 and β-arrestin2 (Aplin et al., 2008; 2009; Hunyady and Catt, 2006).
The two β-arrestin isoforms share 78% sequence identity and regulate the majority of 7TM receptors (Gurevich and Gurevich, 2006). The binding of β-arrestins to AT1 receptors hinders these receptors from further interaction with G proteins. This process is termed desensitization. Besides terminating G protein-dependent signalling, β-arrestin1 and β-arrestin2 also function as scaffolds, as they mediate internalization and initiate a second ‘wave’ of signalling involving many kinases and signalling molecules (Lefkowitz and Shenoy, 2005; Zhai et al., 2005; Aplin et al., 2007; Xiao et al., 2007; Hansen et al., 2008).
The two β-arrestin isoforms may have different, and in some cases opposite, properties in terms of kinase activation and internalization (Ahn et al., 2004; Kuo et al., 2006; Fan et al., 2007; Kumar et al., 2007; Sneddon and Friedman, 2007). It has been shown that down regulation of β-arrestin2 leads to a decrease in AT1 receptor-mediated activation of the extracellular signal-regulated kinase (ERK1/2), while down regulation of β-arrestin1 markedly increases ERK1/2 activation (Ahn et al., 2004). Recently it has also been demonstrated that β-arrestin2 increases smooth muscle cell proliferation and migration, thereby aggravating atherosclerosis, while β-arrestin1 was shown to have the opposite effect (Kim et al., 2008). These findings clearly suggest that it could be beneficial to design drugs working as biased agonists that selectively recruit either β-arrestin1 or -2. Furthermore, the different properties of β-arrestin1 and β-arrestin2 in modulating receptor signalling may to some extent reflect different modes of interaction between the receptor and the two β-arrestin isoforms.
Based on their interaction with β-arrestins, the 7TM receptors are divided into two classes. Class A receptors, which include receptors such as the β2 adrenoceptor and vasopressin 1a (V1a) receptor, interact transiently with β-arrestins. In contrast, class B receptors, such as the AT1 and the vasopressin 2 (V2) receptor, interact more stably with β-arrestins (Oakley et al., 2000). Furthermore, class A and B receptors differ in their preference towards the two β-arrestin isoforms. Class A receptors have been shown to bind β-arrestin2 more tightly than β-arrestin1, whereas class B receptors bind β-arrestin1 and 2 equally well (Oakley et al., 2000). However, these studies do not address how β-arrestins affect the receptor conformation or how they affect ligand binding properties.
It has been observed that both G proteins and β-arrestins are able to increase receptor affinity for agonists, as described in the ternary complex model (Samama et al., 1993; Gurevich et al., 1997; Martini et al., 2002; Jorgensen et al., 2005). The interaction between 7TM receptors and β-arrestin has been studied by using fusion proteins, where the C terminus tail of the 7TM receptor was fused directly to the N terminus end of β-arrestin.
In order to study the interactions between AT1A receptors and β-arrestins quantitatively, we used two strategies: β-arrestin recruitment-based Bioluminescence Resonance Energy Transfer (BRET) assay and studies of fusion proteins between AT1A receptors and β-arrestin1 or 2. The results showed that β-arrestin1 and 2 were both recruited to AT1A receptor with similar ligand potencies and efficacies. Furthermore, we found that the AT1A-β-arrestin2 fusion protein displayed a higher affinity for agonists than the AT1a-β-arrestin1 protein, a difference most likely to reflect the induction of different receptor conformations.
Methods
Molecular biology
All constructs are expressed in pcDNA3.1. The wild-type (WT) HA-tagged rat AT1A receptor was made by inserting cDNA encoding the peptide sequence of the HA tag in the N-terminus of the rat AT1A receptor. To create the AT1A-β-arrestin fusion proteins, the coding sequence of HA-tagged rat AT1A receptor lacking the stop codon was digested with NheI and BamHI and inserted into the N terminus end of cDNA sequence of β-arrestin1 and β-arrestin2 respectively. All constructs were verified by sequencing. The AT1A receptor fused to Rluc is described in (Hansen et al., 2004b).
In our experiment we used two different cell types: COS-7 cells and HEK293T cells. These cell lines provide good models for studying binding characteristics and protein recruitment, because they are easily transfected and easy to handle. The assay of IP accumulation and radioligand binding assays include many washing steps, and COS-7 cells were used in these assays, as they adhere more tightly to the wells. For the BRET experiments the HEK293 cells were preferred, because they presentthe receptor better on the cell surface than COS-7 cells
β-arrestin recruitment assay
HEK293T cells (4.5 × 106) were seeded into a p10 dish and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, Gentamycin (0.5 mg·mL−1) and L-glutamine (2 mM). After 24 h, the cells were transfected using Polyethylenimine (PEI). We used 1 µg of AT1A-Rluc and 3ug of either GFP2-β-arrestin1 or GFP2-β-arrestin2.
Forty-eight hours post transfection, HEK293T cells were washed with PBS, detached with PBS/trypsin-EDTA (0.25% trypsin; 1 mM EDTA, Invitrogen), harvested by centrifugation (5 min, 1000× g), resuspended in PBS supplemented with 0.5 mM Ca2+ and 0.5 mM Mg2+ and incubated at room temperature on a shaker (app. 250 r.p.m.) until the time of the experiments. The resuspended cells were distributed in 96-well microplates (black/white optiplate, PerkinElmer) and incubated in the presence or absence of different ligands for 15 min after agonist addition for dose–response curves. This time was chosen after preliminary time-dependent β-arrestin recruitment experiments showed that the BRET response reached a plateau after approximately 10–15 min (data not shown).
