

Abstract
BACKGROUND AND PURPOSE
Recently identified antagonists of the urotensin–II (U-II) receptor (UT) are of limited utility for investigating the (patho)physiological role of U-II due to poor potency and limited selectivity and/or intrinsic activity.
EXPERIMENTAL APPROACH
The pharmacological properties of two novel UT antagonists, GSK1440115 and GSK1562590, were compared using multiple bioassays.
KEY RESULTS
GSK1440115 (pKi= 7.34–8.64 across species) and GSK1562590 (pKi= 9.14–9.66 across species) are high affinity ligands of mammalian recombinant (mouse, rat, cat, monkey, human) and native (SJRH30 cells) UT. Both compounds exhibited >100-fold selectivity for UT versus 87 distinct mammalian GPCR, enzyme, ion channel and neurotransmitter uptake targets. GSK1440115 showed competitive antagonism at UT in arteries from all species tested (pA2= 5.59–7.71). In contrast, GSK1562590 was an insurmountable UT antagonist in rat, cat and hUT transgenic mouse arteries (pKb= 8.93–10.12 across species), but a competitive antagonist in monkey arteries (pKb= 8.87–8.93). Likewise, GSK1562590 inhibited the hU-II-induced systemic pressor response in anaesthetized cats at a dose 10-fold lower than that of GSK1440115. The antagonistic effects of GSK1440115, but not GSK1562590, could be reversed by washout in rat isolated aorta. In ex vivo studies, GSK1562590 inhibited hU-II-induced contraction of rat aorta for at least 24 h following dosing. Dissociation of GSK1562590 binding was considerably slower at rat than monkey UT.
CONCLUSIONS AND IMPLICATIONS
Whereas both GSK1440115 and GSK1562590 represent high-affinity/selective UT antagonists suitable for assessing the (patho)physiological role of U-II, only GSK1562590 exhibited sustained UT residence time and improved preclinical efficacy in vivo.
Keywords: urotensin-II, UT, GSK1562590, GSK1440115, antagonist, blood pressure, G-protein-coupled receptor, residence time, dissociation rate, biochemical efficiency
Introduction
Human urotensin-II (hU-II) is a cyclic undecapeptide hormone and endogenous ligand of the G-protein-coupled U-II receptor, UT (nomenclature follows Alexander et al., 2009). Functionally, hU-II is among ‘the most potent mammalian vasoconstrictors identified to date’ (Ames et al., 1999). In addition to its vasoconstrictor activity, hU-II also modulates cardiac contractility (Russell et al., 2001; Gong et al., 2004; Quaile et al., 2009), cardiomyocyte hypertrophy and fibrosis (Tzanidis et al., 2003; Johns et al., 2004; Onan et al., 2004; Liu et al., 2009), natriuresis (Song et al., 2003; Zhang et al., 2003) and insulin release (Silvestre et al., 2001), raising the possibility that the hU-II/UT system plays a significant role in cardiovascular, renal and metabolic diseases.
Over the past decade, development of non-peptide UT antagonists has allowed investigators to begin to delineate the (patho)physiological role of the hU-II/UT system (see Maryanoff and Kinney, 2010). The most advanced drug candidate is Actelion's Palosuran (ACT-058362; Clozel et al., 2004) but phase 2b clinical trials were ceased in May 2005 due to lack of efficacy in diabetic nephropathy patients (http://www1.actelion.com/documents/publications/Milestones_Company.pdf; Clozel et al., 2006). However, it was recently suggested that this disappointing outcome with Palosuran might be the result of attenuated antagonist potency in intact cells/tissues (Behm et al., 2008) or masking of activity due to clinical co-administration with angiotensin II pathway blockers, as required for ethical reasons (Sidharta et al., 2006; Sidharta et al., 2009).
As the (patho)physiological significance of the hU-II/UT pathway remains ambiguous, the identification of UT antagonists with high selectivity and pan-species activity remains greatly desired. In addition, the association of commercially available drugs which exhibit slow receptor dissociation rates with improved clinical efficacies (see Swinney, 2004 and Copeland et al., 2006 for review) has raised interest in identifying UT antagonists with increased residence time (period of time that antagonist occupies its receptor). While optimizing high-throughput screening hits, we identified two compounds, GSK1440115 and GSK1562590 (Figure 1), which exhibited differential characteristics consistent with rapidly and slowly reversible modes of action respectively. The present study aimed to detail the pharmacological characteristics of both compounds and to test whether or not receptor dissociation rates extrapolated to altered efficacy in vitro, ex vivo and in vivo. Although both compounds are high-affinity/selective UT antagonists suitable for interrogating the (patho)physiological role of U-II, GSK1562590 represents the first UT antagonist with enhanced UT receptor residence time, resulting in extended duration of pharmacodynamic activity ex vivo.
Figure 1.
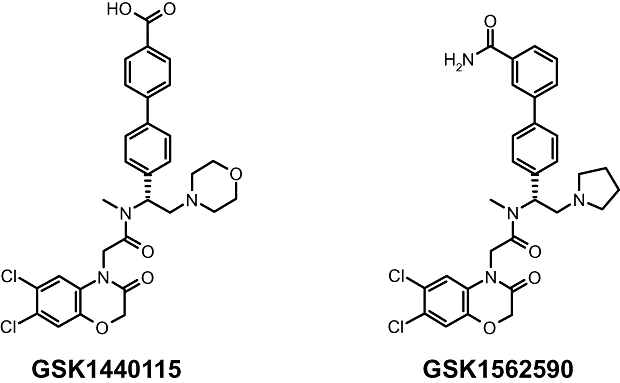
Structures of the novel UT antagonists (A) GSK1440115 (4′-[(1R)-1-[[(6,7-dichloro-3-oxo-2,3-dihydro-4H-1,4-benzoxazin-4-yl)acetyl](methyl)amino]-2-(4-morpholinyl)ethyl]-4-biphenylcarboxylic acid, trifluoroacetate) and (B) GSK1562590 (4′-[(1R)-1-[[(6,7-dichloro-3-oxo-2,3-dihydro-4H-1,4-benzoxazin-4-yl)acetyl](methyl)amino]-2-(1-pyrrolidinyl)ethyl]-3-biphenylcarboxamide, hydrochloride).
Methods
All studies were performed in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities in accordance with institutional guidelines.
Radioligand binding at recombinant UT
[125I]hU-II competition binding assays were performed using membranes isolated from human osteosarcoma (U2OS) cells, transiently expressing rat or human UT, or human embryonic kidney (HEK293) cells, stably expressing mouse, cat or monkey UT (Ames et al., 1999; Behm et al., 2006). Following a 30 min incubation of membranes with polystyrene WGA-SPA beads (1:50, ww−1) in assay buffer [20 mM Tris (pH 7.4) 5 mM MgCl2 and 0.1% BSA] at room temperature, the bead : UT membrane mixture was combined with [125I]hU-II (0.6 nM final concentration) and added to 384-well Proxy plates (Perkin Elmer, Shelton, CT) containing 0.1 nM–10 µM GSK1440115 or GSK1562590 or DMSO vehicle (1% final). Plates were incubated for 2 h (monkey and mouse UT) to 3 h (human, cat and rat UT) at room temperature and then read using scintillation counting (Viewlux or TopCount; Perkin Elmer). Non-specific binding was defined using 1 µM unlabelled hU-II.
Radioligand binding at native UT in human SJRH30 cells
Whole cell [125I]hU-II competition binding assays were performed at native human UT using intact SJRH30 cells (human rhabdomyosarcoma cell line; Douglas et al., 2004b). Assays were performed with 200 pM [125I]hU-II in the presence or absence of 1 pM-1 µM unlabelled hU-II, GSK1440115 or GSK1562590 in Dulbecco's phosphate-buffered saline (DPBS+; 0.7 mM CaCl2, 10 mM MgCl2, 1.4 mM glucose, 0.2% BSA). Assay plates were incubated at 37°C for 30 min. Following three washes with DPBS+, the cells were lysed with 2N NaOH and the resulting lysate was counted using a gamma counter (Wallac Wizard™; Perkin Elmer). Non-specific binding was defined using 1 µM unlabelled hU-II.
Reversibility of ligand binding at rat, monkey and human UT
Rat, monkey and human UT membranes were incubated with DMSO or 10 nM GSK1562590 for 30 min at 25°C. Following this equilibration period, incubation mixtures were diluted in buffer [25 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 2 mM Na-EGTA, 0.1 mg·mL−1 bacitracin] and centrifuged (47 000×g, 20 min, 4°C). The membrane pellets were then resuspended in buffer following which [125I]hU-II binding site density (Bmax) and affinity (KD) values were determined (t= 0). At t= 0.5, 1 or 2 h following the initial resuspension, a subset of membranes were ‘washed’ a second time and subjected to Bmax and KD determination. Bmax and KD values were determined by homologous competition binding (Wallac Wizard™) whereby multiple concentrations of unlabelled hU-II were used to compete for binding with a fixed concentration of [125I]hU-II (Douglas et al., 2005).
Secondary pharmacological profiling
The secondary pharmacological properties of 1 µM GSK1440115 and 1 µM GSK1562590 (concentrations >400- and 1900-fold above the human recombinant UT Ki values respectively) were initially characterized using a panel of 87 binding assays for distinct mammalian G-protein-coupled receptor, enzyme, ion channel and neurostransmitter uptake targets (Cerep, Paris, France): adenosine (A1,2A,3), adrenoceptors (non-selective α1,2, α1A,1B,2B,2C, β1,2,3), angiotensin (AT1,2), central benzodiazepine (BZD), bradykinin (B2), cannabinoid (CB1,2), cholecystokinin (CCK1), calcitonin gene-related peptide (CGRP), dopamine (D1,2S,3,4.4), endothelin (ETA,B), GABA (non-selective, GABAA), AMPA, kainate, NMDA, galanin (Gal2), Il-8 (CXCR2), histamine (H1,2), imidazoline (I2), leukotriene (BLT1), melanocortin (MC4), melatonin (MT1), muscarinic (M1-4), neuronal nicotinic, neuropeptide Y (Y1,2), neurotensin (NTS1), opioid (δ2, κ, µ, σ), P2X, 5-HT (5-HT1A,1B,1D,2A,2C,3,4e,5A,6,7), tachykinin (NK2,3), vasoactive intestinal peptide (VPAC1), vasopressin (V1a), oestrogen (ERa,b), progesterone (PR) and androgen (AR) receptors, Ca2+ (L-type), K+ (KV, SKCa), Na+ and Cl– channels, and noradrenaline and dopamine transporters. All assays were performed using human orthologues except non-selective α, and α1A adrenoceptors, σ opioid, AMPA, BZD, GABA, kainate, NMDA, I2, P2X, 5-HT1B, Ca2+, KV, SKCa, Na+, Cl- (rat), κ opioid (guinea-pig) and 5-HT1D (bovine) receptors. Dose–response curves were obtained for several of the more significant interactions (>50% activity) observed during the initial 1 µM screen.
Human κ opioid receptor (κ) agonism
κ opioid receptor agonism was measured using a [35S]GTPγS binding assay for human κ opioid receptor (κ) stably expressed in hamster CHO cell membranes. Assays were performed in the presence of DMSO vehicle (1%), GSK1440115 or GSK1562590, in a buffer containing 20 mM HEPES, 10 mM MgCl2, 100 mM NaCl (adjusted to pH 7.4 with KOH) and 10 µM GDP. Incubations were performed for 3–5 h (room temperature) at which point [35S]GTPγS SPA binding was evaluated using a Wallac 1430 ViewLux plus microplate imager.
Human tachykinin (hNK2) receptor antagonism
Inhibition of EC80 neurokinin A (NKA)-mediated Ca2+-mobilization was assessed using fluo-4-loaded, intact U2OS cells expressing human recombinant NK2 receptor by FLIPR (fluorescence imaging plate reader; Molecular Devices, Sunnyvale, CA) analysis in the presence of DMSO vehicle (1%), GSK1440115 or GSK1562590. Assays were performed in HEPES buffered saline [20 mM HEPES, 145 mM NaCl, 5 mM KCl, 1 mM CaCl2, 5.6 mM glucose (pH 7.3)].
