

Abstract
BACKGROUND AND PURPOSE
Volume-regulated anion channels (VRACs) play an important role in cell-volume regulation. α1-Adrenoceptor stimulation by phenylephrine (PE) suppressed the hypotonic activation of VRAC current in mouse ventricular cells and regulatory volume decrease (RVD) was also absent in PE-treated cells. We examined whether the effects of α1-adrenoceptor stimuli on VRAC current were modulated by phosphatidylinositol signalling.
EXPERIMENTAL APPROACH
Whole-cell patch-clamp method was used to record the hypotonicity-induced VRAC current in mouse ventricular cells. RVD was analyzed by videomicroscopic measurement of cell images.
KEY RESULTS
The attenuation of VRAC current by PE was suppressed by α1A-adrenoceptor antagonists (prazosin and WB-4101), anti-Gq protein antibody and a specific phosphoinositide-specific phospholipase C (PLC) inhibitor (U-73122), but not by antagonists for α1B-, α1D- or β-adrenoceptor, or protein kinase C inhibitors. The inhibition of VRAC by PE was antagonized by intracellular excess phosphatidylinositol 4,5-bisphosphate (PIP2), while intracellular anti-PIP2 antibody (PIP2 Ab) inhibited the activation of VRAC currents. When cells were loaded with phosphatidylinositol 3,4,5-trisphosphate (PIP3) with or without PIP2 Ab, PE little affected the VRAC current. Extracellular m-3M3FBS (an activator of PLC) suppressed VRAC in the absence of PE, and this effect was reversed by intracellular excess PIP2.
CONCLUSIONS AND IMPLICATIONS
Our results indicate that the stimulation of α1A-adrenoceptors by PE inhibited the activation of cardiac VRAC current via PIP3 depletion brought about by PLC-dependent reduction of membrane PIP2 level.
Keywords: chloride channels, adrenoceptors, cell swelling, PIP2, ventricular cells
Introduction
The stimulation of α1-adrenoceptors has been reported to regulate rapid changes in the contractile function and electrophysiological properties of cardiac muscle cells (Fedida et al., 1993; Hein and Michel, 2007). In addition to acute effects, stimulated α1-adrenoceptors also have long-term effects on cardiac structure and function, such as cellular hypertrophy and apoptosis (Woodcock, 2007). The intracellular signalling pathway of α1-adrenoceptors involves coupling to the heterotrimeric G proteins that contain Gq subunits. The Gq subunit is an activator of phosphoinositide-specific phospholipase C (PLC), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG), which in turn results in the mobilization of intracellular Ca2+ and the activation of protein kinase C (PKC) (Hein and Michel, 2007).
Volume-regulated anion channels (VRAC; channel and receptor nomenclature follows Alexander et al., 2009) are widely expressed in mammalian cells and the currents through these channels are thought to play an important role in cell-volume regulation, particularly in the regulatory volume decrease (RVD) (Sarkadi and Parker, 1991; Okada, 1998; Wright and Rees, 1998). As outwardly rectifying chloride current through VRAC has been identified in many types of cardiac cells (Vandenberg et al., 1996; Sorota, 1999; Hume et al., 2000; Duan et al., 2005), it is expected that cardiac VRAC current also contributes to cell-volume regulation. In addition, recent studies have demonstrated that VRAC may relate to several pathological changes in the structure and function of heart including arrhythmia, cardiac ischaemic preconditioning, apoptosis and the adaptive remodelling of the heart during myocardial hypertrophy and heart failure (Duan et al., 2005; Okada et al., 2006).
The relationship between α1-adrenoceptor stimulation and cardiac VRAC current is not clear. Duan et al. (1995) showed that phenylephrine (PE; a non-subtype-selective α1-adrenoceptor agonist) inhibited VRAC current in a PKC-dependent manner in rabbit atrial myocytes. Du and Sorota (2000) reported that PE did not affect VRAC current but that PKC stimulated it in dog atrial cells. The effect of α1-adrenoceptor stimulation on VRAC current of mouse cardiac cells has not yet been studied.
Inositolphospholipids such as PIP2 and phosphatidylinositol 3,4,5-trisphosphate (PIP3) are normally located in the inner leaflet of the plasma membrane, and PIP2 represents less than 1% of membrane phospholipids and PIP3 is present in a negligible amount under resting conditions (Czech, 2000). As described previously, α1-adrenoceptor stimulation can reduce the membrane PIP2 level, and this may in turn reduce the PIP3 level because PIP3 is produced from PIP2 by an action of phosphoinositide 3-kinase (PI3K). PI3K has been given a role in the activation of VRAC and PI3K inhibitors attenuate VRAC current in several non-cardiac cell types (Feranchak et al., 1998; Shi et al., 2002; Vessey et al., 2004; Wang et al., 2004; Browe and Baumgarten, 2006) and in cardiac cells (Browe and Baumgarten, 2006; Ren et al., 2008). We recently showed that VRAC current in mouse ventricular cells was strongly inhibited by intracellular application of anti-PIP2 or anti-PIP3 antibody, and that intracellularly applied PIP3, but not PIP2, could restore the VRAC current which was suppressed by PI3K inhibitors (Yamamoto et al., 2008). These findings suggested that cardiac VRAC is regulated by PIP3 and/or its downstream signalling pathways.
Therefore, in the present study, we attempted to see whether the possible reduction of membrane PIP2 due to α1-adrenoceptor stimulation and the accompanying decrease in PIP3 would affect VRAC current in mouse ventricular cells, by using electrophysiological and pharmacological methods. Our results suggest that α1-adrenoceptor stimulation suppressed the activation of VRAC current via depletion of membrane PIP2 levels, followed by depletion of PIP3. A preliminary account of this work has appeared in abstract form (Ichishima et al., 2009).
Methods
Cell preparation
All animal care and experimental procedures conformed to the Guiding Principles of the Physiological Society of Japan and were approved by the Saga University Animal Care and Use Committee. The animals were allowed free access to food and water and kept on a 12–12 h light/dark cycle. Single ventricular myocytes from mouse hearts were isolated using an enzymatic dispersion technique as previously described (Yamamoto and Ehara, 2006; Yamamoto et al., 2007). Briefly, mice (18–25 g, C-57BL/6J/black inbred, male) were anaesthetized with sodium pentobarbitone (50 mg·mL−1, i.p.). The chest was opened, and the heart was rapidly removed and perfused, by using a modified Langendorff technique, with a physiological saline solution (PSS, see next) warmed to 37°C to wash out blood, and then with a nominally Ca2+-free PSS until the heart ceased to beat, and finally with the Ca2+-free solution containing 0.1% collagenase (CLSII, Worthington, Lakewood, NJ, USA) and 1.0% bovine serum albumin for 20–30 min. The ventricles were removed and cell dissociation was achieved by gentle mechanical agitation of the tissue in high-K+, low-Cl- storage solution (see next), and the dissociated cells were stored in a refrigerator (4°C) for later use (within 8 h). Only rod-shaped myocytes with clear cross-striations and no blebs were used in the experiments.
Electrophysiological techniques
The tight-seal whole-cell patch-clamp technique was used to record whole-cell currents. Patch pipettes (1.5 mm O.D. borosilicate glass electrodes) had a tip resistance of 1–3 MΩ when filled with pipette solution. Voltage-clamp recordings were performed using patch-clamp amplifiers (Model TM-1000; ACT ME, Tokyo, Japan or Axopatch 200B; Axon Instruments, Foster City, CA, USA) and membrane currents were filtered at a frequency of 2 kHz, and sampled at 5 kHz with Digidata 1322A and pCLAMP 9.2 software (Axon Instruments). A 3 M KCl-agar bridge between the bath and the Ag-AgCl reference electrode was used to minimize changes in liquid junction potential. Unless otherwise stated, membrane current recordings were monitored by applying voltage pulses of 400 ms duration to +40 mV from a holding potential of −40 mV every 6 s. When necessary, the current density was calculated by membrane capacitance, which was obtained using pCLAMP 9.0 software. Usually, 5 min was allowed for adequate cell dialysis after membrane rupture, before beginning of the voltage clamp protocol. In the experiments in which cells had to be dialyzed with pipette solutions containing some antibody proteins and phosphatidylinositides (see next), ∼30 min was allowed after membrane rupture before the current recording, expecting a more sufficient cell dialysis. All experiments were performed at 36.5 ± 0.5°C.