DeepBlueC coelenterazine (Coelenterazine 400a, Biosynth) was added 2 s before reading, using an injector at a final concentration of 5 µM. Measurement of Rluc-mediated luminescence and GFP2-mediated emission from each well was made using a Tecan Infinite F500 microplate reader (Tecan Group Ltd, Männedorf, Switzerland). The BRET ratio was determined by calculating the ratio of the light emitted by GFP2 (515 nm) over the light emitted by the Rluc (410 nm). The background BRET signal from Rluc was determined by coexpressing the Rluc construct with empty vector, and the BRET2 ratio generated from this transfection was subtracted from all other BRET2 ratios. The maximum BRET2 response generated by angiotensin II (Ang II) was set to 100% and the response mediated by all the other ligands was normalized to this value.
Immunocytochemistry and confocal microscopy
HEK293 cells were grown in DMEM supplemented with L-glutamine and 10% FBS. On day 1, cells were seeded in 10 cm dishes, 4 × 106 cells per dish in 10 mL medium. On day 2, cells were transfected with 5 µg HA-tagged WT AT1A receptor or AT1A-β-arrestin1 or AT1A-β-arrestin2 using Lipofectamine2000 (Invitrogen) according to the manufacturer's protocol. For transfection, DNA and Lipofectamine2000 were both diluted in Optimem (Invitrogen) and medium was changed on cells 4 h after transfection. On day 3, cells were seeded on poly-L-lysine coated coverslips (Corning) in 2 mL medium. Cells were grown until about 50% confluency and immunocytochemistry was performed.
To visualize receptors expressed on the cell surface, an antibody feeding assay was performed. Cells were incubated for 30 min at 37°C with anti-HA11 antibody (1:1000, Covance), stimulated with Ang II or left untreated for 30 min, washed with PBS and fixed with 4% paraformaldehyde in PBS. Cells were subsequently blocked and permeabilized in blotting solution (50 mM Tris-Cl pH 7.5, 1 mM CaCl2, 3% milk, 0.1% Triton X-100) for 20 min and stained with fluorescently conjugated Alexa anti-IgG1 488 (green) antibody (1:500 in blotting solution, Invitrogen) for 45 min. Coverslips were mounted onto glass slides using mounting media from Vectashield containing 4′,6′-diamidino-2-phenylidole (DAPI).
To visualize total amount of receptor protein, cells were initially washed with PBS, fixed and permeabilized as above, and then incubated with anti-HA-11 antibody (1:1000 in blotting solution) for 1 h. Cells were washed three times in Tris-buffered saline with 1 mM CaCl2, incubated with secondary antibody and mounted as described above. Mounted slides were analysed on an LSM510 laser confocal microscope (Zeiss) using the 63x oil objective.
PI hydrolysis assay
COS-7 cells (2.5 × 106) were seeded in a p10 dish and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, penicillin (50 units·mL−1), streptomycin (50 units·mL−1) and L-glutamine (2 mM). After 24 h, the cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. The day after transfection, cells were split into poly-D-lysine coated 96-well plates (50 000 cells per well) in inositol-free DMEM supplemented with non-essential amino acids, 10% fetal calf serum and myo-[2-3H]inositol (2 µCi·mL−1 medium). The following day, cells were washed with Hanks balanced salt solution (HBSS), supplemented with 1 mM CaCl2, 1 mM MgCl2, 10 mM LiCl (HBSS++) and incubated with HBSS++ for 20 min at 37°C. The buffer was aspirated and cells were incubated with increasing concentrations of ligands for 20 min at 37°C. Ligands were removed, and cells were incubated on ice with formic acid (10 mM) for at least 45 min. 20 µL of the lysis solution was transferred to a solid white 96-well plate, and 80 µL of freshly diluted SPA YSi beads (12 mg·mL−1, GE Healthcare) were added. The plates were shaken vigorously on a shaker for half an hour, and incubated for at least 8 h at room temperature prior to measurement. The IP response was normalized to maximum Ang II response observed for the WT AT1A receptor.
Competition radioligand binding assay
This assay was performed as described previously (Hansen et al., 2004a). Briefly, COS-7 cells were transfected and seeded into 48-well plates as described above for the IP hydrolysis assay. Following day, cells were washed once with cold HBSS, supplemented with CaCl2 (0.9 mM) and MgCl2 (1.05 mM) and cooled at 4°C for 30 min. The HBSS was aspirated and cells were incubated on ice for 4 h with 1 nM [tyrosyl-3,5-3H]Ang II (5-isoleucine) or 10−11 M of [125I]-SI-Ang II and increasing amounts of unlabelled ligand. Each concentration point was performed in duplicate. After incubation, cells were washed twice in cold HBSS, before the addition of 0.5 mL lysis buffer (1.0% Triton X-100, 50 mM Tris/HCl, pH 7.5, 100 mM NaCl and 5 mM EDTA) for 30–60 min at room temperature. The lysis buffer was transferred to scintillation tubes containing 8 mL of Ultima Gold (PerkinElmer).
Data analysis
Data are shown as means ± SEM. Results were analysed by non-linear regression using Prism GraphPad Software. Values for inhibition and dissociation constant (Ki and Kd) were estimated from competition binding by using the equations: Kd= IC50-L and Ki= IC50/(1 + L/Kd), where L is the concentration of radioligand. Values of P < 0.05 were considered to show significant differences between means.
Materials
Ang II, Ang III (Val4)Ang III and telmisartan were all from Sigma-Aldrich (St. Louis, MO, USA). SII-Ang II (Sar-Arg-Val-Ile-Ile-His-Pro-Ile-COOH) was synthesized at Cleveland Clinic, Lerner Research Institute (Cleveland, OH, USA). [3H]-Ang II (tyrosyl-3,5-[3H]Ang II 5-isoleucine) was from GE Healthcare while [125I]-SI-Ang II (Sar-Arg-Val-Tyr-Val-His-Pro-Ile-COOH) was from PerkinElmer.