Radioligand binding at NK2, 5-HT1A and D2S receptors
[3H]8-OH-DPAT and [3H]spiperone competition binding assays were performed using HEK293 cell membranes stably expressing human recombinant 5-HT1A and D2S receptors respectively. The [125I]NKA competition binding assay was performed using NK2 BacMam transduced CHOK1 cells. Briefly, varying concentrations of GSK1562590 (10 nM–30 µM) were added to cell membranes in the presence of 3 nM [3H]8-OH-DPAT, 3 nM [3H]spiperone or 0.15 nM [125I]NKA. Plates were incubated for 1 h at room temperature and SPA binding was determined via scintillation counting. Non-specific binding was defined using 10 µM 8-OH-DPAT (+)butaclamol or [Nle10]-NKA (4–10).
Mammalian in vitro vascular contractility assessment
Proximal descending thoracic aortae were isolated from male Sprague-Dawley rats (400–500 g, Charles River, Raleigh, NC) and hUT transgenic mice (25–35 g; Behm et al., 2008) following induction of anaesthesia (5% isoflurane in O2) and exsanguination. Following sodium pentobarbitone overdose (100 mg·kg−1, i.v.), mesenteric resistance arteries, femoral arteries and thoracic aortae were isolated from adult male cats (4–5 kg; Liberty Research Inc., Waverly, NY) and renal and superior mesenteric arteries were isolated from male cynomolgus monkeys (4–7 kg; Primate Products, Miami, FL; Covance, Alice TX; Charles River, Andover MA; Mannheimer, Homestead, FL).
Conduit arterial rings (approximately 3 mm in length) were denuded of endothelium by rubbing the lumen with a fine forceps and suspended in Krebs solution of the following composition (mM): NaCl (112.0), KCl (4.7), KH2PO4 (1.2), MgSO4 (1.2), CaCl2 (2.5), NaHCO3 (25.0), dextrose (11.0), indomethacin (0.01). Isometric force responses were measured (MLT0201/D transducers; Letica, Barcelona, Spain) under optimal resting tension (1.0 g, 2.0 g, 2.0 g and 0.5 g in rat, cat, monkey and transgenic mouse vessels respectively; Douglas et al., 2000; Behm et al., 2004). Cat endothelium-intact mesenteric resistance arteries (125–150 µm internal diameter) were mounted on a wire myograph (Danish myotechnologies, Aarhus, Denmark) under 0.5 g optimal resting tension. Changes in isometric force were recorded digitally (ADInstruments Chart 5.0 software, Colorado Springs, CO, USA). Following 1 h equilibration, vessels were treated with 60 mM KCl and 1 µM phenylephrine (subsequent responses were normalized to KCl). Once the contractile response to phenylephrine reached a plateau, carbachol (10 µM) was added in order to evaluate functional endothelial integrity.
Arteries were pretreated (30 min) with vehicle (0.1% DMSO), GSK1440115 (300–10 000 nM) or GSK1562590 (0.001–1000 nM), following which cumulative concentrations of hU-II (0.01–10 000 nM) were added to the tissue baths at half-log increments. Contractile responses to each concentration of hU-II were allowed to reach a plateau (∼10–15 min) before the addition of subsequent concentrations, and only one concentration–response curve was generated per tissue. In separate experiments, selectivity studies were performed with GSK1440115 (3 µM) and GSK1562590 (0.3 µM) using non-UT spasmogens (KCl, phenylephrine and endothelin-1).
Reversibility of UT antagonism in rat isolated aorta: in vitro washout studies
Cumulative concentration–response curves to hU-II (0.1 nM–3 µM) were generated following a 30 min pretreatment with vehicle (0.1% DMSO), GSK1440115 (1000 nM) or GSK1562590 (0.3 nM). Separate tissues were washed repeatedly for 1.5–24 h with fresh Krebs solution (not containing antagonist) before generating the hU-II concentration–response curves.
Reversibility of UT antagonism in rat isolated aorta: ex vivo studies
Male Sprague-Dawley rats (400–500 g) were dosed via oral gavage with vehicle (5% DMSO, 20% hydroxylpropyl-beta-cyclodextran) or GSK1562590 (1 mg·kg−1). Following time periods ranging from 2–48 h, rats were anaesthetized with inhaled isoflurane (5% in O2) and killed by cervical dislocation and exsanguination. Rings of the proximal descending thoracic aorta were suspended in tissue baths for generation of hU-II concentration–response curves (0.1 nM–10 µM) as described above. Blood was collected just before death for determining plasma drug concentrations.
Haemodynamic assessment in the anaesthetized cat
Haemodynamic measurements were made in anaesthetized cats as previously described (Behm et al., 2004). Briefly, male and female cats (1–4 kg) were initially anaesthetized with ketamine (3 mg·kg−1, i.m.) followed by isoflurane (5% in O2) and artificially ventilated via a tracheal cannula (Model 665 ventilator, Harvard Apparatus, Holliston, MA). Femoral arteries were catheterized for the measurement of mean, systolic and diastolic arterial blood pressure. A Swan-Gantz catheter was inserted into the left femoral vein and advanced into the pulmonary artery to measure cardiac output by thermodilution (Cardiomax, Columbus, OH). A pressure transducer was advanced into the left ventricle via the carotid artery. A lead II ECG was also recorded via limb lead electrodes. Pressure, ECG and blood flow signals were pre-amplified (Astro-Medical, West Warwick, RI) prior to digitization (500 Hz) using a computerized data acquisition system (CA recorder, DISS, Integrated Telemetry Systems, Dexter, MI). ECG intervals were subsequently analysed using computerized pattern recognition analysis (Emka Technologies, Falls Church, VA). Following surgical instrumentation, anaesthesia was maintained with α-chloralose (65 mg·kg−1, i.v. bolus) and haemodynamics and blood gases were allowed to stabilize (∼20 min). At 60 min prior to hU-II administration (1 nmol·kg−1, i.v. bolus), cats were pretreated with vehicle (5% DMSO, 20% aqueous Cavitron™[hydroxypropyl-β-cyclodextrin]), GSK1440115 (0.3, 1, 3 and 10 mg·kg−1) or GSK1562590 (0.01, 0.03, 0.1, 1 and 10 mg·kg−1) via a 30 min i.v. infusion (0.167 mL·min−1).
Quantification of plasma drug levels
Plasma levels of GSK1440115 and GSK1562590 were determined using liquid chromatography/tandem mass spectrometric (LC/MS/MS) detection. Briefly, drug levels were quantified via selected reaction monitoring of the transition from ionized drug (m/z = 598 and 582 for GSK1440115 and GSK1562590 respectively) to the fragment ion (m/z = 511 and 511 respectively) using atmospheric pressure chemical ionization on an Applied Biosystems API 5000 (Foster City, CA).
Data analysis
Unless stated otherwise, all values are mean ± standard error of the mean and n represents the total number of animals studied or individual experiments performed. Competition binding curves were analysed by non-linear regression (GraphPad Prism, La Jolla, CA) using the equation by Cheng & Prusoff (1973):
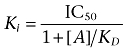 |
where [A] represents the concentration of competing ligand (GSK1440115 or GSK1562590), IC50 the concentration of competing ligand that inhibits radiolabel binding by 50% and KD the equilibrium dissociation constant of the radioligand.
Concentration-dependent contractility curves were fitted to a logistic equation as previously described (Douglas et al., 2005):
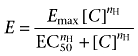 |
where E is the contractile response, [C] the concentration of agonist, EC50 the concentration of agonist required to produce a half maximal response, nH the Hill coefficient and Emax the maximum contractile response.
Empirical measurements of competitive antagonist potencies (pA2, determined using a single concentration of antagonist) were calculated using the Schild equation (Jenkinson et al., 1998):
where DR is the dose-ratio (ratio of equiactive concentrations of agonist in the presence and absence of antagonist) and [B] is the concentration of antagonist.
Competitive antagonist affinities generated using multiple concentrations of antagonist [the equilibrium dissociation constant pKb and associated 95% confidence intervals (CI)] were calculated using non-linear regression (Clark) analysis (Lew and Angus, 1997):
where [B] is the antagonist concentration and Log c is the difference between the antagonist pKb and the agonist control curve pEC50.
Noncompetitive antagonist affinities (pKb) were determined using the method of Gaddum where equiactive concentrations of agonist in the absence or presence of the noncompetitive antagonist were compared in a linear regression (Gaddum et al., 1955; Kenakin, 2006). The resulting slope was used to calculate the equilibrium constant Kb using the following equation:
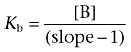 |
Where [B] is the antagonist concentration and slope is calculated from the double reciprocal plot of equiactive concentrations of agonist in the presence and absence of antagonist.
Areas under the blood pressure responses following hU-II administration were calculated by applying the trapezoid rule. Statistical comparisons were made using paired, two-tailed t-tests or anova (with Dunnett's or Bonferroni multiple comparisons post-tests) and values were considered significantly different when P≤ 0.05.
Materials
GSK1440115 (4′-[(1R)-1-[[(6,7-dichloro-3-oxo-2,3-dihydro-4H-1,4-benzoxazin-4-yl)acetyl](methyl)amino]-2-(4-morpholinyl)ethyl]-4-biphenylcarboxylic acid, trifluoroacetate; Figure 1) and GSK1562590 (4′-[(1R)-1-[[(6,7-dichloro-3-oxo-2,3-dihydro-4H-1,4-benzoxazin-4-yl)acetyl](methyl)amino]-2-(1-pyrrolidinyl)ethyl]-3-biphenylcarboxamide, hydrochloride; Figure 1) were synthesized at GlaxoSmithKline (King of Prussia, PA). hU-II, [125I] hU-II (Tyr9 monoiodinated) and endothelin-1 were synthesized by California Peptide Research Inc. (Napa, CA), Perkin Elmer (Shelton, CT) and American Peptide Company, Inc. (Sunnyvale, CA) respectively. Leadseeker WGA-SPA beads were from Amersham (Arlington, Heights, IL). Isoflurane, ketamine and sodium pentobarbital were from Abbott Laboratories (North Chicago, IL), Fort Dodge Animal Health (Ford Dodge, IA) and Vortech Pharmaceuticals (Dearborn, MI) respectively. α-chloralose (Spectrum Chemical, New Brunswick, NJ) was freshly prepared as a sterile saline solution (40 mg·mL−1) containing 25 mg·mL−1 of sodium bicarbonate (JT Baker, Phillipsburg, NJ) and 20 mg·mL−1 sodium tetraborate decahydrate (Sigma, St. Louis, MO). Carbachol, indomethacin, phenylephrine were from Sigma. All other reagents were of analytical grade.
Results
Radioligand binding at recombinant UT
GSK1440115 (pKi= 7.34–8.64) and GSK1562590 (pKi= 9.14–9.66) functioned as high affinity ligands at all recombinant mammalian UT orthologues studied, including mouse, rat, cat, monkey and human. Overall, GSK1562590 was a 5- to 101-fold more potent ligand across species as compared with GSK1440115. Hill slopes approximated unity for GSK1440115 and GSK1562590 (ranging from 0.70 to 0.94 and 0.95 to 1.47 respectively), indicating non-cooperative interactions with a single class of binding sites (Table 1). Both compounds consistently displaced the radioligand [125I]hU-II by 100%.
Table 1.
GSK1440115 and GSK1562590 binding affinities to recombinant and native mammalian UT
| pKi | nH | n | |||||
|---|---|---|---|---|---|---|---|
| UT orthologue | GSK1440115 | GSK1562590 | GSK1440115 | GSK1562590 | GSK1440115 | GSK1562590 | Relative affinity |
| Mouse (recombinant) | 7.34 ± 0.07 | 9.34 ± 0.02 | 0.93 ± 0.04 | 1.47 ± 0.05 | 4 | 13 | 101-fold |
| Rat (recombinant) | 8.49 ± 0.25 | 9.66 ± 0.04* | 0.70 ± 0.09 | 0.95 ± 0.05 | 4 | 10 | 25-fold |
| Cat (recombinant) | 8.45 ± 0.15 | 9.64 ± 0.03† | 0.78 ± 0.05 | 1.39 ± 0.03 | 4 | 13 | 22-fold |
| Monkey (recombinant) | 7.53 ± 0.12 | 9.14 ± 0.03 | 0.94 ± 0.05 | 1.05 ± 0.03 | 4 | 13 | 48-fold |
| Human (recombinant) | 8.64 ± 0.06 | 9.28 ± 0.0† | 0.88 ± 0.02 | 1.07 ± 0.02 | 12 | 33 | 5-fold |
| Human (native; SJRH30) | 8.38 ± 0.11 | 9.46 ± 0.06 | 0.81 ± 0.08 | 1.09 ± 0.15 | 4 | 4 | 15-fold |
pKi > 9.79 observed in n= 5 additional experiments.
pKi > 9.79 observed in n= 2 additional experiments.