Solutions used
The PSS for cell preparation contained (mM): 120 NaCl, 10 glucose, 4.5 KCl, 5.0 MgCl2, 1.5 CaCl2, 20 taurine, 1.0 NaHPO4, 10 β-hydroxybutyric acid, 10 HEPES; pH 7.4 adjusted with NaOH; 300 mOsm with mannitol. Ca2+-free PSS was prepared by simply omitting CaCl2 from the PSS. The high-K+, low-Cl- solution for cell storage contained (mM): 70 potassium glutamate, 20 KCl, 1.0 MgCl2, 10 KH2PO4, 10 taurine, 10 EGTA, 10 glucose, 0.1% albumin, 10 β-hydroxybutyric acid and 10 HEPES; pH 7.2 with KOH; 300 mOsm with mannitol. For recording of Cl- currents, the extracellular and intracellular solutions were chosen to maximize recording of Cl- currents and to reduce possible contamination with cation currents and Ca2+-dependent currents. The standard extracellular hypotonic solution (HYPO) contained (mM): 80 N-methyl-D-glucamine (NMDG)-Cl, 20 tetraethylammonium (TEA)-Cl, 1.0 MgCl2, 0.5 CaCl2, 0.5 BaCl2, 0.5 CdCl2, 0.01 GdCl3, 5.5 glucose, 10 HEPES, 0.01 nicardipine; pH 7.4 adjusted with CsOH (∼5 mM); total (CI)o= 105 mM; 210 mOsm with mannitol. Appropriate amounts of mannitol were added to this solution to make the standard extracellular isotonic (310 mOsm) solution (ISO). The standard intracellular pipette solution contained (mM): 105 NMDG, 90 L-aspartic acid, 40 TEA-Cl, 5 NaCl, 5 Mg-ATP, 0.1 Tris-GTP, 5 EGTA, 5 HEPES; pH 7.2 adjusted with NMDG; total [Cl-]i= 45 mM; 290 mOsm with mannitol. In the experiments shown in Figure 2A–C, external solutions with 45 mM [Cl-]o were prepared by replacing 60 mM of NMDG-Cl with the same concentration of NMDG-aspartate. Pipette solutions with 105 mM [Cl-]i were also prepared. They were made by replacing 60 mM of L-aspartic acid in the standard pipette solution with the same concentration of HCl. In some experiments, pipette solutions contained 20 mM EGTA or 5 mM BAPTA in place of 5 mM EGTA.
Figure 2.
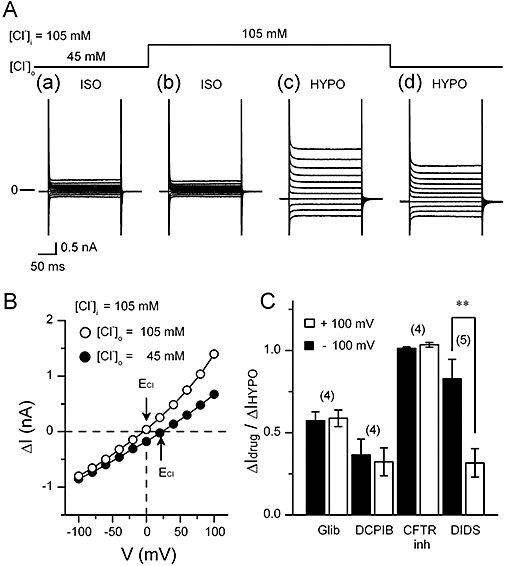
[Cl-]o-dependence and the sensitivity to some Cl- channel inhibitors of the hypotonic solution (HYPO)-induced current. In the experiments shown in (A) and (B), pipette solution contained 105 mM Cl- (105 mM [Cl-]i) instead of the usual amount (45 mM Cl-). (A) Typical current recordings in isotonic solution (ISO) and HYPO with different [Cl-]o. The cell was initially exposed to (a) isotonic 45 mM [Cl-]o solution, and subsequently to (b) isotonic 105 mM [Cl-]o. Then the cell was exposed to (c) hypotonic 105 mM [Cl-]o, and subsequently to (d) hypotonic 45 mM [Cl-]o. Currents were recorded by applying 400 ms voltage-clamp steps to membrane potentials between −100 and +100 mV in +20 mV steps from a holding potential of −40 mV every 2 s. (B) shows the I-V relationship of HYPO-induced (difference) currents obtained from the data shown in (A). The curve for 105 mM [Cl-]o was derived from b and c in A and that for 45 mM [Cl-]o, from a and d. Arrows indicate the ECl value predicted for each condition. (C) Comparison of the current inhibition by Cl- channel inhibitors, Glibenclamide (Glib, 100 µM, non-specific Cl channel inhibitor), DCPIB (10 µM, specific volume-regulated anion channel inhibitor), CFTRinh-172 (10 µM, specific CFTR inhibitor) and DIDS (100 µM, non-specific Cl channel inhibitor). ΔIdrug/ΔIHYPO shows the ratio of the current elicited in inhibitor-containing HYPO to that elicited in inhibitor-free HYPO (see Methods). The current data observed at +100 and −100 mV are shown. Numbers in parentheses are the number of the cells examined. *significantly different with P < 0.05.
Videomicroscopy and image analysis
The measurement of the cell area was performed independently of the electrophysiological measurements. The method to measure the cell area on the videomicroscopic image was the same as described previously (Yamamoto et al., 2001; 2004;). Briefly, microscopic images of the cell on an inverted microscope (Diaphot; Nikon, Tokyo, Japan) were recorded with a CCD video camera (CS3330; Tokyo Electronic, Tokyo, Japan) mounted on the side port of the microscope, which was equipped with a ×40 objective and a ×1 relay lens. Video images were captured at a 640 × 480 resolution and fed into a computer (DELL Dimension 4100; Dell Computer Corporation, Round Rock, TX, USA) through an 8-bit frame grabber (LG-3; Scion, Frederick, MD, USA). Number of the pixels included within the captured cell-image was detected by custom-made macro programs on Scion-Image software ver.4.03 (Scion). The chamber was continuously perfused with bathing solutions at the rate of 1.0–1.2 mL·min−1, and the area of cell-image was calculated every 6 s during the course of the experiments. It should be noted that the cell area is only a rough index of the cell volume. The validity of this method to estimate the cell volume has been discussed in a previous paper (Yamamoto et al., 2001).
Data analysis
The blocking effects of Cl- channel inhibitors shown in Figure 2C were calculated from the equation: ΔIdrug/ΔIHYPO= (Idrug−IISO)/(IHYPO−IISO), where IHYPO is the maximal amplitude of HYPO-induced current, Idrug is the minimal HYPO-induced current amplitude after application of the inhibitors and IISO is the current amplitude in isotonic solutions. Concentration–response curves (Figure 2B) for extracellular PE were analyzed by using a Hill equation: response =Imax/(1 + ([A]/IC50)nH), where Imax is the maximum current, [A] is the agonist concentration, IC50 is the agonist concentration producing half maximum response and nH is the Hill coefficient. Data are expressed as mean ± SEM; n indicates the number of cells. Statistical comparisons were performed either by one-way analysis of variance (anova) with a post hoc test (Scheffé's multiple comparison test) for group data, or by paired or unpaired Student's t-test when only two groups were compared. Statistical comparisons of the time course of changes in cell area between two groups were made according to two-way repeated measures anova. A two-tailed probability of <0.05 is taken to indicate statistical significance.
Materials
Chemicals used were: PE (Sigma, St. Louis, MO, USA), glibenclamide (Sigma), 4-(2-butyl-6,7-dichlor-2-cyclopentyl-indan-1-on-5-yl) oxybutyric acid (DCPIB; Sigma), 5-([4-carboxyphenyl]methylene)-2-thioxo-3-([3-trifluoromethyl]phenyl)-4-thiazolidinone (CFTRinh-172; Sigma), 4,4′-diisothiocyanostilben-2,2′-disulphonate (DIDS; Sigma), U-73122 (Sigma), U-73343 (Calbiochem, LaJolla, CA, USA), bisindolylmaleimide-I (BIM; Sigma), staurosporine (Sigma), chelerythrine (Sigma), prazosin (Sigma), WB-4101 (Sigma), L-765 314 (Sigma), BMY-7378 (Sigma), propranolol (Sumitomo Pharmaceuticals, Osaka, Japan), m-3M3FBS (Calbiochem), o-3M3FBS (Tocris, Ellisville, MO, USA), 1-oleoyl-2-acetyl-sn-glycerol (OAG; Sigma) and Pertussis toxin (PTX; WAKO, Osaka, Japan). When used, monoclonal anti-PIP2 antibody (Assay Designs, Ann Arbor, MI, USA), polyclonal anti-Gq/11 antibody (sc-392; Santa Cruz Biotechnology, Santa Cruz, CA, USA), PIP2 (dic 8, Echelon Biosciences, Salt Lake City, UT, USA), or PIP3 (dic 8, Echelon Biosciences) was added to the pipette solution. PIP2 and PIP3 were dissolved in the control pipette solution at a concentration of 10 µM. PIP2 (dic 8) and PIP3 (dic 8) are the short forms of PIP2 and PIP3 which are water-soluble at up to 1 mM. Anti-PIP2 antibody or anti-Gq/11 antibody was used at a 1:40 or 1:100 dilution respectively. Heat-inactivated antibodies (100°C for 20 min) were prepared as control when necessary. The osmolarity of all solutions was measured using freezing point depression osmometers (Model OM-801; Vogel, Giessen, Germany).
Results
Representative current traces elicited by various voltage steps during isotonic and hypotonic solutions in a mouse ventricular cell are shown in Figure 1A. A small basal background current (Figure 1Aa) was observed in the initial ISO. Exposure of the cell to HYPO increased the whole cell currents at both positive and negative voltages. The increased current in HYPO displayed a very weak time-dependent inactivation at positive potentials (Figure 1Aa) and was reduced by returning to ISO (Figure 1Ac). The current–voltage (I-V) relationship of the HYPO-induced current (difference current) showed a moderate outward rectification with the reversal potential (Erev) of about −20 mV (Figure 1B), a value close to the predicted Cl- equilibrium potential (ECl=−21 mV) under the present condition ([Cl-]o/[Cl-]i= 105 mM/45 mM). These properties of the HYPO-induced current appeared to match those reported for the VRAC current in mouse ventricular cells (Gong et al., 2004; Yamamoto-Mizuma et al., 2004b; Yamamoto et al., 2009). The mean-I-V relationships of whole-cell currents obtained in ISO and HYPO (n= 15) are shown in Figure 1C.