Results
Recruitment of β-arrestins 1 and 2 and G protein coupling to AT1A receptors
To characterize the interaction between β-arrestin1 and 2 and AT1A receptors, we co-expressed the C terminally Rluc-tagged AT1A receptor (AT1A-Rluc) with N terminally GFP2-tagged β-arrestin1 and 2 (GFP2-β-arrestin1 and GFP2-β-arrestin2), respectively, and measured the ligand-induced recruitment of β-arrestins using a BRET2 assay (Figure 1A and B). The expression of GFP2-β-arrestin1 and GFP2-β-arrestin2 was comparable in these experiments when measured by GFP2 as was the AT1-Luc expression when measured by coelenterazine h luminescence as described previously (data not shown and Jensen et al., 2002). We chose to study agonists with different potencies and efficacies with respect to Gq protein activation, measured by IP hydrolysis assay (Figure 1C). Accordingly, we used the following ligands: Ang II (full agonist with high potency); Ang III and (Val4) Ang III (full agonists with low potency), SII-Ang II (biased agonist with low binding affinity and no agonist ability on G protein activation) (Holloway et al., 2002; Wei et al., 2003; Hansen et al., 2008) and finally the high-affinity antagonist, telmisartan.
Figure 1.
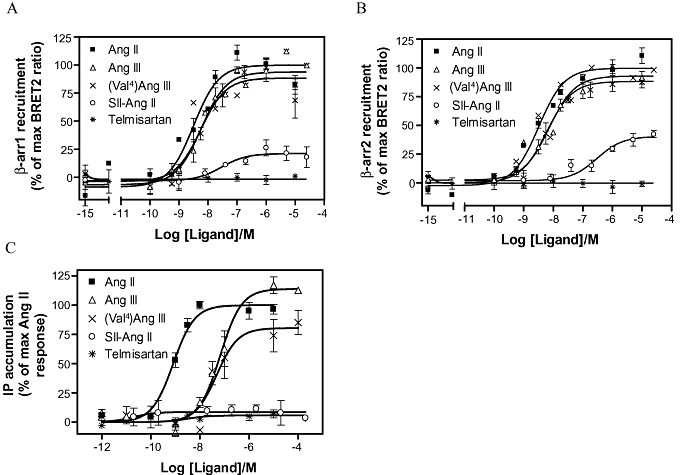
Ligand-induced β-arrestin1 and 2 recruitment and G protein coupling to WT AT1A receptor. (A) and (B) The BRET2 response measured between the AT1A-luc and GFP2-β-arrestin1 (β−arr1) and GFP2-β-arrestin2 (β-arr2), as a function of ligand concentration. The following ligands were used: Ang II, Ang III (Val4)Ang III, SII-Ang II and telmisartan. (C) The AT1A receptor-mediated Gq protein coupling was measured using a IP hydrolysis assay in response to ligand stimulation. Values are reported as a percentage of the maximum response to Ang II. Potencies and relative efficacies are reported in Table 1 and experimental details are described in the Methods section. AT1, angiotensin II type 1; BRET2, Bioluminescence Resonance Energy Transfer 2; IP, inositol phospholipid; WT, wild type.
As depicted in Figure 1A and B, β-arrestin1 and 2 were recruited equally well to the AT1A receptor in response to the tested ligands; the corresponding pEC50 values and efficacies are shown in Table 1. As observed for the Gq protein activation, both Ang III and (Val4)Ang III were low-potency full agonists with respect to β-arrestin1 and 2 recruitment (Figure 1). In our experiments, the biased agonist SII-Ang II showed only weak ability to recruit β-arrestin1 and 2, and was also unable to induce G protein activation. Several studies show that SII-Ang II can act as both a full or a partial agonist with respect to β-arrestin recruitment (Holloway et al., 2002; Wei et al., 2003; Rajagopal et al., 2006). This discrepancy remains to be fully elucidated but it has been shown that the expression of specific G protein kinases may influence the result (Kim et al., 2005). Telmisartan produced no response in any of the assays, which was consistent with previous observations (Hansen et al., 2008).
Table 1.
AT1A receptor-mediated β-arrestin recruitment and G protein coupling
| β-arrestin1 recruitment | β-arrestin2 recruitment | Gq coupling | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligands | pEC50 | Relative efficacy | n | pEC50 | Relative efficacy | n | pEC50 | Relative efficacy | n |
| Ang II | 8.4 ± 0.3 | 100 ± 0.0 | 7 | 8.5 ± 0.2 | 100 ± 0.0 | 7 | 9.0 ± 0.1 | 100 ± 0.0 | 5 |
| Ang III | 8.3 ± 0.2 | 90.5 ± 12.7 | 7 | 8.2 ± 0.4 | 86.1 ± 14.4 | 7 | 7.0 ± 0.1 | 122 ± 6.8 | 3 |
| (Val4)Ang III | 8.3 ± 0.3 | 86.6 ± 18.2 | 7 | 8.3 ± 0.4 | 89.9 ± 12.4 | 7 | 7.5 ± 0.2 | 64.2 ± 4.4 | 3 |
| SII-Ang II | 7.5 ± 1.2 | 27.4 ± 19.4 | 7 | 6.8 ± 0.7* | 38.2 ± 14.2 | 7 | NR | 3 | |
| Telmisartan | NR | 7 | NR | 7 | NR | 3 | |||
The average pEC50± SEM and relative efficacy ± SEM values are reported. Relative efficacies are calculated as a percentage of maximum Ang II response. Dose-response curves are shown in Figure 1. Statistical analysis was performed by one-tailed paired t-test to determine significant differences between GFP2-β-arrestin1 and GFP2-β-arrestin2 recruitment (one-tailed, paired Student's t-test). *P < 0.05.
AT1, angiotensin II type 1; NR, no response.