GSK1440155 and GSK1562590 consistently displaced the radioligand [125I]hU-II by 100%. All values are expressed as mean ± SEM.
Radioligand binding at native UT in human SJRH30 cells
In accordance with the pKi values determined for human recombinant UT, both GSK1440115 and GSK1562590 potently inhibited [125I]hU-II binding to endogenously expressed human UT in intact SJRH30 cells. Similar to the fivefold difference observed at the recombinant receptor, GSK1562590 was 15-fold more potent than GSK1440115 (pKi values of 9.46 ± 0.06 and 8.38 ± 0.11 respectively; n= 4) (Table 1).
Secondary pharmacology profiling
The only ‘non-UT’ assay in which >30% inhibition was observed with 1 µM GSK1440115 was at the κ opioid receptor (99% inhibition of [3H]U-69593 binding in guinea-pig cerebellum; Table 2). In accord with these observations, GSK1440115 was a full agonist of the human κ opioid receptor (κ), providing an pEC50 of 5.63 ± 0.27 (>1000-fold selectivity relative to human UT pKi value; Table 3) in an [35S]GTPγS binding assay.
Table 2.
Secondary in vitro pharmacological properties of GSK1440115 and GSK1562590 (% inhibition of radioligand binding at 1 µM)
| % inhibition at 1 µM | ||||
|---|---|---|---|---|
| Target | Species | Radioligand | GSK1440115 | GSK1562590 |
| κ opioid | guinea-pig | [3H]U-69593 | 99 | 100 |
| NK2 | human | [125I]NKA | 29 | 81 |
| GABAA | rat | [3H]muscimol | 27 | <20 |
| 5-HT1A | human | [3H]8-OH-DPAT | 22 | 90 |
| 5-HT2A | human | [3H]ketanserin | 21 | 22 |
| µ opioid | human | [3H]DAMGO | 20 | 49 |
| D2S | human | [3H]spiperone | <20 | 82 |
| α2C | human | [3H]RX821002 | <20 | 50 |
| α1A | rat | [3H]prazosin | <20 | 47 |
| α1B | human | [3H]prazosin | <20 | 44 |
| DA transporter | human | [3H]BTCP | <20 | 38 |
| α1 (non-selective) | rat | [3H]prazosin | <20 | 36 |
| M3 | human | [3H]4-DAMP | <20 | 29 |
| NE transporter | human | [3H]nisoxetine | <20 | 22 |
| M2 | human | AF-DX 384 | <20 | 20 |
| δ opioid | human | [3H]DADLE | <20 | 20 |
| All other targets | – | – | <20 | <20 |
All values are expressed as mean (n= 2).
5-HT1A/2A, 5-hydroxytryptamine receptors; α1/1A/1B/2C, α-adrenoceptors, D2S, dopamine receptor; DA transporter, dopamine transporter; GABAA, gamma-aminobutyric acid receptor; M2/3, muscarinic receptors; NE transporter, noradrenaline transporter; NK2, tachykinin B receptor.
Table 3.
Secondary in vitro pharmacological properties of GSK1440115 and GSK1562590 (potency determination)
| Potency | Selectivity relative to human UT pKi | |||
|---|---|---|---|---|
| Recombinant human receptor target | GSK1440115 | GSK1562590 | GSK1440115 | GSK1562590 |
| D2S (pKi) | – | 7.02 ± 0.03 (n= 3) | – | 194 |
| κ opioid (pEC50) | 5.63 ± 0.27 (n= 7) | 6.94 ± 0.05 (n= 9) | 2113 | 242 |
| 5-HT1A (pKi) | – | 6.83 ± 0.03 (n= 3) | – | 444 |
| NK2 (pKi) | – | 5.71 ± 0.25 (n= 2) | – | 356 |
| NK2 (pKb) | 5.31 ± 0.04 (n= 2) | 5.65 ± 0.08 (n= 2) | 1988 | 4596 |
GSK1440115 and GSK1562590 did not antagonize the κ opioid receptor (pKb < 5.12). Relative affinities/activities for GSK1440115 and GSK1562590 are expressed relative to their respective human UT pKi values. All values are expressed as mean ± SEM.
The initial selectivity profile of 1 µM GSK1562590 suggested appreciable interactions (>30% inhibition) with aminergic receptors (5-HT, dopamine, α-adrenoceptors), peptidergic receptors (opioid, tachykinin) and the dopamine transporter (Table 2). Less than 30% inhibition was seen in all other assays tested. Several of the more significant interactions (>50% inhibition at 1 µM) were profiled in more detail. Accordingly, GSK1562590 functioned as a ligand for 5-HT1A and D2S receptors, and as a κ opioid receptor agonist and a NK2 antagonist (Table 3), but still with ≥194-fold selectivity for the human UT (0.5 nM UT pKi).
Functional potency in rat isolated aortae
GSK1440115 (0.3–3.0 µM) antagonized hU-II-induced contraction of the rat aorta in a concentration-dependent manner with a pKb of 7.36 (7.18–7.54 95% CI; Figure 2A–B, Table 4). Clark analysis was consistent with a competitive, surmountable mode of action [nH= 0.97 (0.84–1.09 95% CI)].
Figure 2.
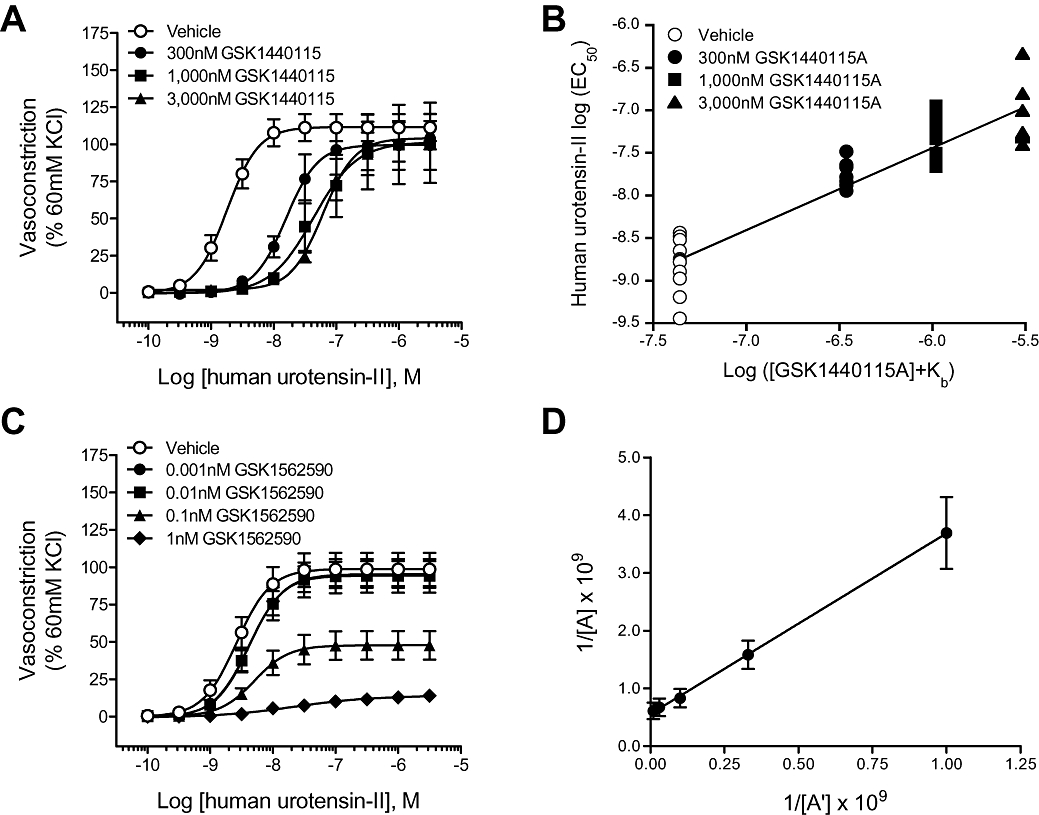
Inhibition of hU-II-induced contraction of rat isolated aortae by GSK1440115 and GSK1562590. (A) GSK1440115 elicited parallel, rightward shifts in the hU-II concentration–response curve. (B) Clark plot (global nonlinear regression analysis) revealed pA2= 7.36 (7.18–7.54 95% CI) and nH= 0.97 (0.84–1.09 CI) consistent with competitive hU-II antagonism. In contrast to GSK1440115 (C) GSK1562590 suppressed the maximum contractile response to hU-II in a concentration-dependent manner (half-log unit values of GSK1562590 were removed for ease of interpretation). (D) Double reciprocal plot of equiactive concentrations of hU-II in the absence (A) and presence (A') of 0.1 nM GSK1562590 is linear (consistent with noncompetitive antagonism) with a slope of 3.13 ± 0.51, indicating a pKb of 10.21 ± 0.11.
Table 4.
Concentration-dependent inhibition of hU-II-induced contraction of rat isolated aorta by GSK1440115 and GSK1562590
| GSK1440115 (nM) | hU-II Emax (% KCl) | hU-II pEC50 | Dose-ratio |
|---|---|---|---|
| Vehicle (n= 13) | 111 ± 9 | 8.80 ± 0.08 | – |
| 300 (n= 8) | 100 ± 17 | 7.76 ± 0.05*** | 10.2 ± 1.9 |
| 1000 (n= 12) | 100 ± 26 | 7.37 ± 0.07*** | 30.9 ± 5.2 |
| 3000 (n= 8) | 110 ± 12 | 7.09 ± 0.13*** | 52.7 ± 13.3 |
| GSK1562590 (nM) | hU-II Emax (% KCl) | hU-II pEC50 | Dose-ratio |
| Vehicle (n= 11) | 98 ± 11 | 8.55 ± 0.07 | – |
| 0.001 (n= 10) | 95 ± 9 | 8.38 ± 0.06 | 1.8 ± 0.3 |
| 0.003 (n= 10) | 74 ± 11 | 8.19 ± 0.12 | 4.1 ± 1.5 |
| 0.01 (n= 11) | 94 ± 11 | 8.35 ± 0.05 | 1.7 ± 0.2 |
| 0.03 (n= 10) | 70 ± 11 | 8.46 ± 0.06 | 1.4 ± 0.2 |
| 0.1 (n= 10) | 48 ± 10** | 8.29 ± 0.02 | 2.1 ± 0.4 |
| 0.3 (n= 10) | 38 ± 9** | 7.93 ± 0.07*** | 6.0 ± 1.9 |
| 1 (n= 10) | 14 ± 2** | 7.51 ± 0.17*** | 22.6 ± 8.5 |
All values are expressed as mean ± SEM. Statistical comparisons for both pEC50 and Emax values were performed using anova analysis with a Dunnett's post-test where
P < 0.01 and
P < 0.001
versus vehicle control values. Global nonlinear regression (Clark) analysis of the competitive UT antagonist GSK1440115 resulted in a pA2 of 7.36 (7.18–7.54 95% CI; Figure 2A–B). Linear regression analysis of 0.1 nM GSK1562590 using the method of Gaddum resulted in a linear plot (consistent with competitive antagonist) with a slope of 3.13 ± 0.51, equating to a pKb of 10.21 ± 0.11 (Figure 2D).
In contrast to GSK1440115, which functioned as a competitive hU-II antagonist, GSK1562590 suppressed the maximum contractile response to hU-II (Figure 2C; Table 4). Consistent with non-competitive antagonism, the slope of the double reciprocal plot of equiactive concentrations of agonist in the presence and absence of 0.1 nM GSK1562590 was linear with a slope of 3.13 ± 0.51, equating to a pKb of 10.21 ± 0.11 (Figure 2D). As such, GSK1562590 was >2-orders of magnitude more potent as a UT antagonist in the rat isolated aorta than GSK1440115.
Pretreatment of rat isolated aorta with either GSK1440115 or GSK1562590 did not alter basal contractile tone. Similarly, both compounds were devoid of intrinsic contractile activity in all other tissues/species studied.
Functional potency in cat isolated arteries
GSK1440115 (1 and 10 µM) antagonized hU-II-induced contraction in cat isolated conduit arteries (femoral artery and thoracic aorta) in a competitive manner (surmountable inhibition with no suppression of Emax; Figure 3A,C; Table 5). Similarly, competitive antagonism was also evident with GSK1440115 (1 µM) in cat mesenteric resistance arteries (Figure 3E; Table 5).
Figure 3.