Figure 1.
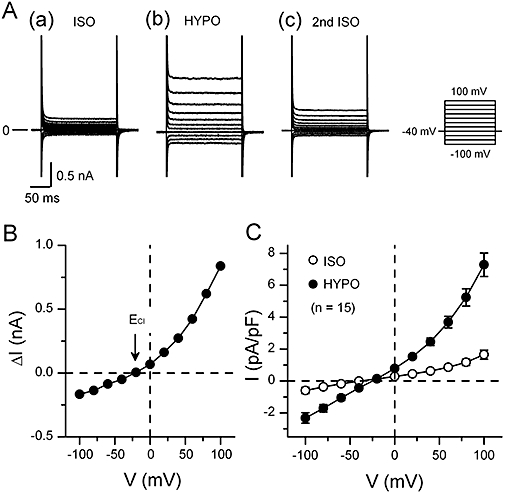
Hypotonicity-induced currents in mouse ventricular cells. (A) Typical whole-cell current recordings in cells exposed to isotonic and hypotonic solutions. The bath solution was changed from isotonic solution (a; ISO) to hypotonic one (b; HYPO) and then back to ISO (c; 2nd ISO). Currents were recorded by applying 400 ms voltage-clamp steps to membrane potentials between −100 and +100 mV in +20 mV steps from a holding potential of −40 mV every 2 s as shown in the right of Ac. (B) shows the I-V relationship of HYPO-induced current (difference current) obtained by subtracting the currents in ISO (a in A) from those in HYPO (b in A). In these experiments, [Cl-]o/[Cl-]i ratio was 105 mM/45 mM with which the predicted Cl- equilibrium potential (ECl) was −21 mV (arrow). (C) The mean I-V relationships (n= 15) of whole-cell currents obtained in ISO and in HYPO.
To confirm that the HYPO-induced current was a Cl- current, we measured it under the conditions of different [Cl-]i and [Cl-]o. In the experiment shown in Figure 2A, [Cl-]i was 105 mM and the cell was first exposed to (a) isotonic 45 mM [Cl-]o solution, and subsequently to (b) isotonic 105 mM [Cl-]o solution. Then the cell was exposed to (c) hypotonic 105 mM [Cl-]o, and subsequently to (d) hypotonic 45 mM [Cl-]o. At both low and high [Cl-]o, application of HYPO increased the whole-cell current (Figure 2Ac,d). The I-V relationships of HYPO-induced (difference) currents at each [Cl-]o are shown in Figure 2B. The HYPO-induced current obtained under symmetrical [Cl-] condition ([Cl-]o/[Cl-]i= 105 mM/105 mM) exhibited a weak outward rectification with Erev of about 0 mV, while the current obtained at low [Cl-]o (45 mM) showed a barely visible outward rectification with Erev of about +20 mV (Figure 2B). The Erev values obtained were close to the ECl values predicted for high and low [Cl-]o at 105 mM [Cl-]i respectively (0 mV for 105 mM [Cl-]o and +21 mV for 45 mM [Cl-]o). Similar results were obtained in three other cells. These observations strongly support the view that the current induced by HYPO in mouse ventricular cells is a Cl- current.
Next, we examined the effect of anion channel inhibitors on the HYPO-induced current. The results are shown in Figure 2C. A widely used anion channel inhibitor, DIDS (100 µM, n= 5; IC50 for VRAC inhibition at +100 mV was 5 µM; Greenwood and Large, 1998), reduced the magnitude of HYPO-induced current to 31.7 ± 8.6% of control at +100 mV, and to 82.8 ± 11.9% of control at −100 mV. Thus, the effect of DIDS on the HYPO-induced current was voltage-dependent, in agreement with the previous study (Sorota, 1994; Yamamoto-Mizuma et al., 2004b). With another anion channel inhibitor, glibenclamide (100 µM, n= 4; IC50 value for VRAC inhibition was 60 µM; Sakaguchi et al., 1997), the current magnitude was 58.8 ± 5.1% of control at +100 mV and 57.3 ± 5.4% of control at −100 mV. A selective VRAC inhibitor, DCPIB (10 µM, n= 4; IC50 value for VRAC inhibition was 4 µM; Decher et al., 2001), strongly suppressed the HYPO-induced current, its magnitude being reduced to 32.3 ± 8.4% of control at +100 mV and to 36.5 ± 9.5% of control at −100 mV. However, the current was insensitive to a thiazolidinone-derived selective inhibitor of the cystic fibrosis transmembrane conductance regulator (CFTR), CFTRinh-172 (n= 4; IC50 value of CFTRinh-172 for CFTR inhibition was 0.2 µM; Ma et al., 2002)
The results obtained so far support the view that the increase in the whole cell current observed here in HYPO in mouse ventricular cells is due to the activation of VRAC current, as noted in the previous studies (Gong et al., 2004; Yamamoto-Mizuma et al., 2004b; Yamamoto et al., 2009). Subsequently, we designated the HYPO-induced current as VRAC current. One of the biophysical properties of cardiac VRAC current is the outward rectification under symmetrical [Cl-] condition (Hume et al., 2000; Yamamoto-Mizuma et al., 2004b). However, the VRAC current exhibited only a weak outward rectification in the present study (Figure 2B). The nature of the discrepancy is unclear, but the difference in the experimental procedures (e.g. the bath temperature employed) could be a factor.
In Figure 3A,B, we show the effect of the α1-adrenoceptor agonist, PE, on the HYPO-induced activation of VRAC current. In the presence of 100 µM PE, the amplitude of VRAC current (Figure 3Ab,B) at positive and negative potentials was small compared with that observed in control cells (Figure 1A,B). Figure 3C shows the time course of changes in the amplitude of whole-cell current observed during exposure to HYPO in the absence and presence of PE. In HYPO without PE, the whole cell current gradually increased, reaching a steady level in about 15 min. This result is similar to the previous reports (Yamamoto et al., 2008; 2009;). In the presence of PE, exposure of the cells to HYPO increased the whole cell current only slightly. PE itself did not appear to have any significant effect on the basal current in ISO before HYPO. Figure 3D shows the concentration dependence of the inhibitory effect of PE on VRAC current. Fitting the curve to a Hill equation yielded IC50 (1.2 µM) and nH (0.91) values. This IC50 value was similar to that reported for α1-adrenoceptor responses in several mammalian ventricular functions (Yokoyama et al., 1998; Luo et al., 2007), but was much smaller than the value reported for the PE -induced inhibition of VRAC in rabbit atrial cells (72 µM; Duan et al., 1995). The inhibition of VRAC current by PE appeared to fully develop at about 100 µM of PE in mouse cells (Figure 3D). In subsequent experiments, we mostly used 100 µM PE for its effect on VRAC.
Figure 3.
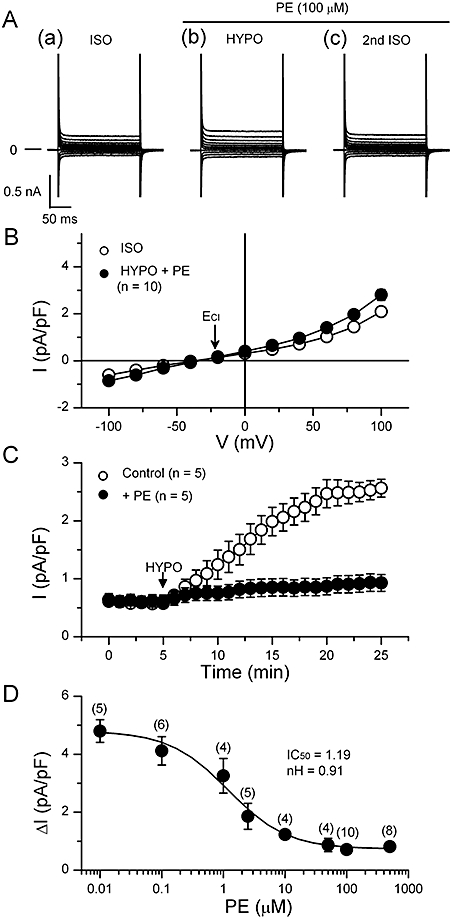
Suppression of the hypotonic activation of volume-regulated anion channel (VRAC) current by phenylephrine in mouse ventricular cells. (A) shows representative records of the currents elicited by the voltage protocol illustrated in Figure 1. Phenylephrine (PE; 100 µM) was present in hypotonic solution (HYPO) and 2nd isotonic solution (ISO). (B) The mean I-V relationships of the currents obtained in ISO and in HYPO with PE (n= 10). The arrow indicates the predicted ECl (−21 mV). (C) Time course of activation of VRAC currents recorded in the absence (open circles) and presence (closed circles) of 100 µM PE. The whole cell current was recorded at +40 mV, while 400 ms voltage steps to +40 mV were applied to the cell from a holding potential of −40 mV every 6 s. Data obtained from five different cells were averaged for each curve. The bath solution was changed from ISO to HYPO at arrow. (D) Concentration dependence of the inhibitory effect of PE on the activation of VRAC current. Hypotonicity-induced (difference) currents (ΔI) at +100 mV are plotted for various PE concentrations. Each point represents mean of data obtained in several different cells, and the curve was obtained by fitting the data points to the Hill equation, yielding IC50 and Hill coefficient (nH) as shown.