Subcellular localization of fusion constructs
To further analyse the interaction between AT1A receptors and β-arrestins, we constructed fusion proteins between AT1A receptors and β-arrestin1 and 2 respectively (Figure 2). The localization of the AT1A receptor fusion proteins was visualized using immunocytochemistry and confocal microscopy on HEK293 cells transiently expressing HA-tagged WT AT1A receptors, AT1A-β-arrestin1 or AT1A-β-arrestin2 constructs. Two different approaches were applied to visualize cell surface expressed proteins and total proteins respectively. To visualize receptors expressed on the cell surface, the antibody feeding assay was performed (Whistler et al., 2002). In this assay, live cells were incubated with anti-HA11 antibody. As the cells are not permeabilized, this antibody incubation will only label receptors that are on the cell surface during the time of incubation. As depicted in Figure 2A, WT AT1A receptors were predominantly localized on the cell surface in the basal state, indicating low levels of internalization. In contrast, the HA-tagged AT1A-β-arrestin1 and AT1A-β-arrestin2 showed a cytoplasmic distribution under the same conditions (Figure 2A), indicating that these receptors constitutively internalize and/or recycle. This observation is in good agreement with the established role of β-arrestins as scaffolding molecules for clathrin-coated pit components, whereby the receptor is internalized (Krueger and Dipalma, 1997). Treatment with saturating amounts of Ang II for 30 min. resulted in primarily cytoplasmic staining, suggesting significant internalization of the WT AT1A receptor (Figure 2B). The fusion proteins were also localized intracellularly in the Ang II-treated cells (Figure 2B).
Figure 2.
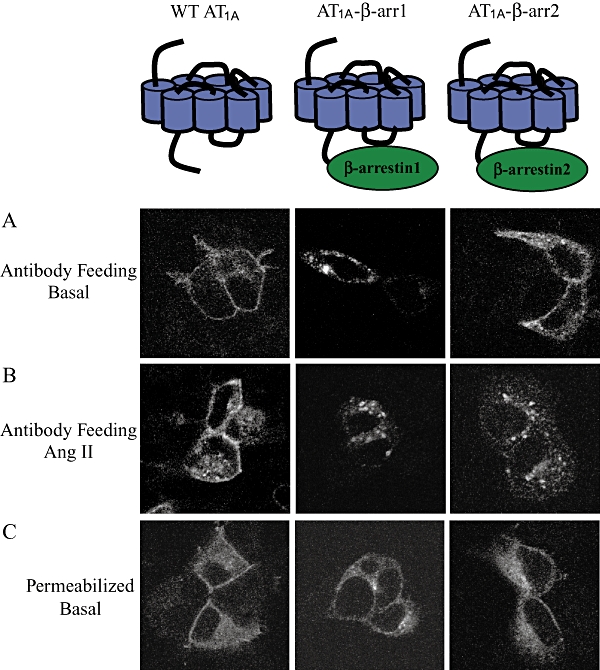
Subcellular localization of HA-tagged WT AT1A receptor, AT1A-β-arrestin1 and AT1A-β-arrestin2 in transiently transfected HEK-293 cells. (A) Antibody feeding assay of unstimulated cells. This procedure selectively stains receptors that are located on the cell surface during incubation with the anti-HA antibody. (B) Antibody feeding assay stains showing cells treated with 10−7 M Ang II for 30 min. (C) Preparations showing whole cell localization of WT AT1A receptor and the fusion constructs in permeabilized cells. AT1, angiotensin II type 1; WT, wild type.
To visualize the total amount of receptor protein, cells were fixed, permeabilized and then treated with anti-HA11 antibody. As shown in Figure 2C, WT AT1A receptors along with the AT1A-β-arrestin fusion proteins were distributed both in the membrane and in the cytoplasm.
G protein-mediated signal transduction by AT1A-β-arrestin1 and AT1A-β-arrestin2 is attenuated
To determine if β-arrestin fusion to AT1A receptor resulted in complete attenuation of Gq signalling, we compared the ability of WT AT1A receptors and fusion proteins to activate IP hydrolysis. As illustrated in Figure 3A, AT1A-β-arrestin1 and AT1A-β-arrestin2 fusion proteins almost abolished IP hydrolysis, compared with WT AT1A receptors. Efficacies and pEC50 values are shown in Table 2. Similarly, the IP hydrolysis in response to the low-affinity agonists, Ang III and (Val4)Ang III, was totally abolished by AT1A-β-arrestin1 and AT1A-β-arrestin2 (Figure 3B and C) and neither the biased agonist SII-Ang II nor the antagonist telmisartan induced G protein signalling of the fusion proteins (Figure 3D and E). Although, we cannot completely exclude the possibility that part of the uncoupling results from a lower cell surface expression, these results are in agreement with previous observations where the ability of β-arrestin fusion proteins to activate G proteins was abolished (Martini et al., 2002; Jorgensen et al., 2005), suggesting that the coupling between the AT1A receptors and β-arrestins is functional.
Figure 3.
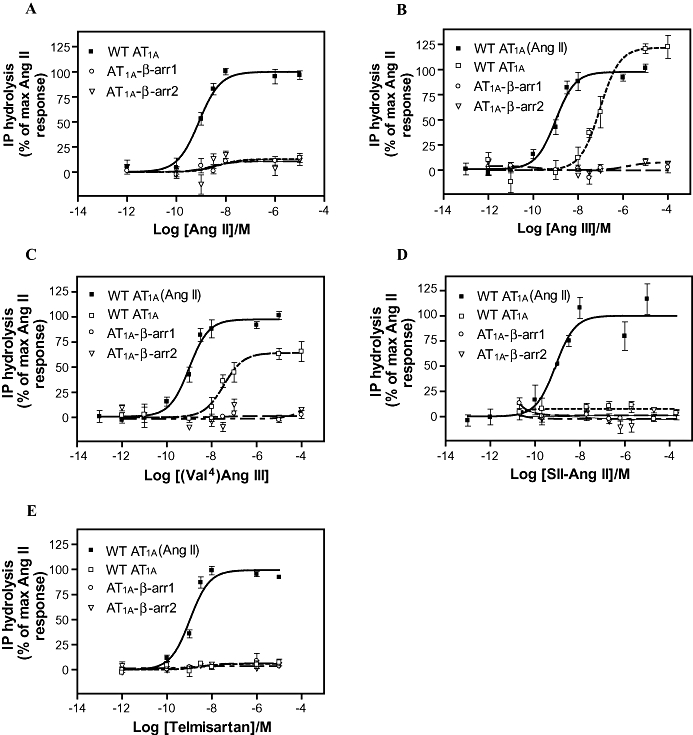
Ligand-induced IP hydrolysis of the AT1A-β-arrestin fusion proteins. Dose–response curves for Ang II (A) Ang III (B) (Val4)Ang III (C) SII-Ang II (D) and telmisartan (E) induced IP hydrolysis by WT AT1A receptors, AT1A-β-arrestin1 and AT1A-β-arrestin2. The relative efficacies of fusion proteins are calculated as a percentage of the maximal Ang II response observed for the WT AT1A receptor from experiments performed in parallel. Potencies and relative efficacies are reported in Table 2. AT1, angiotensin II type 1; IP, inositol phospholipid; WT, wild type.