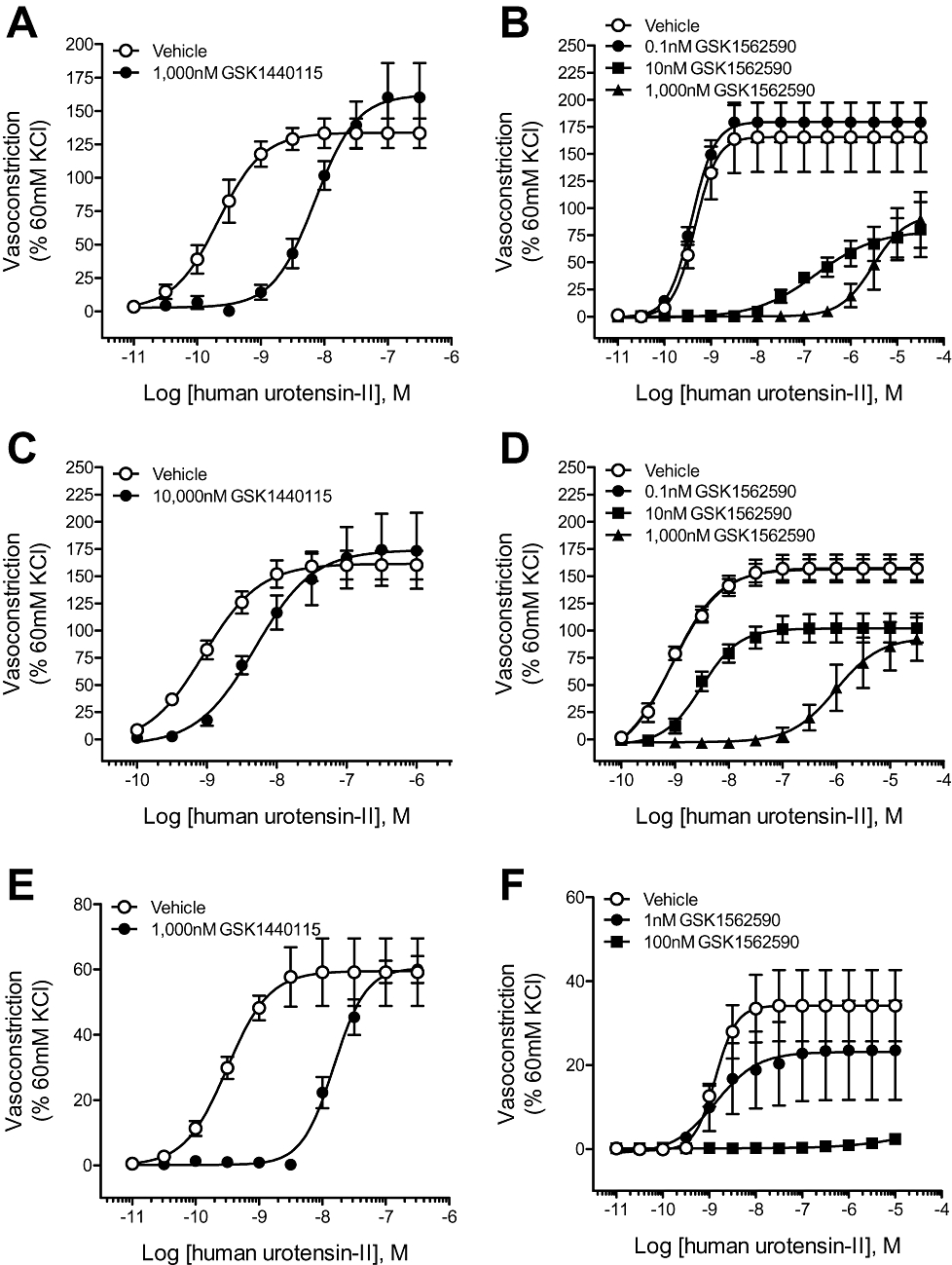
Inhibitory effects of GSK1440115 and GSK1562590 on hU-II-induced contraction of cat isolated arteries. GSK1440115 competitively inhibited (no suppression of the hU-II maximum contractile response) contractions elicited with hU-II in cat isolated (A) thoracic aortae (C) femoral arteries and (E) mesenteric resistance arteries. In contrast to GSK1440115 and consistent with its effects in rat isolated arteries, GSK1562590 suppressed the maximal response to hU-II in cat isolated (B) thoracic aortae (D) femoral arteries and (F) mesenteric resistance arteries.
Table 5.
Inhibition of hU-II-induced contraction of isolated arteries from cat, monkey and human UT transgenic mouse by GSK1440115
| hU-II Emax (% KCl) | hU-II pEC50 | |||||
|---|---|---|---|---|---|---|
| [GSK1440115] nM | Vehicle-treated control | GSK1440115-treated | Vehicle-treated control | GSK1440115-treated | pA2 | |
| Cat | ||||||
| Femoral artery (n= 5) | 1 000 | 134 ± 11 | 161 ± 26 | 9.69 ± 0.15 | 8.28 ± 0.15 | 7.39 ± 0.16 |
| Thoracic aorta (n= 5) | 10 000 | 161 ± 13 | 173 ± 34 | 9.03 ± 0.08 | 8.33 ± 0.07 | 5.59 ± 0.12 |
| Mesenteric resistance artery (n= 3) | 1 000 | 59 ± 10 | 60 ± 4 | 9.56 ± 0.18 | 7.84 ± 0.13 | 7.71 ± 0.06 |
| Monkey | ||||||
| Renal artery (n= 4) | 10 000 | 133 ± 34 | 109 ± 14 | 9.46 ± 0.08 | 7.82 ± 0.12 | 6.63 ± 0.16 |
| Superior mesenteric artery (n= 4) | 10 000 | 178 ± 19 | 158 ± 37 | 9.27 ± 0.16 | 7.80 ± 0.23 | 6.46 ± 0.09 |
| Human UT transgenic mouse | ||||||
| Thoracic aorta (n= 5) | 10 000 | 128 ± 7 | 158 ± 16 | 9.15 ± 0.06 | 6.73 ± 0.07 | 7.41 ± 0.06 |
All values are expressed as mean ± SEM. Statistical comparisons of Emax values were performed using paired, two-tailed t-tests and no values were determined different from vehicle control values (P > 0.05). Competitive antagonist affinities (pA2) were determined using the Schild equation (Jenkinson et al., 1998).
Consistent with data generated in rat aortae, GSK1562590 suppressed the maximal contractile response to hU-II in cat isolated arteries (Figure 3B,D,F; Table 6). Noncompetitive antagonist affinity values (pKb) determined via the method of Gaddum indicated GSK1562590 was >two orders of magnitude more potent in cat isolated conduit arteries than GSK1440115 (Figure 3B,D,F; Table 6).
Table 6.
Inhibitory effects of GSK1562590 on hU-II-induced contraction of cat and human UT transgenic mouse isolated arteries
| hU-II Emax (% KCl) | hU-II pEC50 | |||||
|---|---|---|---|---|---|---|
| [GSK1562590] nM | Vehicle-treated control | GSK1562590-treated | Vehicle-treated control | GSK1562590-treated | pKb | |
| Cat | ||||||
| Femoral artery (n= 3–4) | 0.1 | 166 ± 32 | 180 ± 18 | 9.36 ± 0.05 | 9.44 ± 0.04 | – |
| 10 | 148 ± 38 | 81 ± 27 | 9.36 ± 0.07 | 6.84 ± 0.31 | 10.12 ± 0.27 | |
| 1000 | 166 ± 32 | 93 ± 26 | 9.36 ± 0.05 | 5.45 ± 0.17 | 9.88 ± 0.29 | |
| Thoracic aorta (n= 4) | 0.1 | 157 ± 13 | 156 ± 10 | 8.94 ± 0.08 | 8.95 ± 0.07 | – |
| 10 | 157 ± 13 | 102 ± 13** | 8.94 ± 0.08 | 8.44 ± 0.06 | 8.93 ± 0.23 | |
| 1000 | 157 ± 13 | 93 ± 19** | 8.94 ± 0.08 | 5.88 ± 0.19 | 9.09 ± 0.28 | |
| Mesenteric resistance artery (n= 4) | 1 | 34 ± 8 | 23 ± 12 | 8.89 ± 0.05 | 8.75 ± 0.19 | – |
| 100 | 34 ± 8 | 0* | 8.89 ± 0.05 | -ND | >7.0 | |
| Human UT transgenic mouse | ||||||
| Thoracic aorta (n= 5) | 3 | 68±5 | 0*** | 8.81±0.08 | -ND | >8.5 |
All values are expressed as mean ± SEM. Statistical comparisons of Emax values were performed using paired, two-tailed t-tests or anova analysis with a Dunnett's post-test where
P < 0.05,
P < 0.01 and
P < 0.001
versus vehicle control values. Noncompetitive antagonist affinities (pKb) were determined using the method of Gaddum where equiactive concentrations of agonist in the absence or presence of the noncompetitive antagonist were compared in a linear regression (Gaddum et al., 1955; Kenakin, 2006).
Functional potency in monkey isolated arteries
GSK1440115 (10 µM) antagonized hU-II-induced contraction in monkey renal and superior mesenteric arteries in a competitive manner (no suppression of Emax; Figure 4A,C; Table 5). As observed in cat isolated conduit vessels, the affinity of GSK1440115 in monkey conduit arteries (pA2= 6.46–6.63) is 6- to 18-fold lower than those observed in smaller diameter vessels such as the rat aortae (pKb= 7.36) and cat mesenteric resistance vessels (pA2= 7.71).
Figure 4.
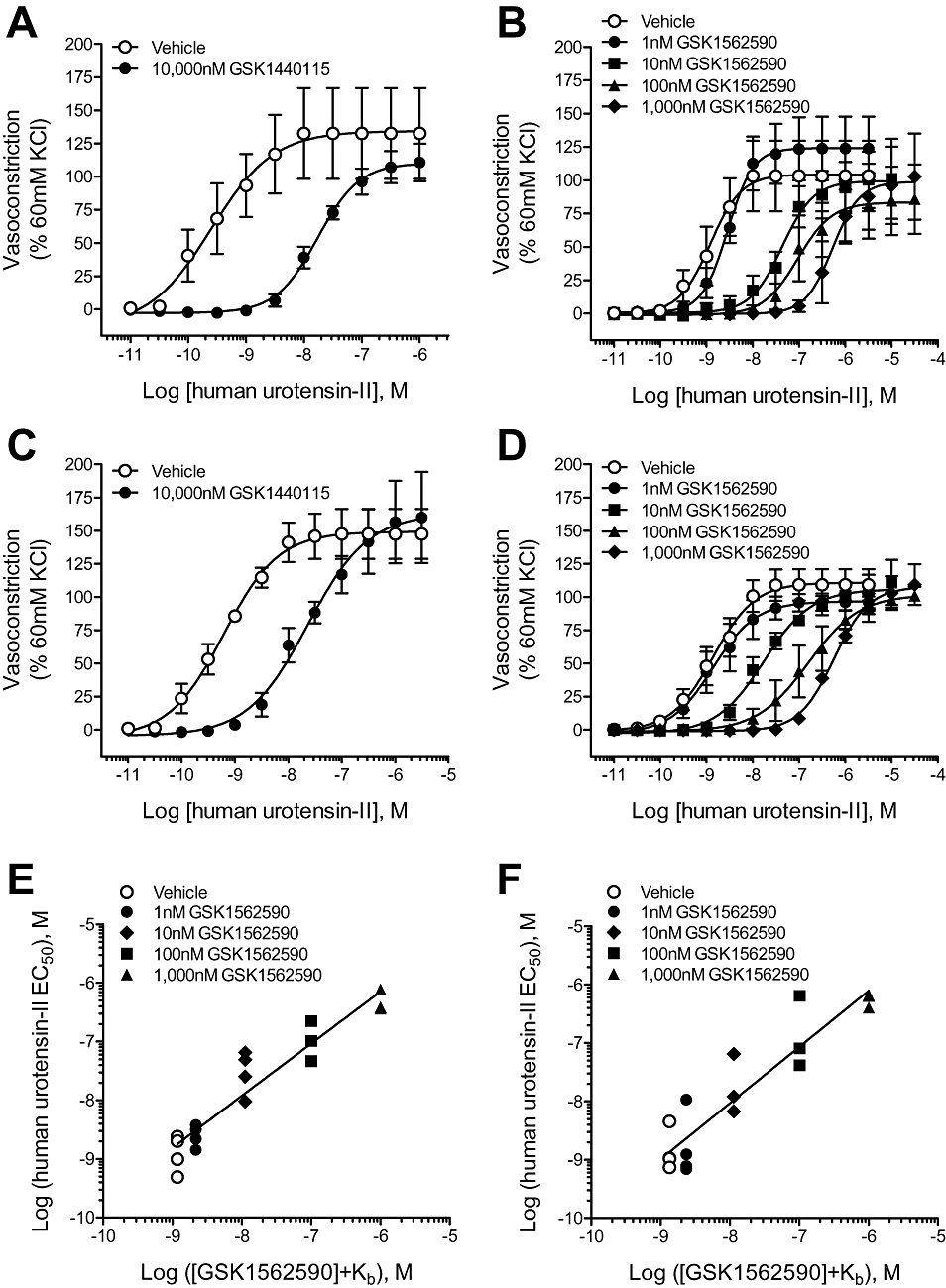
Competitive inhibition of hU-II-induced contraction of monkey isolated arteries by GSK1440115 and GSK1562590. GSK1440115 (10 000 nM) antagonized hU-II-induced contractions in monkey isolated (A) renal and (C) superior mesenteric arteries without suppressing the maximal contractile response. Similarly, GSK1562590 elicited parallel, rightward shifts to the hU-II concentration–response curve in both (B) renal and (D) superior mesenteric arteries, consistent with competitive hU-II antagonism. Clark plot analysis (global nonlinear regression) of GSK1562590 in (E) renal and (F) superior mesenteric arteries revealed pA2 values of 8.93 (8.47–9.39 95% CI) and 8.87 (8.32–9.43 95% CI) and slopes of 0.89 (0.73–1.05 95% CI) and 0.96 (0.75–1.17 95% CI), respectively, consistent with a competitive mode of antagonism.