VRAC current is implicated in the development of RVD in many types of mammalian cells including cardiac cells (Sarkadi and Parker, 1991; Okada, 1998; Wright and Rees, 1998; Yamamoto et al., 2001; 2004;). Therefore, we compared the changes in cell volume during hypotonic challenge between control and PE-treated cells. The relative cell area on captured video images was used as an index of cell volume. Figure 4A shows the time-course of the changes in relative cell area during hypotonic challenge in control cells. Upon switching the bathing solution from ISO to HYPO, the cell area increased, as expected. But the increase peaked about 7 min after the initiation of hypotonic perfusion, and the cell area decreased thereafter, indicating operation of the mechanism of RVD, to which VRAC current contributed (Yamamoto et al., 2004). In the presence of PE, the cells also swelled in response to external hypotonicity (Figure 4B) but there was little RVD in this case (Figure 4B and C). Reapplication of isotonic solution decreased the cell area of the hypotonically swollen cells, as expected (Figure 4A and B). In control cells, the level of cell area attained in the reapplied isotonic solution was smaller than the initial level, showing excessive cell shrinkage (Figure 4A). This undershoot of the relative cell area below the initial level is attributed to a loss of solutes from the intracellular medium during RVD (Okada, 1998; Yamamoto et al., 2004). In contrast, there was no clear undershoot of cell area in the PE-treated cells which showed little RVD (Figure 4B). These results are in accordance with the finding that PE inhibited the development of VRAC current during hypotonic challenge (Figure 3).
Figure 4.
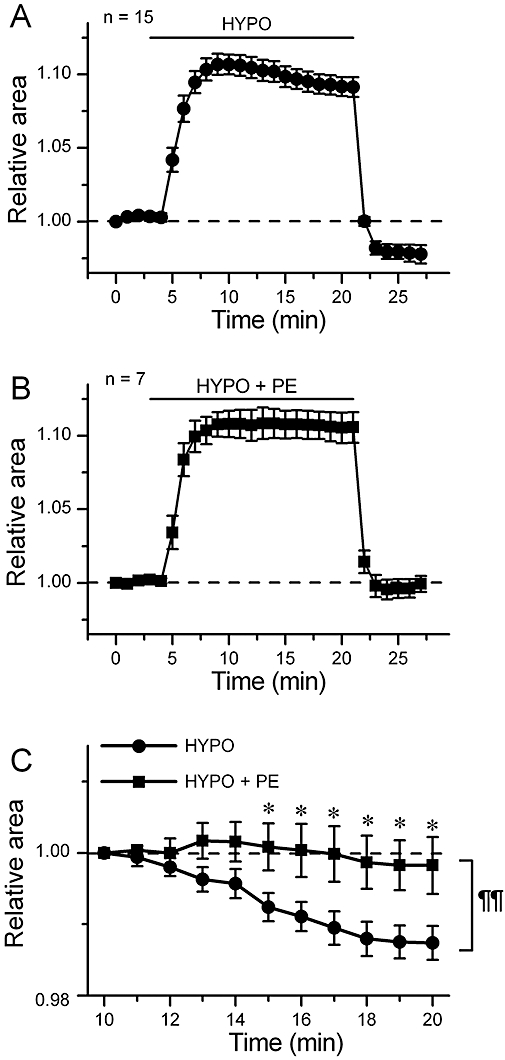
Inhibition of cell volume regulation by phenylephrine (PE). (A) and (B), time course of change of the relative cell area in cells exposed to hypotonic solution (HYPO) in the absence (A, n= 15) and presence (B, n= 7) of 100 µM PE. The cells were initially bathed in isotonic solution (ISO), and then HYPO or HYPO + PE was applied during the period indicated by bar. (C) shows an expanded illustration of a part of A and a part of B. The values are expressed as relative to those obtained 5 min after application of HYPO. *, significantly larger than the time-matched HYPO data with P < 0.05 according to an unpaired t-test. A comparison of curves with repeated measures anova yielded P < 0.01 (¶¶).
Next, we attempted to identify the subtype of α1-adrenoceptor involved in the PE -induced inhibition of VRAC current. Figure 5A shows that 5 µM of prazosin [a nonselective α1-adrenoceptor antagonist (Yamada et al., 1980; O-Uchi et al., 2008)] or 2 µM of WB-4101 [an α1A-adrenoceptor-selective antagonist (Yamada et al., 1980; O-Uchi et al., 2008)], significantly depressed the inhibitory effect of 100 µM PE on VRAC current, whereas 50 nM of L-765 314 [an α1B-adrenoceptor-selective antagonist (Patane et al., 1998; O-Uchi et al., 2008)], 30 nM of BMY-7378 [an α1D-adrenoceptor-selective antagonist (Deng et al., 1996; O-Uchi et al., 2008)] and 1 µM of propranolol [a β-adrenoceptor antagonist (Alexander et al., 1975; Iyadomi et al., 1995)] appeared to be without effect. Similar results were obtained in the cells exposed to a lower concentration (10 µM) of PE (data not shown). These results suggest the involvement of α1A-adrenoceptors in the PE-induced inhibition of VRAC current in mouse ventricular cells. All α1-adrenoceptor subtypes expressed in the heart may be coupled to Gq/11 protein and may regulate the production of PKC via activation of PLC (Xiang and Kobilka, 2003). However, a previous report (Duan et al., 1995) showed that α1-adrenoceptor-mediated inhibition of VRAC current involves the activation of PKC by PTX (a selective Gi protein inhibitor)-sensitive G protein in rabbit atrial cells. To clarify the signalling processes involved in mouse cells, we examined the effects of anti-Gq antibody, PTX, PLC and PKC inhibitors on the PE-induced inhibition of VRAC current.
Figure 5.
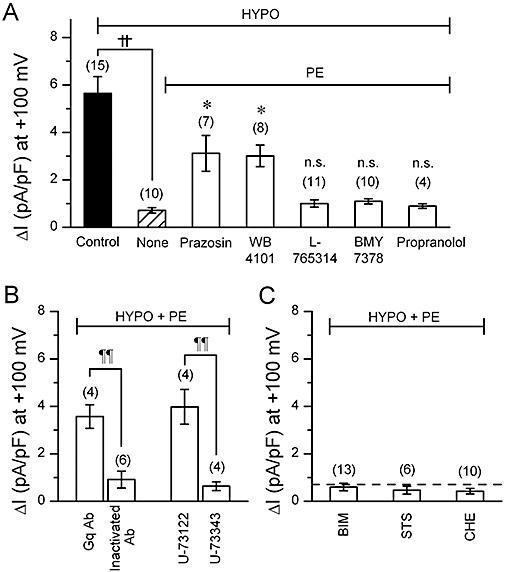
Volume-regulated anion channel (VRAC) current observed in cells with various α1 adrenoceptor–related compounds. (A)–(C), mean density of hypotonicity-induced (difference) currents measured at +100 mV under various conditions. Phenylephrine (PE) concentration was 100 µM throughout. (A) shows the selective effect of α1A-adrenoceptor antagonists on the PE-induced VRAC inhibition. The cells were exposed simply to hypotonic solution (HYPO) (Control) or to PE-containing HYPO. In the latter case, the cells were pretreated with 5 µM prazosin, 2 µM WB-4101, 50 nM L-765314, 30 nM BMY-7378 or 1 µM propranolol for 10 min under isotonic condition, and then exposed to PE-containing HYPO in the continuous presence of these antagonists. Some cells were given no antagonist (None). ††, significantly different with P < 0.01. *significantly larger than None with P < 0.05. n.s., not significantly different from None. (B) shows effects of anti-Gq antibody (Gq Ab) or a phosphoinositide-specific phospholipase C (PLC) inhibitor, U-73122, on PE-induced VRAC inhibition. For Gq Ab experiments, Gq Ab or heat-inactivated Gq Ab was added to the pipette solution at a 1:100 dilution. For the experiments of PLC inhibitor, the procedure of drug application was similar to that adopted in the experiments shown in A, that is, it included a pre-treatment phase of 10 min. U-73122 (5 µM) or its inactive analogue, U-73343 (5 µM) was used. ¶¶, significantly different with P < 0.01. (C) shows the lack of effect of PKC inhibitors on PE-induced VRAC inhibition. The procedure of drug application was similar to the above. Bisindoylmaleimide (BIM; 100 nM), staurosporine (STS; 10 nM) or chelerythrine (CHE; 10 µM) was used. Number in parentheses indicates number of the cells examined. Dashed line indicates the average magnitude of VRAC current with PE, derived from the data shown as None in (A).