Table 2.
Ligand-induced IP hydrolysis
| Assay | WT AT1A | AT1A-β-arr1 | AT1A-β-arr2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IP3 hydrolysis | pEC50 | Relative efficacy | n | pEC50 | Relative efficacy | n | pEC50 | Relative efficacy | n |
| Ang II | 9.0 ± 0.1 | 100 ± 0.0 | 5 | 7.9 ± 0.5 | 9.5 ± 2.2* | 4 | 8.6 ± 1.0 | 5.2 ± 2.9* | 4 |
| Ang III | 7.0 ± 0.1 | 122 ± 6.8* | 3 | NR | 3 | NR | 3 | ||
| (Val4)Ang III | 7.5 ± 0.2 | 64.2 ± 4.4* | 3 | NR | 3 | NR | 3 | ||
| SII-Ang II | NR | 3 | NR | 3 | NR | 3 | |||
| Telmisartan | NR | 3 | NR | 3 | NR | 3 | |||
Average pEC50 and relative efficacy values are reported. Data are presented as mean ± SEM from n experiments. Relative efficacies were calculated as a percentage of maximal Ang II response observed for the WT AT1A receptors. Dose-response curves are shown in Figure 3.
P < 0.05; significantly different from Ang II responses with WT AT1A receptors (one-tailed, paired Student's t-test).
AT1, angiotensin II type 1; IP, inositol phospholipid; NR, no response; WT, wild type.
The AT1A-β-arrestin fusion proteins show increased affinity for agonists
Previous studies have shown that 7TM receptor-β-arrestin fusion proteins induced significant affinity shifts in agonist binding, indicating that β-arrestins stabilize receptors in a high-affinity conformation (Martini et al., 2002; Jorgensen et al., 2005). Here, we used the same approach to analyse if β-arrestins induce high-affinity conformations of the AT1A receptor, and to analyse whether β-arrestin1 and 2 are equally potent in this regard. In order to investigate whether β-arrestins induce high-affinity conformations of the AT1 receptor, we performed competition binding assays on the AT1A-β-arrestin1 and AT1A-β-arrestin2 fusion proteins in parallel with the WT AT1A receptor, by using the radioligand [3H]-Ang II and increasing amounts of unlabelled Ang II. Surprisingly, we did not observe any apparent affinity shifts with respect to Ang II binding when comparing to the WT AT1A (Figure 4A); pKi values are shown in Table 3. To further analyse if β-arrestins induced high-affinity receptor conformations, we used a different approach, which was inspired by previous studies demonstrating that more significant shifts in Ki values are observed in heterologous competition binding assays than in homologous competition binding assays (Martini et al., 2002; Jorgensen et al., 2005). Therefore, we tested the affinities for Ang II in a heterologous competition assay, with the radioligand [125I]-SI-Ang II, and indeed seven- to eightfold shifts were observed for AT1A-β-arrestin1 and AT1A-β-arrestin2, when compared with WT AT1A receptors (WT AT1A, Ki= 2.22 × 10−8; AT1A-β-arrestin1, Ki= 2.84 × 10−9; AT1A-β-arrestin2, Ki= 3.10 × 10−9) (Figure 4B). The Ki values estimated from this experiment can be affected partially (or completely) by a change in the affinity for SI-Ang II. We have performed a homologous competition assay using labelled SI-Ang II and unlabelled SI-Ang II. Here, the Ki of the fusion constructs was identical to that of the WT AT1 receptors (data not shown). However, this does not completely exclude an affinity shift of the tracer, but it clearly demonstrates that the relationship between SI-Ang II and Ang II binding is affected by fusing the receptor to β-arrestins.
Figure 4.
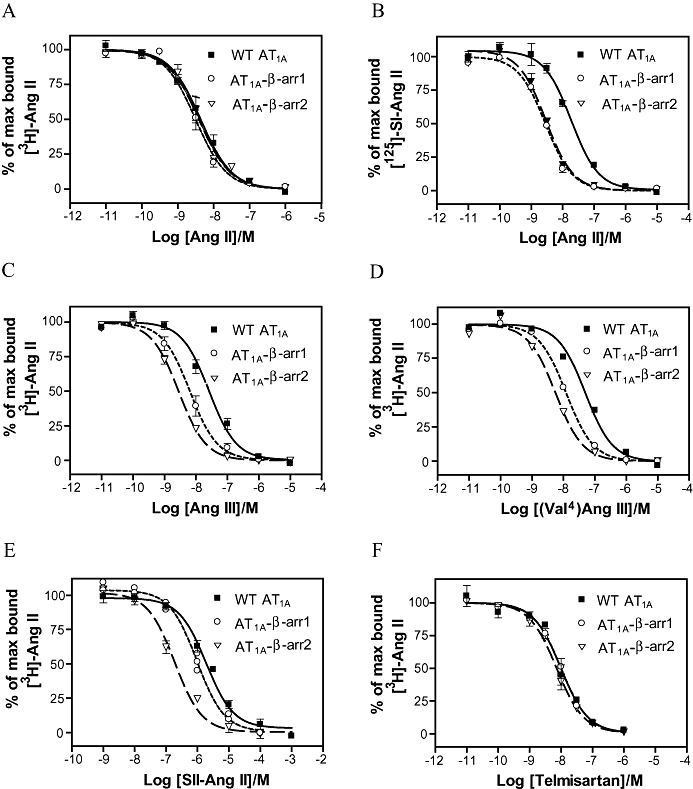
Competition radioligand binding for the WT AT1A receptor, AT1A-β-arrestin1 and AT1A-β-arrestin2. AT1A receptors were transiently expressed in COS-7 cells and competitive radioligand binding assays performed as described. [3H]-Ang II (10−9 M) (A, C–E) or [125I]-SI-ang II (10−11 M) (B) was used as radioligand tracers with the following unlabelled ligands: Ang II (A and B), Ang III (C) (Val4)Ang III (D) SII-Ang II (E) and telmisartan (F). Average pKi values are reported in Table 3. AT1, angiotensin II type 1; WT, wild type.