In contrast to rat and cat arteries where GSK1562590 suppressed the maximum contractile response to hU-II, GSK1562590 acted as a competitive UT antagonist in monkey isolated arteries (Figure 4B,D; Table 6). Global non-linear regression (Clark) analysis revealed pKb values of 8.93 (8.47–9.39 95% CI) and 8.87 (8.32–9.43 95% CI) for GSK1562590 in monkey isolated renal and superior mesenteric arteries [slopes of 0.89 (0.73–1.05 95% CI) and 0.96 (0.75–1.17 95% CI) and Emax unaltered, consistent with a competitive mode of antagonism; Figure 4E–F]. As such, GSK1562590 was >235-fold more potent in monkey isolated arteries than GSK1440115.
Functional potency in hUT transgenic mouse isolated aortae
GSK1440115 (10 µM) competitively inhibited (i.e. surmountable inhibition with no Emax suppression) hU-II-induced contraction of aortae isolated from transgenic mice expressing the human UT with a pA2 of 7.41 ± 0.06 (Figure 5A; Table 5).
Figure 5.
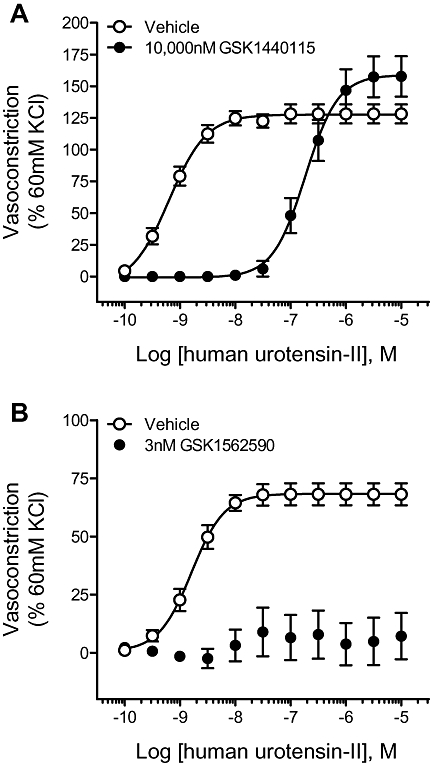
Inhibition of hU-II-induced contraction of isolated aortae from hUT transgenic mice by (A) GSK1440115 and (B) GSK1562590. Whereas pretreatment with 10 000 nM GSK1440115 elicited a parallel, rightward shift in the hU-II concentration–response curve (consistent with competitive hU-II antagonism), 3 nM GSK1562590 abolished the contractile responses to hU-II.
GSK1562590 (3 nM) functioned as a non-competitive antagonist in the hUT transgenic mouse isolated aorta, completely ablating the hU-II-induced contraction (Figure 5B, Table 6). The estimated inhibitor potency (pA2) was >8.5, making GSK1562590 at least an order of magnitude more potent than GSK1440115 in this vascular tissue.
Selective inhibition of hU-II contraction in rat isolated aorta
GSK1440115 (3 µM) did not attenuate the contractile actions of endothelin-1, phenylephrine or KCl on the rat isolated aorta (pA2 < 5.5; Table 7). Similarly, at a concentration of 300 nM [>3 orders of magnitude over the hU-II pKb in this tissue (10.21)], GSK1562590 failed to alter contraction elicited by endothelin-1, phenylephrine or KCl (Table 8). As such, both GSK1440115 and GSK1562590 are selective at inhibiting the contractile actions of hU-II in this preparation.
Table 7.
Selective effects of GSK1440115 (3 µM) on vasoconstrictor-induced contraction of the rat isolated aorta
| Emax (% KCl) | pEC50 | |||
|---|---|---|---|---|
| Spasmogen | Vehicle-treated control | GSK1440115-treated | Vehicle-treated control | GSK1440115-treated |
| KCl | 108 ± 3 | 108 ± 2 | 1.84 ± 0.03 | 1.83 ± 0.03 |
| Phenylephrine | 130 ± 4 | 128 ± 4 | 8.10 ± 0.02 | 7.94 ± 0.09 |
| Endothelin-1 | 138 ± 5 | 129 ± 3 | 8.89 ± 0.08 | 8.89 ± 0.10 |
All values are expressed as mean ± SEM (n= 5). Statistical comparisons of Emax and pEC50 values were performed using paired, two-tailed t-tests and no values were determined different from vehicle control values (P > 0.05).
Table 8.
Selective effects of GSK1562590 (0.3 µM) on vasoconstrictor-induced contraction of the rat isolated aorta
| Emax (% KCl) | pEC50 | |||
|---|---|---|---|---|
| Spasmogen | Vehicle-treated control | GSK1562590-treated | Vehicle-treated control | GSK1562590-treated |
| KCl | 116 ± 2 | 117 ± 2 | 1.83 ± 0.03 | 1.98 ± 0.06 |
| Phenylephrine | 129 ± 5 | 132 ± 4 | 8.07 ± 0.14 | 8.18 ± 0.13 |
| Endothelin-1 | 138 ± 1 | 139 ± 3 | 8.73 ± 0.02 | 8.96 ± 0.05 |
All values are expressed as mean ± SEM (n= 4). Statistical comparisons of Emax and pEC50 values were performed using paired, two-tailed t-tests and no values were determined different from vehicle control values (P > 0.05).
Reversibility of UT antagonism by washout in rat isolated aorta: in vitro studies
Exposure of rat isolated aortae to 0.3 µM GSK1440115 (with no washing) produced a parallel rightward-shift in the hU-II concentration–response curve with an EC50 ratio of 18.9, as expected for a competitive antagonist. The extent of hU-II inhibition was significantly reduced, however, when pretreatment with GSK1440115 was immediately followed by extensive (90 min) washing prior to construction of hU-II concentration–response curves. Under these conditions, a dose ratio of 3.0 was obtained i.e. antagonism with 0.3 µM GSK1440115 was attenuated 84% by washing (Figure 6A,B). This result is consistent with a reversible mode of UT occupancy.
Figure 6.
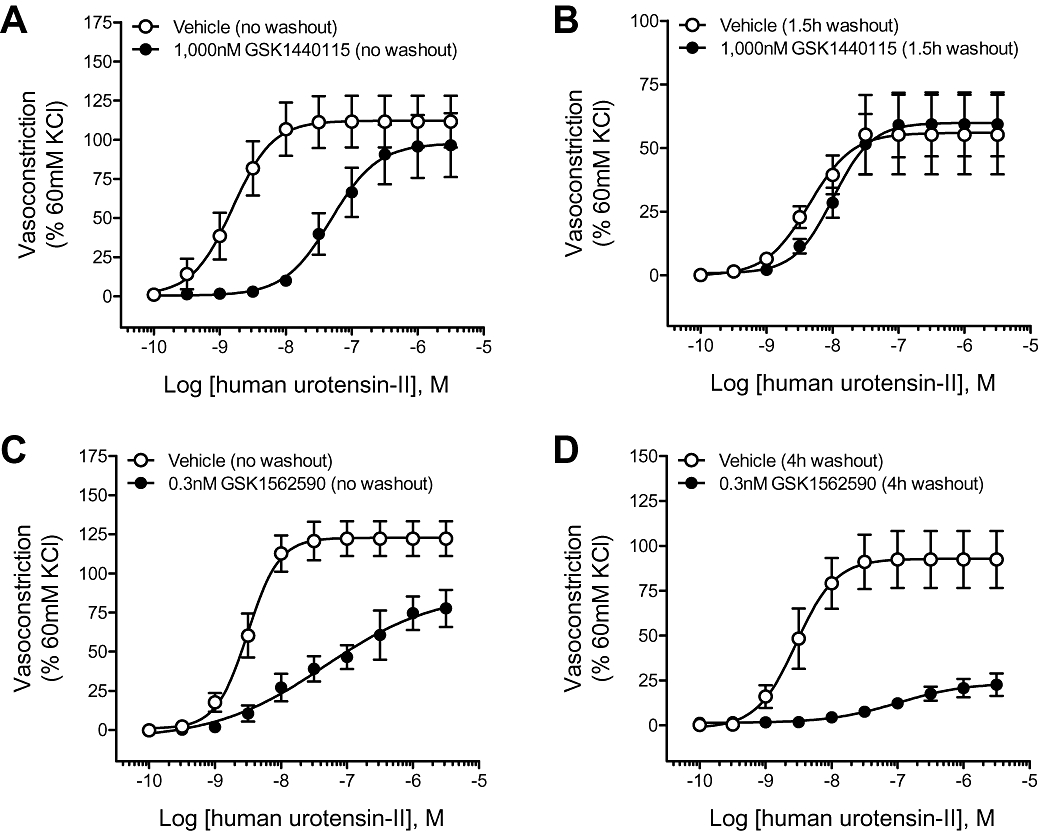
The inhibitory properties of (A, B) GSK1440115 but not (C, D) GSK1562590 were reversed by washout in the rat isolated aorta. Pre-incubation with 1000 nM GSK1440115 elicited parallel, rightward-shifts to the hU-II concentration–response curve (EC50 ratio of 18.9), and these inhibitory effects were reversible (EC50 ratio of 3.0) by repeatedly washing the tissues for 1.5 h with fresh Krebs solution (not containing antagonist) before generating the hU-II concentration–response curves. In contrast to GSK1440115, repeated rinsing of the tissue baths for 4 h failed to reverse the inhibitory effects of GSK1562590.
In contrast to GSK1440115, repeatedly rinsing the tissue with fresh Krebs solution for 2 h to ≤16 h failed to reverse the inhibitory effects of 0.3 nM GSK1562590 in vitro (Figure 6C,D).
Reversibility of UT antagonism in rat isolated aorta: ex vivo studies
As significant GSK1562590 reversibility was not evident within the time constraints of the standard in vitro assay (tissue viability is an issue at ≥16 h), ex vivo studies were performed. In initial studies, rat aortae were isolated 2 h (predetermined Tmax) following oral dosing with 0.01–100 mg·kg−1 GSK1562590. Doses of ≥1 mg·kg−1 significantly inhibited hU-II contraction in isolated aortae assessed ex vivo (e.g. 66% inhibition at 1 mg·kg−1 GSK1562590, p.o.; Figure 7A).
Figure 7.