To see whether Gq/11 protein was involved in the response to PE, we performed the experiments on the cells dialyzed with a pipette solution containing anti-Gq antibody. Our antibody was an affinity-purified polyclonal antibody directed against 19 amino acids within the extreme carboxyl termini of Gq/11, and the inhibitory effect of its intracellular application on the Gq/11-dependent system has been described previously, and anti-Gq/11 antibody itself appeared to have little effect on the basal whole cell current in mouse ventricular cells (Yamamoto et al., 2007). When such dialyzed cells were exposed to HYPO in the presence of PE, a VRAC current of a definite amplitude developed (Figure 5B), indicating attenuation of the inhibitory effect of PE on VRAC current. In contrast, the usual inhibition of VRAC current by PE was observed in the cells loaded with heat-inactivated anti-Gq antibody (Figure 5B). We also tested whether PTX-sensitive G protein was involved in the response to PE. The cells were incubated for over 2 h at room temperature in control storage solution or in a storage solution containing PTX (0.5 µg·mL−1) (Yamamoto-Mizuma et al., 2004a). We observed that the attenuation of HYPO-activated VRAC current by PE in PTX-treated cells (n= 4) was not significantly different from that in control cells (n= 4) (data not shown). These results suggest that Gq/11 proteins but not PTX-sensitive G proteins are essential for the inhibition of VRAC current by α1A-adrenoceptor stimulation in mouse ventricular cells.
In the presence of 5 µM U-73122, a specific PLC inhibitor (IC50 value for PLC inhibition was 1 µM; Bleasdale et al., 1990), a large VRAC current was activated in PE-treated cells (Figure 5B), whereas the usual attenuation of VRAC current by PE occurred in the cells exposed to 5 µM U-73343 (an inactive analogue of U-73122). Further, we could not detect any PKC-dependent regulation in the PE-induced inhibition on VRAC current. As shown in Figure 5C, three different PKC inhibitors [100 nM bisindolylmaleimide-I (Yamamoto-Mizuma et al., 2004b), 10 nM staurosporine (Bewick et al., 1999) and 10 µM chelerythrine (Gong et al., 2004)] had little effect on the PE-induced attenuation of VRAC current. Thus, the inhibitory effect of PE on VRAC current observed in mouse cells appeared to involve PLC activation but not PKC activation.
Our next experiments investigated how the PLC activity was related to the observed response to PE. It is well-known that PLC hydrolyzes PIP2 into DAG and IP3, which may result in a decrease in the membrane PIP2 level (Xiang and Kobilka, 2003). Changes in the PIP2 level may lead to changes in the PIP3 level, because PIP3 is generated from PIP2 by an action of PI3K. Therefore, we attempted to see whether changes in the membrane PIP2 or PIP3 levels played a role in the PE-induced inhibition of VRAC current in mouse heart. For this purpose, pipette solutions containing water-soluble form of PIP2 or PIP3, and/or anti-PIP2 antibody (PIP2 Ab) were prepared and used for cell dialysis.
Figure 6A,B shows the results of the experiments in which the effect of PE on VRAC current was examined in the cells loaded with PIP2. The intracellular application of excess PIP2 (10 µM) itself did not induce any significant changes in the background current level under isotonic conditions for at least 30 min (on the left in Figure 6B). PE slightly inhibited the VRAC current under hypotonic condition in the cells loaded with PIP2 (Figure 6A,B). Thus, the magnitude of VRAC current obtained in hypotonic solution was similar in the presence or absence of PE in these cells (Figure 6B). These findings are compatible with the idea that, under physiological conditions, stimulation of α1A-adrenoceptors by PE results in a decrease in the membrane PIP2 level via activation of PLC, that the decreased PIP2 level is a factor causing the depression of VRAC current, and that intracellular excess PIP2 can cancel the PIP2-depleting action of PE.
Figure 6.
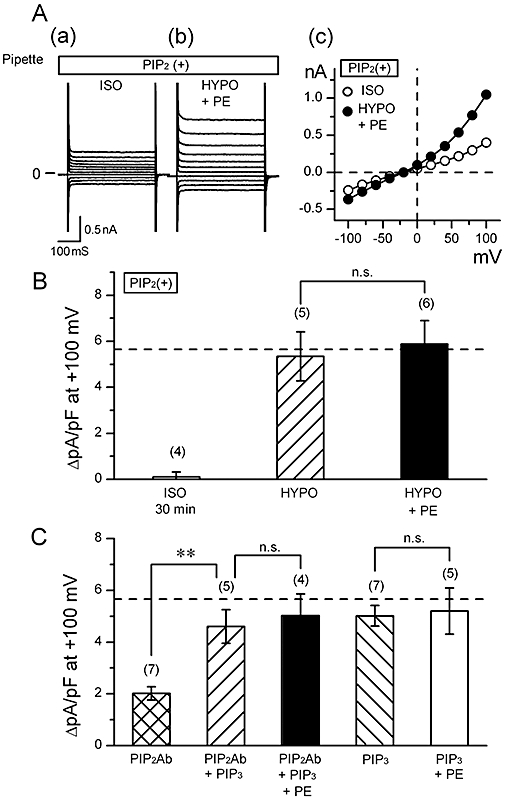
Effect of phosphatidylinositides on phenylephrine (PE)-induced suppression of volume-regulated anion channel (VRAC current). (A) Raw current traces in response to various voltage steps (a and b) and the corresponding I-V relationships (c), obtained in a cell loaded with 10 µM PIP2. Data were obtained in isotonic solution (ISO; a) and hypotonic solution (HYPO) containing 100 µM PE (HYPO + PE; b). Currents were elicited by applying 400 ms voltage-clamp steps to membrane potentials between −100 and +100 mV in +20 mV steps from a holding potential of −40 mV every 2 s. (B) Mean density of hypotonicity-induced (difference) current at +100 mV obtained in cells dialyzed with 10 µM PIP2. Data were obtained in the absence (HYPO) and presence (HYPO + PE) of 100 µM PE. The difference between the current magnitudes observed 5 and 30 min after the beginning of whole cell recording under isotonic condition is also shown on the left (ISO 30 min). n.s., not significantly different. (C) Mean density of hypotonicity-induced (difference) current at +100 mV obtained in cells dialyzed with some phosphatidylinositide-related compounds. Three types of pipette solution were prepared; they contained anti-PIP2 antibody (PIP2Ab), PIP3, or both PIP2Ab and PIP3. PIP2Ab was added to the pipette solution at a 1:40 dilution, and PIP3, at a concentration of 10 µM. Some cells were extracellularly exposed to 100 µM PE (+PE). **, significantly different with P < 0.01. n.s., not significantly different. Dashed line in (B) and (C) indicates the control magnitude of VRAC current derived from the data in Figure 4A. Number in parentheses indicates number of the cells examined.
Then the question arises as to how PIP2 can be related to the activation of VRAC current. Our previous study (Yamamoto et al., 2008) showed that the development of VRAC current was suppressed in the cells loaded with PIP2 Ab which was expected to inactivate PIP2, and that VRAC current was restored if the cells were loaded with PIP3 (10 µM) in addition to PIP2 Ab. The present study confirmed these findings (first and second columns of Figure 6C), supporting the idea that the activation of VRAC current requires the presence of PIP3 which is produced from PIP2 by an action of PI3K. Moreover, PE could not suppress the development of VRAC current in the cells with PIP2 Ab and PIP3 (third column of Figure 6C). PE was similarly ineffective on VRAC current in the cells dialyzed with PIP3 alone (on the right in Figure 6C). It appears, therefore, that VRAC current can develop in the presence or absence of PIP2 if PIP3 is present, and that the presence of exogenous excess PIP3 counteracts the effect of PE on this current in the presence or absence of PIP2. It should be noted that loading with PIP3 had little influence on the amplitude of the VRAC current (fourth column of Figure 5C).
An intracellular excess of exogenous PIP2 counteracted the inhibitory effect of PE on VRAC current (Figure 6A,B). Such effects of PIP2 to restore VRAC current was observed also in the cells in which PLC was directly activated without stimulation of α1A-adrenoceptors. When the cells were exposed to m-3M3FBS (a putative PLC activator), the development of VRAC current was suppressed (on the left in Figure 7A). Thus, this agent appeared to mimic the action of PE. The concentration of m-3M3FBS used (25 µM) was a maximal dose for the hydrolysis of PIP2 (Bae et al., 2003). Furthermore, like PE, m-3M3FBS exerted little effect on VRAC current if the cells had been loaded with excess PIP2 (second column of Figure 7A). The depression of VRAC current by m-3M3FBS was not affected by the application of BIM to the cell (third column of Figure 7A), supporting that PKC activation was not involved in the depressant effect of the PE -PLC system on VRAC current. An inactive analogue of m-3M3FBS, o-3M3FBS, did not inhibit VRAC current, as expected (fourth column of Figure 7A). These findings are compatible with the view that PLC-dependent reduction of membrane PIP2 level, which should result in PIP3 depletion, inhibited the activation of VRAC current.
Figure 7.