Table 3.
Radioligand competition binding of the WT AT1A receptor (WT AT1AR) and the fusion constructs with β-arrestins 1 or 2 (β-arr1, β-arr2)
| Assay | WT AT1AR | AT1AR-β-arr1 | AT1AR-β-arr2 | |||||
|---|---|---|---|---|---|---|---|---|
| Competition binding | pKi | n | pKi | Fold shift | n | pKi | Fold shift | n |
| Ang II | 7.7 ± 0.1 | 3 | 8.5 ± 0.0 | 8 | 3 | 8.5 ± 0.1 | 8 | 4 |
| Ang III | 7.6 ± 0.1 | 5 | 8.4 ± 0.1*,† | 6 | 4 | 8.6 ± 0.1* | 10 | 5 |
| (Val4)Ang III | 7.4 ± 0.2 | 4 | 8.0 ± 0.2*,† | 4 | 3 | 8.3 ± 0.3* | 9 | 4 |
| SII-Ang II | 5.8 ± 0.1 | 4 | 6.0 ± 0.1† | NS | 3 | 6.8 ± 0.1* | 9 | 3 |
| Telmisartan | 8.1 ± 0.1 | 5 | 8.1 ± 0.1 | NS | 3 | 8.3 ± 0.0 | NS | 5 |
Data represent mean pKi± SEM from n experiments. Fold shift denotes the ratio of Ki of the WT AT1A receptor to fusion constructs. Curves are depicted in Figure 4.
P < 0.05; significantly different from Ki values of WT AT1A receptor.
P < 0.05, significant differences between Ki values of AT1A-β-arrestin1 and 2 (one-tailed, paired Student's t-test).
AT1, angiotensin II type 1; NS, not significant (P > 0.05); WT, wild type.
When analysing the agonists Ang III (Val4)Ang III, and SII-Ang II and using [3H]-Ang II as the tracer ligand, we also observed that β-arrestin fusion proteins increased the ligand binding affinity compared with WT AT1A receptors. These results indicate that β-arrestins stabilize the receptor in a conformation that has a high affinity for the agonists investigated, which is most likely to be caused by a specific interaction between the two molecules. On the other hand, we cannot completely exclude the possibility that it is caused by ‘non-specific’ interactions or as a consequence of tagging the receptor on the C-terminal tail. However, tagging the AT1 receptor or other 7TM receptors in the C-terminal tail with GFP2 does not affect binding affinity of the receptor, which makes this explanation less likely (Jensen et al., 2002; Jorgensen et al., 2005).
Importantly, we also observed significant differences in how β-arrestin1 and 2 affect ligand binding abilities. When competing with the low-affinity agonists Ang III and (Val4)Ang III, larger affinity shifts were observed for AT1A-βarrestin2 (nine- to 10-fold affinity shift) than for AT1A-βarrestin1 (four- to sixfold affinity shift). This is illustrated in Figure 4C–D and summarized in Table 3. As illustrated in Figure 4E and Table 3, the interaction of AT1A receptors with β-arrestin2 also increased the affinity for the biased agonist SII-Ang II up to ninefold, in contrast to AT1A-βarrestin1 where no affinity shift was observed. This observation shows that β-arrestin1 and β-arrestin2 induce different conformations in the AT1 receptor and may reflect that β-arrestin2 interacts more stably with the AT1A receptor, than β-arrestin1.
As expected no change in the binding affinity for the antagonist telmisartan was observed for either fusion proteins (Figure 4F). Thus, our data showed that both β-arrestins stabilized the AT1A receptor in a conformation with higher agonist binding affinity and, furthermore, that β-arrestin2 induce larger affinity shifts than β-arrestin1.
Discussion and conclusions
Increasing evidence suggest that the functions of β-arrestin1 and 2 in 7TM receptor signalling may differ. Therefore, we wanted to test whether the two β-arrestin isoforms interacted differently with the receptor. A comparative study on AT1A receptor interaction with β-arrestin1 and 2 was carried out with ligands of different binding affinities and signalling properties, such as agonists, biased agonists and antagonists with varying affinities for the receptor. In this study, we demonstrated that β-arrestin1 and 2 were recruited to the AT1A receptor in response to agonists with comparable potencies and efficacies. Thus, both β-arrestin isoforms interact equally well with the AT1A receptors in the BRET2 assay, which is consistent with previous studies, demonstrating that Class B receptors do not show any preference towards a specific β-arrestin isoform (Oakley et al., 2000).