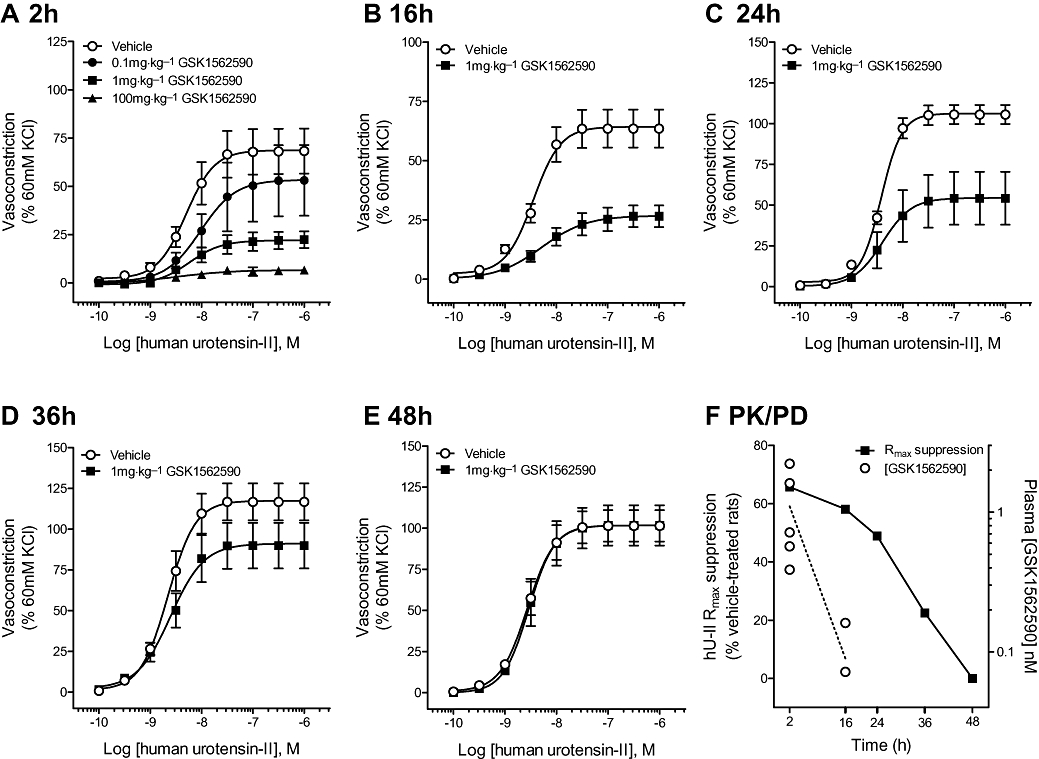
Inhibitory effects of GSK1562590 (1 mg·kg−1, p.o.) on hU-II-induced contraction of rat aorta assessed ex vivo. (A) Rat aorta isolated 2 h (Tmax) following oral dosing of GSK1562590 (0.1–100 mg·kg−1) exhibited dose-dependent inhibition of hU-II contraction. The maximal contractile responses to hU-II were significantly inhibited by 1 mg·kg−1 GSK1562590 at (B) 16 h and (C) 24 h following oral dosing. No significant hU-II inhibition was observed in tissues harvested (D) 36 h and (E) 48 h following oral administration of 1 mg·kg−1 GSK1562590, consistent with a reversible mode of action. (F) Comparison of plasma drug levels and pharmacodynamic duration of action suggest a slow UT koff for GSK1562590.
In a follow-on temporal study, the maximal contractile responses to hU-II were significantly attenuated ex vivo for up to 36 h after dosing at 1 mg·kg−1 GSK1562590, p.o. (Figure 7B–C; Table 9). However, no hU-II inhibition was observed in vessels harvested 36–48 h following oral administration of 1 mg·kg−1 GSK1562590 (Figure 7D–E; Table 9).
Table 9.
Inhibitory effects of GSK1562590 (1 mg·kg−1, p.o.) on hU-II-induced contraction of rat aorta ex vivo
| hU-II pEC50 | hU-II Emax (% 60 mM KCl) | ||||
|---|---|---|---|---|---|
| Time (h) | Plasma concentration (nM) | Vehicle | GSK1562590 | Vehicle | GSK1562590 |
| 2 | 1.10 ± 0.35 | 8.35 ± 0.07 | 8.07 ± 0.18 | 66 ± 37 | 23 ± 9* |
| 16 | <0.12 ± 0.04† | 8.52 ± 0.05 | 8.27 ± 0.09* | 64 ± 8 | 27 ± 5* |
| 24 | <0.07 | 8.46 ± 0.02 | 8.29 ± 0.09 | 106 ± 6 | 54 ± 16* |
| 36 | <0.07 | 8.65 ± 0.05 | 8.61 ± 0.05 | 117 ± 11 | 91 ± 14 |
| 48 | <0.07 | 8.56 ± 0.05 | 8.49 ± 0.07 | 102 ± 9 | 102 ± 12 |
All values are expressed as mean ± SEM (n= 5–7).
Plasma concentrations below limit of detection (0.07 nM) for four out of six animals. Statistical comparisons of Emax and pEC50 values were made using unpaired, two-tailed t-tests where
P < 0.05 versus vehicle.
At 1 mg·kg−1p.o., plasma levels of ∼1 nM were attained at 2 h (approximately 10-fold over the Kb in the rat isolated aorta). Interestingly, GSK1562590 plasma levels were extremely low or undetectable at ≥16 h (<70 pM [lowest limit of quantification]; Figure 7E; Table 9). As such, plasma exposure underestimates the pharmacodynamic duration of action of GSK1562590, consistent with a slow koff of GSK1562590 from rat UT. The inhibition was selective for hU-II as neither KCl nor phenylephrine contraction was altered at any time point studied (data not shown). Although GSK1440115 was not profiled under these conditions, an alternative non-pyrrolidine (i.e. morpholino) analog, GSK1575988, which functioned as a competitive, reversible UT antagonist in rat isolated aorta, was profiled under these ex vivo conditions and caused minimal (i.e. insignificant) inhibition at T2h (data not shown).
Reversibility of GSK1562590 binding to rat, monkey and human UT
To verify that the slow in vitro and ex vivo‘washout’ from rat tissues directly correlated with a slow dissociation of GSK1562590 from the rat UT, radioligand binding reversibility studies were performed using recombinant rat, monkey and human UT.
Compared with treatment with vehicle, 10 nM GSK1562590 attenuated the observed Bmax for [125I]hU-II binding by 5.2–38.3% (Table 10), 7.6–10.2% and 11.9% (data not shown) at washout t= 0, 0.5 and 1 h, respectively, for cell membranes expressing recombinant rat UT (n= 2). In contrast, GSK1562590 binding to membranes enriched with recombinant monkey receptor was fully reversible, exhibiting Bmax values of 92.5 ± 5.2% (Table 10) and 94.2 ± 5.6% (data not shown) at washout t= 0 and 2 h, respectively, compared with vehicle-treated membranes (n= 4). Binding of GSK1562590 (10 nM) to membranes containing recombinant human UT was also reversible, but slower than that observed for the monkey receptor, exhibiting a Bmax of 38.5–61.2% of vehicle-treated membranes immediately after washing (t= 0; n= 2; Table 10). GSK1562590 (10 nM) did not significantly alter the KD values for [125I]hU-II for any of the three UT orthologues studied (data not shown).
Table 10.
Reversible interaction of GSK1562590 (10 nM) binding at rat, monkey and human recombinant UT
| UT cell membrane | [125I]hU-II Bmax at washout t= 0 (% vehicle) | Time for [125I]hU-II Bmax >90% vehicle |
|---|---|---|
| Rat UT (n= 2) | 5.2–38.3% | >1 h |
| Monkey UT (n= 4) | 92.5 ± 5.2% | 0 h |
| Human UT (n= 2) | 38.5–61.2% | <1 h |
All values are expressed as mean ± SEM, where n represents the number of individual experiments performed in duplicate. UT membranes were incubated with DMSO or 10 nM GSK1562590 for 30 min at 25°C following which the incubation mixtures were diluted, centrifuged and resuspended in buffer. Following resuspension, [125I]hU-II KD and Bmax values were determined (t= 0). A subset of membranes were ‘washed’ a second time at various times following the initial resuspension (t= 0.5, 1 or 2 h) to assess GSK1562590 binding reversibility (time for [125I]hU-II Bmax >90% vehicle). KD and Bmax values were determined by homologous competition binding whereby multiple concentrations of unlabelled hU-II were used to compete for binding with a fixed concentration of [125I]hU-II (Douglas et al., 2005). GSK1562590 did not alter the KD values for [125I]hU-II for any of the three UT orthologues studied.
In vivo cat hemodynamics
GSK1440115 did not alter basal hemodynamics or ECG parameters in the anaesthetized cat at concentrations ≤3 mg·kg−1. In contrast, at the highest dose tested (10 mg·kg−1), GSK1440115 reduced basal arterial blood pressure by ∼20 mmHg when compared with vehicle-treated cats, an effect accompanied by reduced left ventricular systolic pressure and left ventricular relaxation (-dP/dt) and elevated QRS and ST segments (P < 0.05). GSK1440115 attenuated the in vivo pressor response to 1 nmol·kg−1 hU-II at a minimally active pharmacological dose of 1 mg·kg−1. Surprisingly, significant inhibition of hU-II effects was also observed using 10 mg·kg−1 GSK1440115 but not at the intermediate dose of 3 mg·kg−1 (Figure 8A,C).
Figure 8.
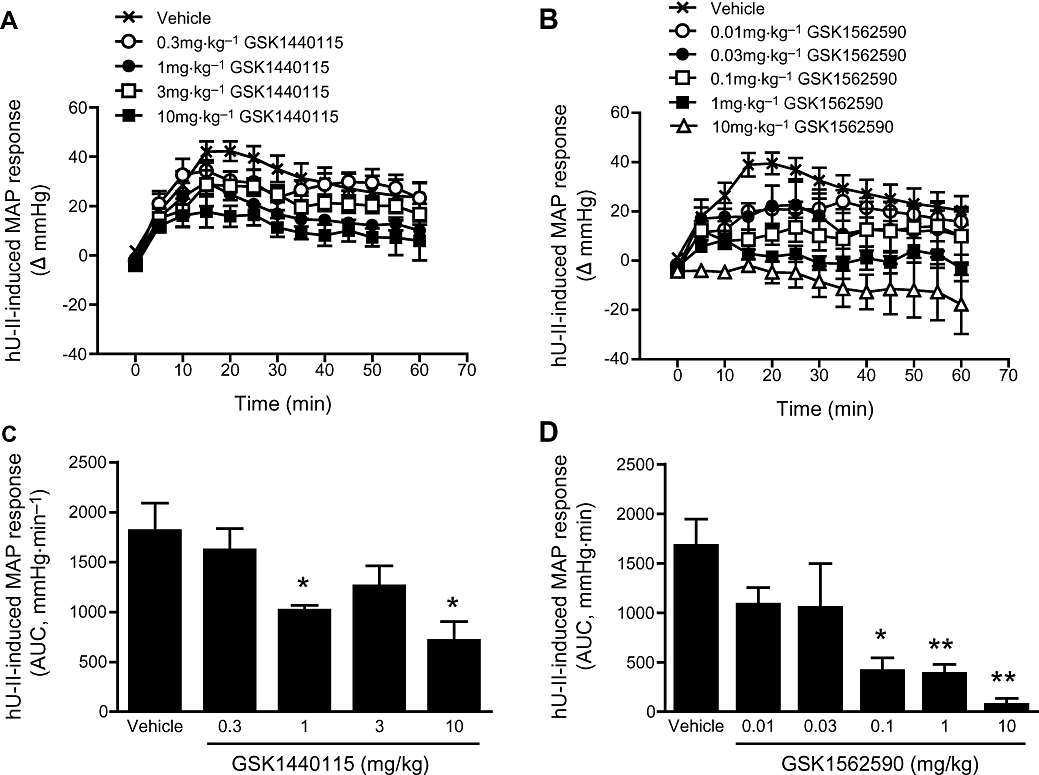
Inhibitory effects of GSK1440115 and GSK1562590 to exogenous hU-II challenge in the anaesthetized cat. The pressor response to hU-II (1 nmol·kg−1, i.v. bolus) was dose-dependently inhibited by (A) GSK1440115 and (B) GSK1562590. Area under the mean arterial blood pressure responses for (C) GSK1440115 and (D) GSK1562590 indicated minimally active doses of 1 and 0.1 mg·kg−1 respectively.
In contrast with GSK1440115, GSK1562590 (0.01–10 mg·kg−1) did not alter any basal haemodynamic or ECG parameters at any dose studied. GSK1562590 blocked the pressor actions of hU-II at 0.1 mg·kg−1, a 10-fold lower dose than that required for GSK1440115 (Figure 8B,D).
The plasma levels achieved following administration of both compounds were approximately dose linear (data not shown).