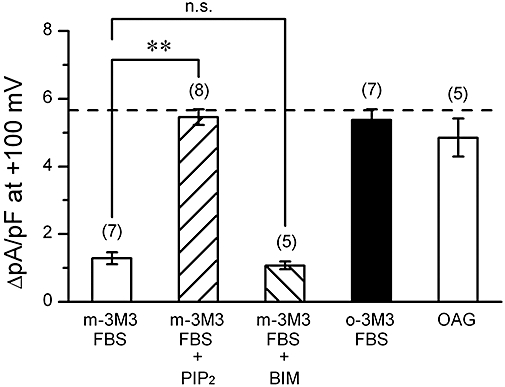
Volume-regulated anion channel (VRAC) current observed in cells with various phospholipase C (PLC) activators. Mean density of hypotonicity-induced (difference) currents measured at +100 mV under various conditions. Data were obtained in cells exposed to m-3M3FBS (a direct PLC activator; 25 µM), o-3M3FBS (inactive analogue of m-3M3FBS; 25 µM) or 1-oleoyl-2-acetyl-sn-glycerol (OAG; a membrane-permeable DAG analogue; 50 µM). Some of the cells exposed to m-3M3FBS had been dialyzed with 10 µM PIP2 (+PIP2). Some cells were co-exposed to m-3M3FBS and 100 nM BIM (+BIM). Dashed line indicates the control magnitude of VRAC current derived from the data in Figure 4A. **significantly different with P < 0.01. n.s., not significantly different. Number in parentheses indicates number of the cells examined.
The activation of PLC may produce DAG and IP3 by hydrolyzing PIP2. It is known that DAG directly activates some subtypes of canonical transient receptor potential channels independently of PKC activation in ventricular cells (Nishida and Kurose, 2008). Additional experiments were performed to see whether DAG had any role in the activation of VRAC current, using a membrane-permeable DAG analogue, 1-oleoyl-2-acetyl-sn-glycerol (OAG). We observed that 50 µM OAG (Kraft, 2007) did not suppress the activation of VRAC current in HYPO (on the right in Figure 7) in mouse ventricular cells. On the other hand, IP3 may act on the intracellular Ca2+ store and stimulate the release of Ca2+. It is unlikely, however, that changes in the intracellular Ca2+ level played a role in the regulation of VRAC current, because the present data were obtained from the cells which were mostly dialyzed with pipette solutions containing 5 mM EGTA. Furthermore, the results were similar if the pipette solutions contained higher concentration (20 mM) of EGTA (n= 4) or 5 mM of BAPTA (n= 4), a faster Ca2+ chelator (data not shown).
Discussion and conclusions
The stimulation of α1-adrenoceptors by PE inhibited the hypotonic activation of VRAC current in mouse ventricular cells. We attempted to elucidate the underlying mechanism of this action of PE and our results have been interpreted in the following scheme. The stimulation of α1A-adrenoceptors activates PLC through a Gq protein-dependent process and the activated PLC hydrolyzes membrane PIP2, which results in a depletion of PIP2. The latter in turn leads to a depletion of membrane PIP3, because PIP2 is the substrate for PI3K-mediated production of PIP3. The depletion of PIP3, by an unknown mechanism, interferes with the activation of VRAC current.
The requirement of PIP3 for the development of VRAC current in mouse cardiac cells has been suggested in a previous study (Yamamoto et al., 2008). Here, the development of VRAC current was suppressed in the cells loaded with PIP2 Ab and that depressed VRAC current was restored when the cells were loaded with PIP2 Ab and PIP3 (Figure 6C). Moreover, the development of VRAC current was inhibited by intracellular application of PI3K inhibitors (LY294002 and wortmannin) or anti-PIP3 antibody, while intracellular excess PIP3, but not PIP2, could restore the VRAC current which was suppressed by PI3K inhibitors (Yamamoto et al., 2008). These findings were taken to suggest that the presence of PIP3, which is generated from PIP2 by PI3K, is important for the activation of VRAC current under physiological conditions. As to the inhibitory effect of PE on VRAC current, this agonist could not suppress this current in the cells loaded with excess PIP2 or PIP3 (Figure 6B,C). This finding is explained by the scheme outlined previously. The excess PIP2 or PIP3 may cancel the action of PE to deplete PIP2, and hence PIP3 (see previous). However, considering that endogenous PIP2 and PIP3 are thought to be located in the inner leaflet of the plasma membrane and that the exogenous PIP2 and PIP3 molecules were water-soluble short forms of these compounds, a question arises as to whether such PIP2 and PIP3 derivatives, applied through a pipette, acted on VRAC after incorporation into the membrane or directly from the cytoplasm. This point remains to be clarified.
The concentration of PIP3 required for the physiological activation of VRAC current is uncertain. The restoration of VRAC current by 10 µM PIP3 (Figure 6C) does not necessarily mean that PIP3 acts on the VRAC system in this concentration range, under physiological conditions. Further, if the stimulation of α1A-adrenoceptors by PE results in a decrease in membrane PIP3 level, it is still uncertain to what extent PE can reduce the PIP3 level in swollen cells. Obviously, further studies are required to quantify the dynamic changes in the membrane PIP2 and PIP3 levels during cell swelling and during α1A-adrenoceptor stimulation. It must be clarified also how PIP3 or related compounds can interact with the VRAC. PIP2 appeared to act as a precursor of PIP3, having no direct effect on the VRAC system [(Yamamoto et al., 2008); see also Figure 6]. It should be noted, however, that PIP2 may exert a direct action on other cardiac membrane currents. It has been reported that membrane PIP2 plays essential roles in the α1-adrenoceptor mediated regulation of the membrane K+ currents [acetylcholine-activated K+ current (Cho et al., 2001; Meyer et al., 2001); ATP-sensitive K+ current (Haruna et al., 2002)] in the heart. In these α1-adrenoceptor modulations of K+ currents, PIP2 is thought to act as a signalling molecule influencing the channel activity.
The inhibitory effect of PE on VRAC was generally observed in the cells which were dialyzed with PIP2- and PIP3-free pipette solutions. This may indicate that the endogenous PIP2 and PIP3, which are located in the inner leaflet of the membrane, cannot be readily washed out by dialysis with solutions lacking these compounds. In line with this view, Meyer et al. (2001), using the cells dialyzed with normal, that is, PIP2-free, pipette solutions, observed that the stimulation of PLC-coupled receptors induced a slow inhibition of G protein-gated, inwardly rectified, K+ current which was due to a depletion of membrane PIP2, and that supplementing the pipette solution with PIP2 (0.5 mM) significantly reduced the current inhibition.
Duan et al. (1995) reported that VRAC current in rabbit atrial myocytes was inhibited by PE via the activation of PTX-sensitive α1A-adrenoceptors. Furthermore, PKC inhibitors blocked the inhibitory effect of PE on VRAC current, while PMA (a PKC activator) itself inhibited VRAC current. They concluded that PE inhibits VRAC current by interacting with an α1A-adrenoceptor mechanism that is coupled to PKC via a PTX-sensitive G protein in rabbit atrial cells. In our study using mouse ventricular cells, the inhibitory effect of PE on VRAC current appeared to be mediated by α1A-adrenoceptors coupled to Gq protein which were insensitive to PTX, and PKC inhibitors did not block the response to PE.
In addition, there is a large difference in the IC50 values for the PE-induced inhibition of VRAC current. Duan et al. obtained an IC50 value of 72 µM at 30°C, and this value is more than one order of magnitude higher than that obtained in the present study (1.2 µM at 37°C). The nature of these discrepancies between the results of Duan et al. and ours is unknown but the difference of tissue, species and temperature could be a factor. The different results of Du & and Sorota (2000) who showed that PE did not affect VRAC current in dog atrial cells, and that PKC activity stimulated in this current, might also reflect the tissue and species difference.
We mostly used a maximal concentration (100 µM) of PE to examine its effect on VRAC, though the IC50 value of PE for VRAC inhibition was about 1.2 µM (Figure 3D). As the PE-induced inhibition of VRAC was suppressed by application of an α1-adrenoceptor antagonist (5 µM prazosin) and an α1A-adrenoceptor-selective antagonist (2 µM WB-4101) (Figure 5A), PE must have acted on α1A-adrenoceptors to reduce VRAC current. However, the high concentrations of PE might induce some complications, by exerting non-specific effects on VRAC. The incomplete suppression by the above antagonists of the PE-induced VRAC inhibition (Figure 5A) might raise the possibility that PE at high concentrations had an inhibitory effect on VRAC, independent of α1A-adrenoceptors. Alternatively, using the high concentration (100 µM) of PE may have effectively competed with the antagonists, as the concentration of the latter was low (5 µM for prazosin and 2 µM for WB-4101). Further studies are necessary to clarify this point. Because the presence of 1 µM propranolol did not affect the PE-induced inhibition of VRAC (Figure 5A), at least the activation of β-adrenoceptors by high concentrations of PE (Wagner et al., 1974), if any, did not appear to be involved in the observed responses to PE.
In the present study, we examined the effect of PE on VRAC by exposing the cells to HYPO in the presence of PE. The experiments with a different protocol, where PE should be applied to the cells after the VRAC current had developed in advance in HYPO, remain to be done, because they would provide more detailed information on the mechanism underlying the PE action on VRAC. Our preliminary examinations with the latter protocol showed that PE could only slightly attenuate the activated VRAC current. This might indicate a complicated feature of the activation process of VRAC as influenced by HYPO and the PE -phosphatidylinositide system. We are now extending our study to confirm and to analyze the said finding.