As there were no apparent differences in β-arrestin1 and 2 recruitment to AT1A receptors, we constructed fusion proteins between these receptors and β-arrestin1 and 2 respectively. Fusion proteins have been useful tools to study the interaction between β-arrestins (or G proteins) and other 7TM receptors (Seifert et al., 1999; Milligan, 2000; Martini et al., 2002; Jorgensen et al., 2005). Fusing two interacting proteins offer the obvious advantage that it brings two proteins of interest in close proximity. Furthermore, fusing two proteins of interest will, ideally, abolish the interaction with other signalling molecules and create a stoichiometry of 1:1 between the fused proteins. With the fusion receptor strategy, we demonstrate that the fusion of AT1A to β-arrestins mimics the natural AT1A/β-arrestin interaction and that fusion proteins can be used to disclose small differences in protein-protein interactions. First we showed that the subcellular localization of fusion proteins was predominantly cytoplasmic even in the absence of agonist stimulation, suggesting constitutive coupling between AT1A receptors and β-arrestins (Figure 2). Second, we showed that fusion of β-arrestin1 and β-arrestin2 to AT1A receptors abolished coupling to Gq. Although, we cannot completely exclude that part of the uncoupling results from a lower cell surface expression, similar findings have been made for other 7TM/β-arrestin fusion receptors, such as the NK1 or the GLP-1 receptor (Martini et al., 2002; Jorgensen et al., 2005). Third, both β-arrestin1 and β-arrestin2 constrain the AT1A receptor in a high-affinity conformation. The results show that β-arrestins increase AT1A receptor affinity for all agonists, compared with WT AT1A receptors (Figure 4B–E, and Table 3), while no affinity shift was evident in response to the antagonist telmisartan (Figure 4F, and Table 3). Thus, the high-affinity AT1A receptor conformation stabilized by β-arrestins is only formed in presence of agonists which is in good agreement with the alternative ternary complex model (Samama et al., 1993; Gurevich et al., 1997).
Ang II, Ang III and (Val4)Ang III were shown to be full agonists on β-arrestin recruitment (Figure 1A and B), and both β-arrestin1 and 2 were able to increase the AT1A receptor affinity for these agonists up to 10-fold, compared with WT receptors. Interestingly, β-arrestin2 induced higher affinity shifts than β-arrestin1, which could reflect the fact that β-arrestin2 formed a more stable complex with AT1A receptors than β-arrestin1. On the other hand, we were not able to detect these differences in the BRET2 assay, where β-arrestin1 and 2 were both recruited to AT1A receptors with similar potencies and efficacies for the tested ligands. Nevertheless, this observation still demonstrates that β-arrestin1 and 2 interact with the AT1A receptor in different ways, most likely reflecting a stabilization of different conformations within the fusion proteins.
The discrepancy between BRET2 results and radioligand binding experiments may be surprising but can be explained by differences between the two assays. In the binding assay, steady-state or equilibrium is almost reached, whereas the BRET assay is a functional assay where equilibrium is not reached, due to the constant dynamics inside the cell. Furthermore, the temperature at which the experiments are performed is different. The BRET assay is carried out at room temperature whereas binding is performed at 4°C. The differences could also arise because the affinity shifts we observe in the binding experiments are not caused by a tighter interaction between the two proteins, but instead by the fact that a different state is generated. As equilibrium is not reached in the BRET assay, this conformational change does not necessarily transform into an actual shift in EC50 values.
As mentioned previously, the AT1 receptor signals through two major pathways: the G protein-dependent signalling pathway and the G protein-independent signalling pathway, which is mediated by β-arrestins. The two signalling pathways of the AT1 receptor have been linked to different biological phenotypes. The G protein-dependent signalling pathway has been associated with effects such as cell death and fibrosis, while the G protein-independent pathway has been associated with cell survival and cell renewal (Zhai et al., 2005; Aplin et al., 2008). Thus, differential activation of the AT1 receptor by a biased agonist may be favourable in certain pathological states. Our present finding demonstrates that it even may be possible to develop drugs that selectively recruit β-arrestin1 or 2, which could be an advantage in certain pathological states. A recent study has demonstrated that β-arrestin1 and 2 have opposing roles in the development of atherosclerosis (Kim et al., 2008); β-arrestin2 was found to aggravate atherosclerosis by inducing smooth muscle cell proliferation and migration, while β-arrestin1 had the opposite effect. Thus, selective inhibition of β-arrestin2 could be a novel therapeutic target in treatment of atherosclerosis.
In conclusion, the present study shows that fusion proteins are valuable tools to detect the differences between β-arrestin1 and 2 interactions with 7TM receptors. Both β-arrestin1 and β-arrestin2 fused to AT1A receptors stabilized the receptor in a high-affinity conformation. Interestingly, β-arrestin2 induced a receptor conformation with a higher agonist-binding affinity than β-arrestin1. Thus, in our study we demonstrate that β-arrestin1 and 2 interact with the AT1A receptor in different ways, which suggest that it could be possible to design AT1 receptor biased agonists with the ability to recruit either of the β-arrestins selectively.
Acknowledgments
We thank Birte Kofoed for excellent technical assistance. This work was sponsored by The Danish National Research Foundation, The Koebmand i Odense Johan og Hanne Weimann f. Seedorffs legat, Aase og Ejnar Danielsens Foundation, The Novo Nordisk Foundation, The Danish Heart Foundation and the Augustinus Foundation.
Glossary
Abbreviations
- AT1
angiotensin II type 1
- ERK
extracellular signal-regulated kinase
- IP
inositol phosphate
- 7TM
7 transmembrane
Conflict of interest
None.