Discussion and conclusions
Delineation of the (patho)physiological role of the U-II system has been hampered by the absence of potent and selective UT antagonists. Indeed, the lack of efficacy observed with Palosuran (ACT-058362; Clozel et al., 2004; 2006; http://www1.actelion.com/documents/publications/Milestones_Company.pdf) in patients with diabetic nephropathy (phase IIb) was clouded by its low antagonist potency in whole cells and tissues (Behm et al., 2008). As such, drug discovery programmes continue to focus on identifying potent and selective UT antagonists suitable for assessment in both preclinical species and man. The present study describes two compounds, GSK1440115 and GSK1562590, which fit these criteria.
Initial radioligand binding characterization of GSK1440115 and GSK1562590 indicated that both compounds were selective, high-affinity, pan-species active UT ligands, with GSK1562590 exhibiting a 5- to 101-fold greater potency than GSK1440115 across species. Aside from their unique potencies, the compounds were differentiated by their functional effects in rat isolated aorta. GSK1440115 behaved as a traditional competitive antagonist, causing rightward shifts of the hU-II contractile concentration–response curve. In contrast, GSK1562590 elicited insurmountable antagonism, by which the maximal contractile response to hU-II was suppressed in a concentration-dependent manner. As discussed further below, we feel that this property of GSK1562590 might contribute to its enhanced efficacy ex vivo.
We envision several possible causes for the insurmountable antagonism exhibited by GSK1562590 in the rat aorta. Based on experience with pharmacophores from other chemical series, we initially suspected that GSK1562590 might suppress the maximal response to hU-II in a non-selective manner, for instance by altering smooth muscle cell Ca2+ modulation. However, this was not the case as GSK1562590 failed to alter vasoconstriction elicited via non-UT-related spasmogens including KCl, phenylephrine and endothelin-1.
Second, the insurmountable effects of GSK1562590 could be the result of a slow UT dissociation rate or covalent bonding modification of the receptor. As no evidence for covalent bonding has been identified to date, the antagonist is not considered truly insurmountable (irreversible inhibition through covalent bonding; Furchgott, 1966). Rather, we feel that our data are most consistent with a slowly reversible binding of GSK1562590 to UT. The phenomenon of slowly reversible inhibition was originally observed by Rang (1966) and has subsequently been used to describe the kinetics of action of several ‘non UT’ G-protein-coupled receptor antagonists including the AT1 antagonists candsartan (Fierens et al., 1999) and telmisartan (Maillard et al., 2002) and the histamine H1 antagonist desloratidine (Anthes et al., 2002). GSK1562590 now represents the first identified slowly reversible, small molecule UT antagonist.
Although the exact molecular interaction responsible for the reduced dissociation rate (koff) of GSK1562590 has not been identified, a key binding interaction of many GPCRs (including UT) involves a salt-bridge between an amino side-chain carboxylate and the ligand (Ji et al., 1998; Ishiguro, 2004; Grieco et al., 2009). It is therefore reasonable to hypothesize that this interaction may be accentuated with GSK1562590 due to the presence of the more basic pyrrolidine residue compared with the less basic morpholine in GSK1440115. Indeed, comparison of numerous pairs of pyrrolidine/morpholine analogs having otherwise identical chemical structures has demonstrated that without exception, the pyrrolidine analogues are 10–1000-fold more potent than their morpholine counterparts (McAtee et al., 2008). In addition, a set of analogues in this chemical series with basic moieties other than pyrrolidine and morpholine exhibited a strong correlation between the basicity of the nitrogen functionality and UT binding affinity (data not shown).
Furthermore, the observation that GSK1562590 failed to cause a parallel shift of the hU-II concentration–response curve in the rat isolated aorta, at any concentration tested, is consistent with the rat aorta being devoid of spare receptors for U-II (Itoh et al., 1987; Douglas et al., 2004a). If spare receptors for U-II were present in this tissue, lower concentrations of GSK1562590, as an insurmountable antagonist, would be expected to elicit a parallel shift of the hU-II concentration–response curve and only when the number of receptors was reduced further by subsequent applications of GSK1562590 would there be an appreciable Emax reduction (Jenkinson, 1996). Our results therefore show that the nature of insurmountable antagonism observed in rat aorta likely reflects the combination of (a) a slow dissociation rate of GSK1562590 from rat UT and (b) a lack of spare receptors for U-II.
Overall, GSK1440115 appeared to function as a competitive UT antagonist in arteries from all species tested and the antagonism was fully surmountable by hU-II with a generally right-ward shift in agonist potency. However, in the absolute sense, GSK1440115 antagonism might depart from simple competitive binding interactions. For example, the highest concentration of GSK1440115 tested in the rat aorta (3 µM) caused no further shift to the hU-II concentration–response curve (i.e.Figure 2A). There are many potential explanations for deviation from pure competitive antagonism, such as reduced compound solubility at higher concentrations and differences in absolute mechanism between tissues, which we have yet to fully define for GSK1440115. In addition, antagonist potency was not consistent between tissues (e.g. Figure 3A,C,E). Indeed, concentrations of GSK1440115 ≤3 µM were not significantly inhibitory in the cat thoracic aorta (data not shown), and we caution that potencies might be underestimated in some tissues where only high concentrations of GSK1440115 (≥3 µM) elicited inhibitory effects. However, this phenomenon is not specific to GSK1440115, as other competitive UT antagonists also exhibit reduced potency in larger diameter arteries such as the cat thoracic aorta (Behm et al., 2006; Behm et al., 2008).
In contrast to the overall competitive nature of antagonism by GSK1440115 in arteries across species, the mode of antagonism observed with GSK1562590 was species-dependent with the compound functioning as an insurmountable UT antagonist in rat, cat and hUT transgenic mouse arteries but as a competitive antagonist in monkey arteries. Consistent with the functional data, the observed rank order of receptor dissociation rates for GSK1562590 from each receptor was: rat ≥ human > monkey UT. Thus, the insurmountable inhibitory effects of GSK1562590 likely reflect non-equilibrium conditions in rat, cat and hUT transgenic mouse arteries, where compound dissociation rates are much longer than the competing interaction. Unfortunately, additional studies aimed to further quantify receptor dissociation rates by increasing washout periods or temperature failed due to significant, time-dependent UT membrane degradation.
Tissue-based in vitro and ex vivo‘washout’ studies provided further support for an extended duration of action for GSK1562590. Consistent with a competitive mode of antagonism, the inhibitory properties of GSK1440115 in the rat isolated aorta were readily reversed following a 1.5 h washout period. In contrast, no significant GSK1562590 reversibility was evident within the time of this standard in vitro washout assay (≤16 h). In follow-on ex vivo studies where rat aortae were isolated following oral dosing with GSK1562590, hU-II contraction was significantly inhibited in tissues from animals treated with GSK1562590 for up to 24 h. These results were surprising as plasma levels were undetectable at ≥16 h (<70 pM) and the tissues were suspended in Krebs buffer in the absence of any inhibitor for ∼4 h before the hU-II concentration–response curves were generated. These data indicate that plasma exposure underestimates the pharmacodynamic duration of action of GSK1562590, consistent with extended UT residence time.
Consistent with the in vitro data, the compounds were readily differentiated in the anaesthetized cat, where GSK1562590 blocked the pressor actions of hU-II at 0.1 mg·kg−1 (plasma levels ≤6 ng·mL−1), a 10-fold lower dose than that required for GSK1440115 (1 mg·kg−1, plasma levels of ≤63 ng·mL−1). Also, in contrast to GSK1440115, GSK1562590 failed to elicit any baseline blood pressure or ECG disturbances at any dose studied. Based on these observations, GSK1562590 not only exhibits higher efficacy but also greater selectivity as compared with GSK1440115 in this model.
Strikingly, significant suppression of the maximal contractile response for hU-II in rat isolated aorta was observed with concentrations of GSK1562590 as low as 0.1 nM, making it > two orders of magnitude more potent than GSK1440115 in this tissue. In addition, as compared with GSK1440115, which generally exhibited a ∼10-fold loss in functional activity, minimal differences in potency between binding and functional assays was noted with GSK1562590. This constitutes a ‘biochemical efficiency’, which is defined as the ratio of a compounds binding affinity (pKi) to its functional response (pIC50, pKb, etc.) (Swinney, 2004), of approximately 0.1 and 1 for GSK1440115 and GSK1562590 respectively. Thus, as a consequence of an efficient non-equilibrium mechanism, GSK1562590 exhibits greater biochemical efficiency than GSK1440115, a characteristic that has been hypothesized to increase both the therapeutic window and efficacy of a drug (Swinney, 2004; Copeland et al., 2006). Indeed, it is estimated that the percentage of marketed drugs with good biochemical efficiency (>0.4) is as high as 88% (Swinney, 2004). Interestingly, palosuran, which failed to show efficacy in diabetic nephropathy patients, exhibited a dramatic loss in UT potency in tissues, equivalent to biochemical efficiencies ranging from 0.1 to as low as 0.001 (Behm et al., 2008).
Drugs with extended residence times (slow dissociation rates) have been linked to a beneficial extended duration of action in vivo. For example, the extremely slow dissociation rates of tiotropium versus ipratropium from the muscarinic (M3) receptor and candesartan versus losartan from the AT1 receptor are thought to contribute to their sustained and improved clinical actions (Laciurciére and Asmar, 1999; Van Noord et al., 2002). Indeed, it has been recently proposed that compounds with this characteristic represent ‘ultimate physiological inhibitors’, where reversal of inhibition is dependent solely on target recycling by the organism (Copeland et al., 2006). In addition, one can hypothesize that a slow receptor off-rate might be even more important for UT antagonists to counteract the (a) pseudo-irreversible binding characteristics of U-II (Ames et al., 1999) and (b) the high levels of circulating ‘U-II-like’ immunoreactivity (∼3 nM, ∼2 orders of magnitude above the human UT Kd; Heller et al., 2002). We now add GSK1562590 to this list of ligands with extended residency.
In summary, both GSK1440115 and GSK1562590 represent high-affinity/selective UT antagonists suitable for interrogating the (patho)physiological role of the U-II system. In contrast to GSK1440115 which functions as a standard, rapidly reversible, competitive antagonist, GSK1562590 represents the first UT antagonist identified with enhanced UT residence time, resulting in extended duration of pharmacodynamic activity ex vivo. As compounds with enhanced residence time have been linked to improved therapeutic windows and increased efficacy, GSK1562590 constitutes an optimized tool for determining the (patho)physiology of the U-II pathway.
Acknowledgments
This paper is dedicated to the memory of our friend and colleague, Dr Stephen A. Douglas. The authors wish to thank D. Zhang and P.M. Eidam for compound scale-up, the MP CEDD DMPK group (specifically H.E. Fries and C.A. Evans) for bioanalytical support, Systems Research (specifically Z. Wu, L.H. Carballo, A.H. Ayscue, M.S. Jiang, E. Bettini and B. Oliosi) for technical assistance in assessing compound selectivity and C.P.A. Doe and G.P. Stankus for performing the cat exogenous U-II challenge model. We also thank Drs D.P. Brooks, J.G. Gleason and E.H. Ohlstein for meaningful discussion and insightful comments.