α1A-Adrenoceptors are members of Gq protein-coupled receptor (GqPCR) family. Endothelin (ET) and angiotensin II (AT-II) are cardioactive signalling molecules whose receptors (ET receptor and AT1 receptor) are also members of GqPCR family. Activation of VRAC is controlled by a signalling cascade of AT-II and reactive oxygen species involving epidermal growth factor receptor kinase and PI3K in rabbit ventricular cells (Browe and Baumgarten, 2004; 2006; Ren et al., 2008). In dog atrial cells, ET-2 enhances VRAC current by a mechanism which is dependent on mitogen-activated protein kinase (Du and Sorota, 2000). Thus, the activation of several types of GqPCR appears to modulate cardiac VRAC, but the downstream signalling pathway may differ depending on the specific receptor type. It is yet unknown whether the stimulation of ET or AT1 receptors influences VRAC or induces any VRAC-like current in mouse cardiac cells, and this issue may be a subject of future study. It should be noted, however, that the stimulation of the P2Y receptor, another member of GqPCR family, did not induce VRAC-like current in the ventricular cells from CFTR-deficient mice (Yamamoto-Mizuma et al., 2004a).
The maintenance of cellular content and volume is essential for the normal function of the cell, and the activation of Cl- transport is a key step in RVD under conditions of extracellular hypotonicity or intracellular hypertonicity (Sardini et al., 2003). VRAC current directly contributes to the RVD in cardiomyocytes and the adaptive remodelling of heart during cardiac hypertrophy and heart failure (Duan et al., 2005). On the other hand, α1-adrenoceptors have been shown to be required for physiological cardiac hypertrophy in mouse models (O'Connell et al., 2003) and the stimulation of α1-adrenoceptors induced hypertrophic changes in neonatal rat ventricular cells (LaMorte et al., 1994). As discussed by Duan et al. (2005), the volume regulation of cardiac cells exerted by VRAC current could be cardioprotective under the pathological conditions which would lead to myocardial hypertrophy. As stress on working hearts is usually accompanied by elevated sympathetic tone, it is tempting to consider that an attenuation of VRAC by α1-adrenoceptor stimulation and the associated loss of RVD facilitate the development of cardiac hypertrophy. Further studies are necessary to clarify these points and to elucidate the pathophysiological roles of α1-adrenoceptor mediated regulation of VRAC currents in cardiac function.
Acknowledgments
This research was partly supported by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture (No. 17500276 to T.E., and Nos. 18790158 and 20500366 to S.Y.).
Glossary
Abbreviations
- AT-II
angiotensin II
- BIM
bisindolylmaleimide-I
- CFTR
cystic fibrosis transmembrane conductance regulator
- CHE
chelerythrine
- DAG
diacylglycerol
- ECl
chloride equilibrium potential
- Erev
reversal potential
- ET
endothelin
- GqPCR
Gq protein-coupled receptor
- HYPO
hypotonic solution
- IP3
inositol 1,4,5-trisphosphate
- ISO
isotonic solution
- OAG
1-oleoyl-2-acetyl-sn-glycerol
- PI3K
phosphoinositide 3-kinase
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PKC
protein kinase C
- PLC
phosphoinositide-specific phospholipase C
- PSS
physiological saline solution
- PTX
Pertussis toxin
- RVD
regulatory volume decrease
- STS
staurosporine
- VRAC
volume-regulated anion channel
Conflicts of interest
None.
References
- Alexander RW, Williams LT, Lefkowitz RJ. Identification of cardiac beta-adrenergic receptors by (-) [3H]alprenolol binding. Proc Natl Acad Sci USA. 1975;72:1564–1568. doi: 10.1073/pnas.72.4.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th Edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae YS, Lee TG, Park JC, Hur JH, Kim Y, Heo K, et al. Identification of a compound that directly stimulates phospholipase C activity. Mol Pharmacol. 2003;63:1043–1050. doi: 10.1124/mol.63.5.1043. [DOI] [PubMed] [Google Scholar]
- Bewick NL, Fernandes C, Pitt AD, Rasmussen HH, Whalley DW. Mechanisms of Na+-K+ pump regulation in cardiac myocytes during hyposmolar swelling. Am J Physiol. 1999;276:C1091–C1099. doi: 10.1152/ajpcell.1999.276.5.C1091. 5 Pt 1. [DOI] [PubMed] [Google Scholar]
- Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ, et al. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther. 1990;255:756–768. [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl- current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–287. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3K and NADPH oxidase in ventricular myocytes. J Gen Physiol. 2006;127:237–251. doi: 10.1085/jgp.200509366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Nam GB, Lee SH, Earm YE, Ho WK. Phosphatidylinositol 4,5-bisphosphate is acting as a signal molecule in α1-adrenergic pathway via the modulation of acetylcholine-activated K+ channels in mouse atrial myocytes. J Biol Chem. 2001;276:159–164. doi: 10.1074/jbc.M004826200. [DOI] [PubMed] [Google Scholar]
- Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–606. doi: 10.1016/s0092-8674(00)80696-0. [DOI] [PubMed] [Google Scholar]
- Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K. DCPIB is a novel selective blocker of ICl,swell and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol. 2001;134:1467–1479. doi: 10.1038/sj.bjp.0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng XF, Chemtob S, Varma DR. Characterization of α1D-adrenoceptor subtype in rat myocardium, aorta and other tissues. Br J Pharmacol. 1996;119:269–276. doi: 10.1111/j.1476-5381.1996.tb15981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du XY, Sorota S. Cardiac swelling-induced chloride current is enhanced by endothelin. J Cardiovasc Pharmacol. 2000;35:769–776. doi: 10.1097/00005344-200005000-00014. [DOI] [PubMed] [Google Scholar]
- Duan D, Fermini B, Nattel S. α-adrenergic control of volume-regulated Cl- currents in rabbit atrial myocytes. Characterization of a novel ionic regulatory mechanism. Circ Res. 1995;77:379–393. doi: 10.1161/01.res.77.2.379. [DOI] [PubMed] [Google Scholar]
- Duan DY, Liu LL, Bozeat N, Huang ZM, Xiang SY, Wang GL, et al. Functional role of anion channels in cardiac diseases. Acta Pharmacol Sin. 2005;26:265–278. doi: 10.1111/j.1745-7254.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- Fedida D, Braun AP, Giles WR. α1-adrenoceptors in myocardium: functional aspects and transmembrane signaling mechanisms. Physiol Rev. 1993;73:469–487. doi: 10.1152/physrev.1993.73.2.469. [DOI] [PubMed] [Google Scholar]
- Feranchak AP, Roman RM, Schwiebert EM, Fitz JG. Phosphatidylinositol 3-kinase contributes to cell volume regulation through effects on ATP release. J Biol Chem. 1998;273:14906–14911. doi: 10.1074/jbc.273.24.14906. [DOI] [PubMed] [Google Scholar]
- Gong W, Xu H, Shimizu T, Morishima S, Tanabe S, Tachibe T, et al. ClC-3-independent, PKC-dependent activity of volume-sensitive Cl channel in mouse ventricular cardiomyocytes. Cell Physiol Biochem. 2004;14:213–224. doi: 10.1159/000080330. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Large WA. Properties of a Cl- current activated by cell swelling in rabbit portal vein vascular smooth muscle cells. Am J Physiol. 1998;275:H1524–H1532. doi: 10.1152/ajpheart.1998.275.5.H1524. 5 Pt 2. [DOI] [PubMed] [Google Scholar]
- Haruna T, Yoshida H, Nakamura TY, Xie LH, Otani H, Ninomiya T, et al. α1-adrenoceptor-mediated breakdown of phosphatidylinositol 4,5-bisphosphate inhibits pinacidil-activated ATP-sensitive K+ currents in rat ventricular myocytes. Circ Res. 2002;91:232–239. doi: 10.1161/01.res.0000029971.60214.49. [DOI] [PubMed] [Google Scholar]
- Hein P, Michel MC. Signal transduction and regulation: are all α1-adrenergic receptor subtypes created equal? Biochem Pharmacol. 2007;73:1097–1106. doi: 10.1016/j.bcp.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiol Rev. 2000;80:31–81. doi: 10.1152/physrev.2000.80.1.31. [DOI] [PubMed] [Google Scholar]
- Ichishima K, Yamamoto S, Ehara T. Regulation of volume-regulated outwardly rectifying anion channels by phosphatidylinositol 3,4,5-triphosphate in mouse ventricular cells. J Physiol Sci. 2009;59(Suppl. 1):127. doi: 10.2220/biomedres.29.307. [DOI] [PubMed] [Google Scholar]
- Iyadomi I, Hirahara K, Ehara T. α-Adrenergic inhibition of the β-adrenoceptor-dependent chloride current in guinea-pig ventricular myocytes. J Physiol. 1995;489:95–104. doi: 10.1113/jphysiol.1995.sp021033. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R. The Na+/Ca2+ exchange inhibitor KB-R7943 potently blocks TRPC channels. Biochem Biophys Res Commun. 2007;361:230–236. doi: 10.1016/j.bbrc.2007.07.019. [DOI] [PubMed] [Google Scholar]
