

Abstract
Oligodendrogliopathy, microglial infiltration, and lack of remyelination are detected in the brains of patients with multiple sclerosis and are accompanied by high levels of the transcription factor p53. In this study, we used the cuprizone model of demyelination, characterized by oligodendrogliopathy and microglial infiltration, to define the effect of p53 inhibition. Myelin preservation, decreased microglial recruitment, and gene expression were observed in mice lacking p53 or receiving systemic administration of the p53 inhibitor pifithrin-α, compared with untreated controls. Decreased levels of lypopolysaccharide-induced gene expression were also observed in vitro, in p53−/− primary microglial cultures or in pifithrin-α-treated microglial BV2 cells. An additional beneficial effect of lack or inhibition of p53 was observed in Sox2+ multipotential progenitors of the subventricular zone that responded with increased proliferation and oligodendrogliogenesis. Based on these results, we propose transient inhibition of p53 as a potential therapeutic target for demyelinating conditions primarily characterized by oligodendrogliopathy.
Keywords: myelin repair, oligodendrocyte, microglia, transcription, myelin, demyelination
Introduction
Multiple sclerosis (MS) is a demyelinating disorder characterized by heterogeneity of clinical and neuropathological signs and disease susceptibility (Lucchinetti et al., 2000). At least two broad categories have been defined based on neuropathological findings: one is characterized by immune-mediated demyelination occurring at perivenous locations, whereas the other one is characterized primarily by myelin loss consequent to oligodendrogliopathy and is associated with extensive microglial infiltration (Barnett and Prineas, 2004). An additional difference between these two neuropathological findings is the presence of remyelination attempts that are frequently detected in the immune-mediated forms and are less prominent in cases with extensive oligodendrogliopathy (Lucchinetti et al., 2000). Very little is known regarding the causes of oligodendroglial death in MS patients; however, increased levels of the pro-apoptotic transcription factor p53 and of its downstream genes have been detected in cases characterized by oligodendrogliopathy, microglial infiltration, and relative lack of remyelination (Kuhlmann et al., 2002; Aboul-Enein et al., 2003; Wosik et al., 2003; Stadelmann et al., 2005). Studies in cultured human oligodendrocytes further identified p53 as a critical pro-apoptotic effector (Ladiwala et al., 1999; Wosik et al., 2003). We thereby hypothesized that this transcription factor could play a very important role in models of primary demyelination consequent to oligodendrocyte dystrophy.
To test this hypothesis, we have adopted the cuprizone model of toxic demyelination, which is characterized by oligodendrocyte apoptosis and microglial infiltration (Hiremath et al., 1998; Matsushima and Morell, 2001). In this model, a reproducible pattern of demyelination can be induced in the dorsal corpus callosum of C57BL/6 mice by administering a cuprizone diet for 6 weeks (Matsushima and Morell, 2001). After 3 weeks of continuous feeding, mature oligodendrocytes die by apoptosis, and this is associated with microglial infiltration in the absence of any breakage of the blood–brain barrier (Hiremath et al., 1998) and increased expression of microglial gene products (Morell et al., 1998; Jurevics et al., 2002). In this study, we demonstrate that elevated levels of p53 can be detected in the corpus callosum of mice during the first 2–3 weeks of cuprizone diet. Using genetic deletion and pharmacological inhibition of p53, we demonstrate that this transcription factor modulates three prominent events characteristic of human and animal models of primary demyelination consequent to oligodendrogliopathy, including extensive oligodendrocytic apoptosis, microglial activation, and lack of remyelination. Thus, we propose the transient inhibition of p53 function as a potential therapeutic target for demyelinating conditions characterized by primary oligodendroglial dysfunction.
Materials and Methods
Animal model of experimental demyelination induced by dietary cuprizone.