References
- Ahn S, Wei H, Garrison TR, Lefkowitz RJ. Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by beta-arrestins 1 and 2. J Biol Chem. 2004;279:7807–7811. doi: 10.1074/jbc.C300443200. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aplin M, Christensen GL, Schneider M, Heydorn A, Gammeltoft S, Kjolbye AL, et al. The angiotensin type 1 receptor activates extracellular signal-regulated kinases 1 and 2 by G protein-dependent and -independent pathways in cardiac myocytes and langendorff-perfused hearts. Basic Clin Pharmacol Toxicol. 2007;100:289–295. doi: 10.1111/j.1742-7843.2007.00063.x. [DOI] [PubMed] [Google Scholar]
- Aplin M, Christensen GL, Hansen JL. Pharmacologic perspectives of functional selectivity by the angiotensin II type 1 receptor. Trends Cardiovasc Med. 2008;18:305–312. doi: 10.1016/j.tcm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Aplin M, Bonde MM, Hansen JL. Molecular determinants of angiotensin II type 1 receptor functional selectivity. J Mol Cell Cardiol. 2009;46:15–24. doi: 10.1016/j.yjmcc.2008.09.123. [DOI] [PubMed] [Google Scholar]
- Burnier M, Brunner HR. Angiotensin II receptor antagonists. Lancet. 2000;355:637–645. doi: 10.1016/s0140-6736(99)10365-9. [DOI] [PubMed] [Google Scholar]
- Fan H, Luttrell LM, Tempel GE, Senn JJ, Halushka PV, Cook JA. Beta-arrestins 1 and 2 differentially regulate LPS-induced signaling and pro-inflammatory gene expression. Mol Immunol. 2007;44:3092–3099. doi: 10.1016/j.molimm.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Gurevich VV. Arrestins: ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- Hansen JL, Haunso S, Brann MR, Sheikh SP, Weiner DM. Loss-of-function polymorphic variants of the human angiotensin II type 1 receptor. Mol Pharmacol. 2004a;65:770–777. doi: 10.1124/mol.65.3.770. [DOI] [PubMed] [Google Scholar]
- Hansen JL, Theilade J, Haunso S, Sheikh SP. Oligomerization of wild type and nonfunctional mutant angiotensin II type I receptors inhibits galphaq protein signaling but not ERK activation. J Biol Chem. 2004b;279:24108–24115. doi: 10.1074/jbc.M400092200. [DOI] [PubMed] [Google Scholar]
- Hansen JL, Aplin M, Hansen JT, Christensen GL, Bonde MM, Schneider M, et al. The human angiotensin AT(1) receptor supports G protein-independent extracellular signal-regulated kinase 1/2 activation and cellular proliferation. Eur J Pharmacol. 2008;590:255–263. doi: 10.1016/j.ejphar.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, et al. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61:768–777. doi: 10.1124/mol.61.4.768. [DOI] [PubMed] [Google Scholar]
- Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol. 2006;20:953–970. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- Jensen AA, Hansen JL, Sheikh SP, Brauner-Osborne H. Probing intermolecular protein-protein interactions in the calcium-sensing receptor homodimer using bioluminescence resonance energy transfer (BRET) Eur J Biochem. 2002;269:5076–5087. doi: 10.1046/j.1432-1033.2002.03218.x. [DOI] [PubMed] [Google Scholar]
- Jorgensen R, Martini L, Schwartz TW, Elling CE. Characterization of glucagon-like peptide-1 receptor beta-arrestin 2 interaction: a high-affinity receptor phenotype. Mol Endocrinol. 2005;19:812–823. doi: 10.1210/me.2004-0312. [DOI] [PubMed] [Google Scholar]
- Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, et al. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci USA. 2005;102:1442–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Zhang L, Peppel K, Wu JH, Zidar DA, Brian L, et al. Beta-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ Res. 2008;103:70–79. doi: 10.1161/CIRCRESAHA.108.172338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger KJ, DiPalma JA. Omeprazole and Barrett's regression: is asymptomatic good enough? Am J Gastroenterol. 1997;92:177–178. [PubMed] [Google Scholar]
- Kumar P, Lau CS, Mathur M, Wang P, DeFea KA. Differential effects of beta-arrestins on the internalization, desensitization and ERK1/2 activation downstream of protease activated receptor-2. Am J Physiol Cell Physiol. 2007;293:C346–C357. doi: 10.1152/ajpcell.00010.2007. [DOI] [PubMed] [Google Scholar]
- Kuo FT, Lu TL, Fu HW. Opposing effects of beta-arrestin1 and beta-arrestin2 on activation and degradation of Src induced by protease-activated receptor 1. Cell Signal. 2006;18:1914–1923. doi: 10.1016/j.cellsig.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Martini L, Hastrup H, Holst B, Fraile-Ramos A, Marsh M, Schwartz TW. NK1 receptor fused to beta-arrestin displays a single-component, high-affinity molecular phenotype. Mol Pharmacol. 2002;62:30–37. doi: 10.1124/mol.62.1.30. [DOI] [PubMed] [Google Scholar]
- Milligan G. Insights into ligand pharmacology using receptor-G-protein fusion proteins. Trends Pharmacol Sci. 2000;21:24–28. doi: 10.1016/s0165-6147(99)01404-2. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, et al. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA. 2006;103:16284–16289. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K, Kobilka BK. GPCR-Galpha fusion proteins: molecular analysis of receptor-G-protein coupling. Trends Pharmacol Sci. 1999;20:383–389. doi: 10.1016/s0165-6147(99)01368-1. [DOI] [PubMed] [Google Scholar]
- Sneddon WB, Friedman PA. Beta-arrestin-dependent parathyroid hormone-stimulated extracellular signal-regulated kinase activation and parathyroid hormone type 1 receptor internalization. Endocrinology. 2007;148:4073–4079. doi: 10.1210/en.2007-0343. [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir MR. Effects of renin-angiotensin system inhibition on end-organ protection: can we do better? Clin Ther. 2007;29:1803–1824. doi: 10.1016/j.clinthera.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Gerber BO, Meng EC, Baranski TJ, von Zastrow M, Bourne HR. Constitutive activation and endocytosis of the complement factor 5a receptor: evidence for multiple activated conformations of a G protein-coupled receptor. Traffic. 2002;3:866–877. doi: 10.1034/j.1600-0854.2002.31203.x. [DOI] [PubMed] [Google Scholar]
- Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci USA. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai P, Yamamoto M, Galeotti J, Liu J, Masurekar M, Thaisz J, et al. Cardiac-specific overexpression of AT1 receptor mutant lacking G alpha q/G alpha i coupling causes hypertrophy and bradycardia in transgenic mice. J Clin Invest. 2005;115:3045–3056. doi: 10.1172/JCI25330. [DOI] [PMC free article] [PubMed] [Google Scholar]