- LaMorte VJ, Thorburn J, Absher D, Spiegel A, Brown JH, Chien KR, et al. Gq- and ras-dependent pathways mediate hypertrophy of neonatal rat ventricular myocytes following α1-adrenergic stimulation. J Biol Chem. 1994;269:13490–13496. [PubMed] [Google Scholar]
- Luo DL, Gao J, Fan LL, Tang Y, Zhang YY, Han QD. Receptor subtype involved in α1-adrenergic receptor-mediated Ca2+ signaling in cardiomyocytes. Acta Pharmacol Sin. 2007;28:968–974. doi: 10.1111/j.1745-7254.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- Ma T, Vetrivel L, Yang H, Pedemonte N, Zegarra-Moran O, Galietta LJ, et al. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J Biol Chem. 2002;277:37235–37241. doi: 10.1074/jbc.M205932200. [DOI] [PubMed] [Google Scholar]
- Meyer T, Wellner-Kienitz MC, Biewald A, Bender K, Eickel A, Pott L. Depletion of phosphatidylinositol 4,5-bisphosphate by activation of phospholipase C-coupled receptors causes slow inhibition but not desensitization of G protein-gated inward rectifier K+ current in atrial myocytes. J Biol Chem. 2001;276:5650–5658. doi: 10.1074/jbc.M009179200. [DOI] [PubMed] [Google Scholar]
- Nishida M, Kurose H. Roles of TRP channels in the development of cardiac hypertrophy. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:395–406. doi: 10.1007/s00210-008-0321-8. [DOI] [PubMed] [Google Scholar]
- O'Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, et al. The α1A/C- and α1B-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest. 2003;111:1783–1791. doi: 10.1172/JCI16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y. Cell volume-sensitive chloride channels. Contrib Nephrol. 1998;123:21–33. doi: 10.1159/000059920. [DOI] [PubMed] [Google Scholar]
- Okada Y, Shimizu T, Maeno E, Tanabe S, Wang X, Takahashi N. Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J Membr Biol. 2006;209:21–29. doi: 10.1007/s00232-005-0836-6. [DOI] [PubMed] [Google Scholar]
- O-Uchi J, Sasaki H, Morimoto S, Kusakari Y, Shinji H, Obata T, et al. Interaction of α1-adrenoceptor subtypes with different G proteins induces opposite effects on cardiac L-type Ca2+ channel. Circ Res. 2008;102:1378–1388. doi: 10.1161/CIRCRESAHA.107.167734. [DOI] [PubMed] [Google Scholar]
- Patane MA, Scott AL, Broten TP, Chang RS, Ransom RW, DiSalvo J, et al. 4-Amino-2-[4-[1-(benzyloxycarbonyl)-2(S)- [[(1,1-dimethylethyl)amino]carbonyl]-piperazinyl]-6, 7-dimethoxyquinazoline (L-765,314): a potent and selective alpha1b adrenergic receptor antagonist. J Med Chem. 1998;41:1205–1208. doi: 10.1021/jm980053f. [DOI] [PubMed] [Google Scholar]
- Ren Z, Raucci FJ, Jr, Browe DM, Baumgarten CM. Regulation of swelling-activated Cl- current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res. 2008;77:73–80. doi: 10.1093/cvr/cvm031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Matsuura H, Ehara T. Swelling-induced Cl- current in guinea-pig atrial myocytes: inhibition by glibenclamide. J Physiol. 1997;505:41–52. doi: 10.1111/j.1469-7793.1997.041bc.x. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardini A, Amey JS, Weylandt KH, Nobles M, Valverde MA, Higgins CF. Cell volume regulation and swelling-activated chloride channels. Biochim Biophys Acta. 2003;1618:153–162. doi: 10.1016/j.bbamem.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Sarkadi B, Parker JC. Activation of ion transport pathways by changes in cell volume. Biochim Biophys Acta. 1991;1071:407–427. doi: 10.1016/0304-4157(91)90005-h. [DOI] [PubMed] [Google Scholar]
- Shi C, Barnes S, Coca-Prados M, Kelly ME. Protein tyrosine kinase and protein phosphatase signaling pathways regulate volume-sensitive chloride currents in a nonpigmented ciliary epithelial cell line. Invest Ophthalmol Vis Sci. 2002;43:1525–1532. [PubMed] [Google Scholar]
- Sorota S. Pharmacologic properties of the swelling-induced chloride current of dog atrial myocytes. J Cardiovasc Electrophysiol. 1994;5:1006–1016. doi: 10.1111/j.1540-8167.1994.tb01143.x. [DOI] [PubMed] [Google Scholar]
- Sorota S. Insights into the structure, distribution and function of the cardiac chloride channels. Cardiovasc Res. 1999;42:361–376. doi: 10.1016/s0008-6363(99)00039-5. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Rees SA, Wright AR, Powell T. Cell swelling and ion transport pathways in cardiac myocytes. Cardiovasc Res. 1996;32:85–97. [PubMed] [Google Scholar]
- Vessey JP, Shi C, Jollimore CA, Stevens KT, Coca-Prados M, Barnes S, et al. Hyposmotic activation of ICl,swell in rabbit nonpigmented ciliary epithelial cells involves increased ClC-3 trafficking to the plasma membrane. Biochem Cell Biol. 2004;82:708–718. doi: 10.1139/o04-107. [DOI] [PubMed] [Google Scholar]
- Wagner J, Endoh M, Reinhardt D. Stimulation by phenylephrine of adrenergic α- and β-receptors in the isolated perfused rabbit heart. Naunyn Schmiedebergs Arch Pharmacol. 1974;282:307–310. doi: 10.1007/BF00501238. [DOI] [PubMed] [Google Scholar]
- Wang GX, McCrudden C, Dai YP, Horowitz B, Hume JR, Yamboliev IA. Hypotonic activation of volume-sensitive outwardly rectifying chloride channels in cultured PASMCs is modulated by SGK. Am J Physiol Heart Circ Physiol. 2004;287:H533–H544. doi: 10.1152/ajpheart.00228.2003. [DOI] [PubMed] [Google Scholar]
- Woodcock EA. Roles of α1A- and α1B-adrenoceptors in heart: insights from studies of genetically modified mice. Clin Exp Pharmacol Physiol. 2007;34:884–888. doi: 10.1111/j.1440-1681.2007.04707.x. [DOI] [PubMed] [Google Scholar]
- Wright AR, Rees SA. Cardiac cell volume: crystal clear or murky waters? A comparison with other cell types. Pharmacol Ther. 1998;80:89–121. doi: 10.1016/s0163-7258(98)00025-4. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Kobilka BK. Myocyte adrenoceptor signaling pathways. Science. 2003;300:1530–1532. doi: 10.1126/science.1079206. [DOI] [PubMed] [Google Scholar]
- Yamada S, Yamamura HI, Roeske WR. Characterization of alpha-1 adrenergic receptors in the heart using [3H]WB4101: effect of 6-hydroxydopamine treatment. J Pharmacol Exp Ther. 1980;215:176–185. [PubMed] [Google Scholar]
- Yamamoto S, Ehara T. Acidic extracellular pH-activated outwardly rectifying chloride current in mammalian cardiac myocytes. Am J Physiol Heart Circ Physiol. 2006;290:H1905–H1914. doi: 10.1152/ajpheart.00965.2005. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Ehara T, Shioya T. Changes in cell volume induced by activation of the cyclic AMP-dependent chloride channel in guinea-pig cardiac myocytes. Jpn J Physiol. 2001;51:31–41. doi: 10.2170/jjphysiol.51.31. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Ishihara K, Ehara T, Shioya T. Cell-volume regulation by swelling-activated chloride current in guinea-pig ventricular myocytes. Jpn J Physiol. 2004;54:31–38. doi: 10.2170/jjphysiol.54.31. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Ichishima K, Ehara T. Regulation of extracellular UTP-activated Cl- current by P2Y-PLC-PKC signaling and ATP hydrolysis in mouse ventricular myocytes. J Physiol Sci. 2007;57:85–94. doi: 10.2170/physiolsci.RP011406. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Ichishima K, Ehara T. Regulation of volume-regulated outwardly rectifying anion channels by phosphatidylinositol 3,4,5-trisphosphate in mouse ventricular cells. Biomed Res. 2008;29:307–315. doi: 10.2220/biomedres.29.307. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Ichishima K, Ehara T. Reduced volume-regulated outwardly rectifying anion channel activity in ventricular myocyte of Type 1 diabetic mice. J Physiol Sci. 2009;59:87–96. doi: 10.1007/s12576-008-0012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto-Mizuma S, Wang GX, Hume JR. P2Y purinergic receptor regulation of CFTR chloride channels in mouse cardiac myocytes. J Physiol. 2004a;556:727–737. doi: 10.1113/jphysiol.2003.059881. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto-Mizuma S, Wang GX, Liu LL, Schegg K, Hatton WJ, Duan D, et al. Altered properties of volume-sensitive osmolyte and anion channels (VSOACs) and membrane protein expression in cardiac and smooth muscle myocytes from Clcn3-/- mice. J Physiol. 2004b;557:439–456. doi: 10.1113/jphysiol.2003.059261. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H, Yasutake M, Avkiran M. α1-adrenergic stimulation of sarcolemmal Na+-H+ exchanger activity in rat ventricular myocytes: evidence for selective mediation by the α1A-adrenoceptor subtype. Circ Res. 1998;82:1078–1085. doi: 10.1161/01.res.82.10.1078. [DOI] [PubMed] [Google Scholar]