All of the experiments were performed in 8-week-old mice. For the experiments on p53 expression levels and pifithrin-α treatment, we used C57BL/6J mice (n = 40). For the immunohistochemical experiments, the microarray gene expression profiling, and the validation by quantitative real-time PCR, we used mice obtained from Trp53Tm1Tyj C57BL/6J heterozygous breeding pairs (n = 24 p53+/+; n = 24 p53−/−). Mouse genotypes were confirmed by tail clipping and PCR using the primers 5′-ACAGCGTGGTGGTACCTTAT-3 (ImRo36), 5′-TATACTCAGAGCCGGCCT-3′ (ImRo37), and 5′-TCCTCGTGCTTTACGGTATC-3′ (neo), yielding a fragment of 375 bp for p53+/+ and 525 bp in p53−/− mice. Eight-week-old male p53−/− and p53+/+ sibling mice were placed on a diet of 0.2% (w/w) cuprizone (Sigma) mixed into milled chow (Harlan Teklad Certified LM-485, code 7012CM), which was available ad libitum. Sham and cuprizone-treated C57BL/6J mice (n = 40) were killed weekly for the time-course study (n = 24); the remaining 16 C57BL/6J, the p53+/+ (n = 24), and p53−/− mice (n = 24) were maintained on the cuprizone diet for the specified time period or received additional treatment as indicated by the specific experimental paradigm. Mice were maintained in sterile pathogen-free conditions according to protocols approved by the Institutional Animal Care and Use Committee of Robert Wood Johnson Medical School/University of Medicine and Dentistry of New Jersey.
Pifithrin treatment in vivo.
The first group of C57BL/6J mice was subject to DMSO (n = 7) or pifithrin-α (n = 7) (2 mg/kg body weight, i.p.) injection every other day after mice were fed with 0.2% cuprizone diet for 2 weeks. After 10 d of continuous injection and feeding treatment, four mice were killed for perfusion with 4% paraformaldehyde (PFA) followed by cryopreservation and immunohistochemical analysis, and the remaining three mice were killed for RNA extraction. The second group of mice was subject to DMSO (n = 4) or pifithrin-α (n = 4) (2 mg/kg body weight, i.p.) injection every other day after mice were fed with 0.2% cuprizone diet for 4 weeks. After 10 d of continuous injection and feeding treatment, four mice were killed for perfusion with 4% PFA for immunohistochemical analysis.
Gene expression profiling using Affymetrix microarrays.
Total RNA was isolated from whole brains of wild-type C57B mice or p53 knock-out mice after 4 weeks of cuprizone treatment using Trizol (Invitrogen) and cleaned up with the RNeasy Mini kit (Qiagen). RNA from each group (n = 3) was pooled in equimolar amount before hybridization with GeneChip Mouse Expression Set 430A Arrays (Affymetrix), which contains probes for detecting 22,600 transcripts. After RNA isolation, all the subsequent technical procedures including quality control, measurement of RNA concentration, cDNA synthesis, biotin labeling of cRNA, hybridization, and scanning of the arrays were performed using standard procedure at the Cancer Institute of New Jersey.
Quantitative real-time PCR.
Total RNA was extracted from mouse brains by using Trizol reagent (Invitrogen) and cleaned using the RNeasy Mini kit (Qiagen). RNA quality was assessed by electrophoresis on denaturing MOPS (4-morpholinepropanesulfonic acid) gels and spectrophotometry. Two micrograms of total RNA were used in 20 μl of reverse transcription (RT) reaction, using the SuperScript RT-PCR kit (Invitrogen). Quantitative real-time PCR was performed using Applied Biosystems SYBR green PCR master mix in the 7900HT Sequence Detection PCR System. Negative controls (samples in which reverse transcriptase was omitted) were amplified individually by PCR using the different primer sets used in the present studies to ensure the absence of genomic DNA contamination. PCR amplification resulted in the generation of single bands. The melting curve of each sample was measured to ensure the specificity of the amplified products. Data were normalized to the internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and analyzed using Pfaffl ΔΔCt method. For primers used in quantitative PCR, see Table 1.
Table 1.