Glossary
Abbreviations
- BacMam
recombinant baculovirus in which the polyhedrin promoter has been replaced with a mammalian promoter
- CHO cells
Chinese hamster ovary cells
- DMSO
dimethylsulphoxide
- DPBS
Dulbecco's phosphate-buffered saline
- FLIPR
fluorescence imaging plate reader
- GPCR
G-protein-coupled receptor
- GSK1440115
4′-[(1R)-1-[[(6,7-dichloro-3-oxo-2,3-dihydro-4H-1,4-benzoxazin-4-yl)acetyl](methyl)amino]-2-(4-morpholinyl)ethyl]-4-biphenylcarboxylic acid trifluoroacetate
- GSK1562590
4′-[(1R)-1-[[(6,7-dichloro-3-oxo-2,3-dihydro-4H-1,4-benzoxazin-4-yl)acetyl](methyl)amino]-2-(1-pyrrolidinyl)ethyl]-3-biphenylcarboxamide hydrochloride
- HEK293
human embryonic kidney cells
- NKA
neurokinin A
- SJRH30
human rhabdomyosarcoma cell line
- SPA
scintillation proximity assay
- U2OS
human osteosarcoma cell line
- (h)U-II
(human) urotensin-II
- (h)UT
(human) urotensin-II receptor
- WGA
wheat germ agglutinin-coated
Conflict of interest
DJB, ARO, HEF, JJM, MAH, JWD, SED, GZW, KBG, CAS, MRH, RNW, MJN and CAL are current employees of GlaxoSmithKline.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames RS, Sarau HM, Chambers JK, Willette RN, Aiyar NV, Romanic AM, et al. Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature. 1999;401:282–286. doi: 10.1038/45809. [DOI] [PubMed] [Google Scholar]
- Anthes JC, Gilchrest H, Richard C, Eckel S, Hesk D, West RE, et al. Biochemical characterization of desloratadine, a potent antagonist of the human histamine H1 receptor. Eur J Pharmacol. 2002;449:229–237. doi: 10.1016/s0014-2999(02)02049-6. [DOI] [PubMed] [Google Scholar]
- Behm DJ, Doe CP, Johns DG, Maniscalco K, Stankus GP, Wibberley A, et al. Urotensin-II: a novel systemic hypertensive factor in the cat. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:274–280. doi: 10.1007/s00210-004-0873-1. [DOI] [PubMed] [Google Scholar]
- Behm DJ, Stankus G, Doe CP, Willette RN, Sarau HM, Foley JJ, et al. The peptidic urotensin-II receptor ligand GSK248451 possesses less intrinsic activity than the low-efficacy partial agonists SB-710411 and urantide in native mammalian tissues and recombinant cell systems. Br J Pharmacol. 2006;148:173–190. doi: 10.1038/sj.bjp.0706716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm DJ, McAtee JJ, Dodson JW, Neeb MJ, Fries HE, Evans CA, et al. Palosuran inhibits binding to primate UT receptors in cell membranes but demonstrates differential activity in intact cells and vascular tissues. Br J Pharmacol. 2008;155:374–386. doi: 10.1038/bjp.2008.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:357–366. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Clozel M, Binkert C, Birker-Robaczewska M, Boukhadra C, Ding SS, Fischli W, et al. Pharmacology of the urotensin-II receptor antagonist palosuran (ACT-058362; 1-[2-(4-benzyl-4-hydroxy-piperidin-1-yl)-ethyl]-3-(2-methyl-quinolin-4-yl)-urea sulfate salt): first demonstration of a pathophysiological role of the urotensin system. J Pharmacol Exp Ther. 2004;311:204–212. doi: 10.1124/jpet.104.068320. [DOI] [PubMed] [Google Scholar]
- Clozel M, Hess P, Qiu C, Ding SS, Rey M. The urotensin-II receptor antagonist palosuran improves pancreatic and renal function in diabetic rats. J Pharmacol Exp Ther. 2006;316:1115–1121. doi: 10.1124/jpet.105.094821. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implication for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Douglas SA, Sulpizio AC, Piercy V, Sarau HM, Ames RS, Aiyar NV, et al. Differential vasoconstrictor activity of human urotensin-II in vascular tissue isolated from the rat, mouse, dog, pig, marmoset and cynomolgus monkey. Br J Pharmacol. 2000;131:1262–1274. doi: 10.1038/sj.bjp.0703690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas SA, Dhanak D, Johns DG. From ‘gills to pills’: urotensin-II as a regulator of mammalian cardiorenal function. Trends Pharmacol Sci. 2004a;25:76–85. doi: 10.1016/j.tips.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Douglas SA, Naselsky D, Ao Z, Disa J, Herold CL, Lynch F, et al. Identification and pharmacological characterization of native, functional human urotensin-II receptors in rhabdomyosarcoma cell lines. Br J Pharmacol. 2004b;142:921–932. doi: 10.1038/sj.bjp.0705743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas SA, Behm DJ, Aiyar NV, Naselsky D, Disa J, Brooks DP, et al. Non-peptidic Urotensin-II Receptor (UT) Antagonists I: in vitro pharmacological characterisation of SB-706375. Br J Pharmacol. 2005;145:620–635. doi: 10.1038/sj.bjp.0706229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierens FL, Vanderheyden PM, De Backer JP, Vauquelin G. Insurmountable angiotensin AT1 receptor antagonists: the role of tight antagonist binding. Eur J Pharmacol. 1999;372:199–206. doi: 10.1016/s0014-2999(99)00205-8. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. Adv Drug Res. 1966;3:21–55. [Google Scholar]
- Gaddum JH, Hameed KA, Hathway DE, Stephens FF. Quantitative studies of antagonists for 5-hydroxytryptamine. Q J Exp Physiol. 1955;40:49–74. doi: 10.1113/expphysiol.1955.sp001097. [DOI] [PubMed] [Google Scholar]
- Gong H, Wang YX, Zhu YZ, Wang WW, Wang MJ, Yao T, et al. Cellular distribution of GPR14 and the positive inotropic role of urotensin II in the myocardium in adult rat. J Appl Physiol. 2004;97:2228–2235. doi: 10.1152/japplphysiol.00540.2004. [DOI] [PubMed] [Google Scholar]
- Grieco P, Carotenuto A, Campiglia P, Gomez-Monterrey I, Auriemma L, Sala M, et al. New insight into the binding mode of peptide ligands at Urotensin-II receptor: structure-activity relationships study on P5U and urantide. J Med Chem. 2009;52:3927–3940. doi: 10.1021/jm900148c. [DOI] [PubMed] [Google Scholar]
- Heller J, Schepke M, Neef M, Woitas R, Rabe C, Sauerbruch T. Increased urotensin II plasma levels in patients with cirrhosis and portal hypertension. J Hepatol. 2002;37:767–772. doi: 10.1016/s0168-8278(02)00295-7. [DOI] [PubMed] [Google Scholar]
- Ishiguro M. Ligand-binding modes in cationic biogenic amine receptors. Chembiochem. 2004;5:1210–1219. doi: 10.1002/cbic.200400068. [DOI] [PubMed] [Google Scholar]
- Itoh H, Itoh Y, Rivier J, Lederis K. Contraction of major artery segments of rat by fish neuropeptide urotensin II. Am J Physiol Regul Integr Comp Physiol. 1987;252:R361–R366. doi: 10.1152/ajpregu.1987.252.2.R361. [DOI] [PubMed] [Google Scholar]
- Jenkinson DH. Classical approaches to the study of drug-receptor interactions. In: Foreman JC, Johansen T, editors. Textbook of Receptor Pharmacology. Boca Raton, FL: CRC Press; 1996. pp. 47–49. [Google Scholar]
- Jenkinson DH, Barnard EA, Hoyer D, Humphrey PPA, Leff P, Shankley NP. Terms and symbols in quantitative pharmacology. In: Girdlestone D, editor. The IUPHAR Compendium of Receptor Characterization and Classification. London: IUPHAR Media; 1998. pp. 6–20. [Google Scholar]
- Ji TH, Grossmann M, Ji I. G protein-coupled receptors. I. Diversity of receptor-ligand interactions. J Biol Chem. 1998;273:17299–17302. doi: 10.1074/jbc.273.28.17299. [DOI] [PubMed] [Google Scholar]
- Johns DG, Ao Z, Naselsky D, Herold CL, Maniscalco K, Sarov-Blat L, et al. Urotensin-II-mediated cardiomyocyte hypertrophy: effect of receptor antagonism and role of inflammatory mediators. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:238–250. doi: 10.1007/s00210-004-0980-z. [DOI] [PubMed] [Google Scholar]
- Kenakin T. A Pharmacology Primer: Theory, Applications, and Methods. Massachusetts: Academic Press; 2006. Orthosteric drug antagonism; pp. 114–119. [Google Scholar]
- Laciurciére Y, Asmar RA. A comparison of the efficacy and duration of action of candesartan cilexetil and losartan as assessed by clinic and ambulatory blood pressure after a missed dose in truly hypertensive patients. Am J Hypertens. 1999;12:1181–1187. doi: 10.1016/s0895-7061(99)00142-9. [DOI] [PubMed] [Google Scholar]
- Lew MJ, Angus JA. An improved method for analysis of competitive agonist/antagonist interactions by non-linear regression. Ann NY Acad Sci. 1997;812:179–181. doi: 10.1111/j.1749-6632.1997.tb48166.x. [DOI] [PubMed] [Google Scholar]
- Liu JC, Chen CH, Chen JJ, Cheng TH. Urotensin II induces rat cardiomyocyte hypertrophy via the transient oxidization of Src homology 2-containing tyrosine phosphatase and transactivation of epidermal growth factor receptor. Mol Pharmacol. 2009;76:1186–1195. doi: 10.1124/mol.109.058297. [DOI] [PubMed] [Google Scholar]
- McAtee JJ, Dodson JW, Dowdell SE, Erhard K, Girard GR, Goodman KB, et al. Potent and selective small-molecule human urotensin-II antagonists with improved pharmacokinetic profiles. Bioorg Med Chem Lett. 2008;18:3716–3719. doi: 10.1016/j.bmcl.2008.05.058. [DOI] [PubMed] [Google Scholar]
- Maillard MP, Perregaux C, Centeno C, Stangier J, Wienen W, Brunner HR, et al. In vitro and in vivo characterization of the activity of telmisartan: an insurmountable angiotensin II receptor antagonist. J Pharmacol Exp Ther. 2002;302:1089–1095. doi: 10.1124/jpet.102.036772. [DOI] [PubMed] [Google Scholar]
- Maryanoff BE, Kinney WA. Urotensin-II receptor modulators as potential drugs. J Med Chem. 2010;53:2695–2708. doi: 10.1021/jm901294u. [DOI] [PubMed] [Google Scholar]
- Onan D, Pipolo L, Yang E, Hannan RD, Thomas WG. Urotensin II promotes hypertrophy of cardiac myocytes via mitogen-activated protein kinases. Mol Endocrinol. 2004;18:2344–2354. doi: 10.1210/me.2003-0309. [DOI] [PubMed] [Google Scholar]
- Quaile MP, Kubo H, Kimbrough CL, Douglas SA, Margulies KB. Direct inotropic effects of exogenous and endogenous urotensin-II: divergent actions in failing and nonfailing human myocardium. Circ Heart Fail. 2009;2:39–46. doi: 10.1161/CIRCHEARTFAILURE.107.748343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang HP. The kinetics of action of acetylcholine antagonists in smooth muscle. Proc R Soc Lond B Biol Sci. 1966;164:488–510. doi: 10.1098/rspb.1966.0045. [DOI] [PubMed] [Google Scholar]
- Russell FD, Molenaar P, O'Brien DM. Cardiostimulant effects of urotensin-II in human heart in vitro. Br J Pharmacol. 2001;132:5–9. doi: 10.1038/sj.bjp.0703811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidharta PN, Wagner FD, Bohnemeier H, Jungnik A, Halabi A, Krawhenbuehl S, et al. Pharmacodynamics and pharmacokinetics of the urotensin II receptor antagonist palosuran in macroalbuminuric, diabetic patients. Clin Pharmacol Ther. 2006;80:246–256. doi: 10.1016/j.clpt.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Sidharta PN, Van Giersbergen PLM, Dingemanse J. Pharmacodynamics and pharmacokinetics of the urotensin II receptor antagonist palosuran in healthy male subjects. J Clin Pharmacol. 2009;49:1168–1175. doi: 10.1177/0091270009341181. [DOI] [PubMed] [Google Scholar]
- Silvestre RA, Rodríguez-Gallardo J, Egido EM, Marco J. Inhibition of insulin release by urotensin II – a study on the perfused rat pancreas. Horm Metab Res. 2001;33:379–381. doi: 10.1055/s-2001-15414. [DOI] [PubMed] [Google Scholar]
- Song W, Ashton N, Balment RJ. Effects of single bolus urotensin II injection in anaesthetized rats. J Physiol. 2003;552P:P107. [Google Scholar]
- Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004;3:801–808. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- Tzanidis A, Hannan RD, Thomas WG, Onan D, Autelitano DJ, See F, et al. Direct actions of urotensin II on the heart: implications for cardiac fibrosis and hypertrophy. Circ Res. 2003;93:246–253. doi: 10.1161/01.RES.0000084382.64418.BC. [DOI] [PubMed] [Google Scholar]
- Van Noord JA, Smeets JJ, Custers FLJ, Korducki L, Cornelissen PJG. Pharmacodynamic steady state of tiotropium in patients with chronic obstructive pulmonary disease. Eur Respir J. 2002;19:639–644. doi: 10.1183/09031936.02.00238002. [DOI] [PubMed] [Google Scholar]
- Zhang AY, Chen YF, Zhang DX, Yi FX, Qi J, Andrade-Gordon P, et al. Urotensin II is a nitric oxide-dependent vasodilator and natriuretic peptide in the rat kidney. Am J Physiol Renal Physiol. 2003;285:F792–F798. doi: 10.1152/ajprenal.00342.2002. [DOI] [PubMed] [Google Scholar]