Sequences of primers for quantitative PCR
| Gene | Forward | Reverse |
|---|---|---|
| BDNF | 5′-CCATAAGGACGCGGACTTGT-3′ | 5′-AAGAGTAGAGGAGGCTCCAAAGG-3′ |
| β2-Microglobulin | 5′-GATACATACGCCTGCAGAGTTAAGC-3′ | 5′-GATCACATGTCTCGATCCCAGTAG-3′ |
| CHKL3 | 5′-ACTATTTTGAAACCAGCAGCCTTT-3′ | 5′-AGATCTGCCGGTTTCTCTTAGTCA-3′ |
| CGT | 5′-TGGTTGACATACTGGATCACTATACTA-3′ | 5′-CGATCACAAATCCACACATATCATT-3′ |
| CNPase | 5′-TGGTGTCCGCTGATGCTTAC-3′ | 5′-CCGCTCGTGGTTGGTATCAT-3′ |
| Gadd45 | 5′-ATTACGGTCGGCGTGTACGA-3′ | 5′-CAGCAGGCACAGTACCACGTTA-3′ |
| GAPDH | 5′-ACCCAGAAGACTGTGGATGG-3′ | 5′-CACATTGGGGGTAGGAACAC-3′ |
| Lysozyme | 5′-GTCACTGCTCAGGCCAAGGT-3′ | 5′-TAGCCAGCCATTCCATTCCT-3′ |
| MSC-2 | 5′-GGGTACTACTGCACAGCAGACAAC-3′ | 5′-GGAATTTTCACGGTGATGTTCA-3′ |
| p21 | 5′-GAGACAACGGCACACTTTGCT-3′ | 5′-CCACAGGCACCATGTCCAA-3′ |
| Sox2 | 5′-AACTTTTGTCCGAGACCGAGAA-3′ | 5′-CCTCCGGGAAGCGTGTACT-3′ |
| Tcf4 | 5′-GTCCTCGCTGGTCAATGAAT-3′ | 5′-CCCTTAAAGAGCCCTCCATC-3′ |
| TNFα | 5′-GCACCACCATCAAGGACTCAA-3′ | 5′-TGCAGAACTCAGGAATGGACAT-3′ |
| p53 | 5′-CTCACTCCAGCTACCTGAAGA-3′ | 5′-AGAGGCAGTCAGTCAGTCTGAGTCA-3′ |
| Mouse IL1β | 5′-GTGAATGCCACCTTTTGACA-3′ | 5′-CAAAGGTTTGGAAGCAGCC-3′ |
Statistical analysis was performed using GraphPad Prism 4.01 software (Graph Pad Software) by ANOVA followed by Bonferroni's multiple comparison test or Friedman test, followed by Dunn's multiple-comparison test. An unpaired Student's t test was used when only two conditions were compared.
Immunohistochemistry, confocal image acquisition, and quantitative analysis.
For immunohistochemistry, animals were anesthetized and perfused intracardially with 4% PFA in 0.1 m phosphate buffer, pH 7.2. Weil myelin staining was performed by the histopathology service NeuroScience Associates. For all the other experiments, the brains were removed, postfixed overnight in the same solution at 4°C, cryopreserved in 30% sucrose in 0.1 m phosphate buffer, and embedded in optimal cutting temperature compound. Frozen sections (20 μm) were cut, incubated in blocking buffer PGBA (0.1 m phosphate buffer, 0.1% gelatin, 1% bovine serum albumin, 0.002% sodium azide) containing 10% normal goat serum (0.5% Triton X-100 was added for cytoplasmic and nuclear antigens) for 30 min, and incubated overnight with the primary antibody diluted in the same blocking buffer. After rinsing in PBS, sections were incubated with the appropriate secondary antibodies conjugated to fluorescein or rhodamine (Southern Biotechnologies, GE Healthcare, Jackson ImmunoResearch, and Vector Laboratories). 4′,6-Diamidino-2-phenylindole (DAPI; 1:1000; Invitrogen) was used as a nuclear counterstain. Immunostained sections were analyzed using a fluorescent microscope (DMRA; Leica) followed by confocal analysis (LSM 510 meta confocal laser-scanning microscope; Carl Zeiss MicroImaging). Confocal images were captured at a 1 μm interval, and stacks of six slices were typically used for the generation of the projections. For the quantification of the in vivo experiments, a minimum of three sections per mouse and three mice for each group were evaluated. The following primary antibodies were used: anti-β-tubulin III (1:500, clone Tuj1; Covance), anti-GFAP (1:1000; Dako), anti-bromodeoxyuridine (BrdU; 1:200; Dako), Sox2 (1:1000; Millipore Bioscience Research Reagents), CC1/APC (mouse monoclonal, 1:50; Oncogene Research Products), myelin basic protein (MBP; mouse monoclonal SMI99, 1:1000 for immunohistochemistry and Western blot analysis; Sternberger Monoclonals), Mac1 (rat monoclonal, 1:100; Millipore Bioscience Research Reagents), and Ki67 (Abcam). After incubation with primary antibodies, the cells were stained with the appropriate secondary antibodies conjugated with fluorophores (Jackson ImmunoResearch) or with biotin (Vector Laboratories). To stain with anti-BrdU antibodies (1:200; Dako), the sections were treated with 2N HCl for 10 min at room temperature, followed by neutralization in 0.1 m borax, pH 8.6, for 10 min at room temperature. After washing with 0.1 m phosphate buffer, the cells were incubated with anti-BrdU antibodies in PGBA containing 0.1% Triton X-100 for at least 3 h at room temperature. Then, the cells were incubated with appropriate secondary antibodies and DAPI. Immunoreactive cells were analyzed using a fluorescence microscope (Leica), and images were captured using a Hamamatsu CCD camera using the Image software.
In vitro experiments in primary cultures of microglial cells from p53+/+ and p53−/− mice and in the BV2 microglial cell line.
Postnatal day 1 cortices from p53+/+ and p53−/− siblings were used for cell culture. Briefly, after removal of the meninges, cortical tissue was minced and dissociated by gentle trituration in NM10 medium (DMEM plus 10% FBS), using fire-polished Pasteur pipettes. Cells were plated into P75 tissue culture flasks coated with 10 ng/ml poly-d-lysine and cultured at 37°C in a humidified incubator with 10% CO2 supplementation. The medium was changed every 2 d. Three weeks later, cells were harvested by shaking at 37°C at 190 rpm overnight. The collected cells were plated into uncoated 10 cm tissue culture dishes. Thirty minutes after plating, the floating cells were discarded by replacing the medium. The following day, cells were treated with 100 ng/ml lypopolysaccharide (LPS) for 6 h, and the RNA was harvested for additional analysis. The immortalized microglia cell line, BV2, was donated by V. Bocchini (University of Perugia, Perugia, Italy); it was generated by infecting primary mouse microglia cultures with a v-raf/v-myc oncogene-carrying retrovirus (J2) (Blasi et al., 1990). BV2 cells were maintained in DMEM/F-12 containing 10% FCS. Confluent cultures were passed by trypsinization. BV2 cells were cultured in DMEM/F-12 plus 10% FBS until ∼75% confluent. Pifithrin-α (20 μm) and/or LPS (100 ng/ml) were added at the same time, and cells were harvested in Trizol 6 h later. Control cells received the same volume of water or DMSO. Total RNA was extracted as reported above. Expression levels of the genes of interest were normalized to GAPDH levels.
In vitro experiments in multipotential neural precursors derived from the adult subventricular zone.
Subventricular zones (SVZs) were dissected from 8-week-old C57BL/6J mice in Leibovitz's L-15 medium (Invitrogen). After digestion with papain (Sigma), cells were mechanically dissociated by pipetting and resuspended in complete medium consisting of DMEM/F-12 (Invitrogen) supplemented with B27 (Invitrogen), 25 mm glucose, 2 μg/ml heparin (Sigma), 20 ng/ml epidermal growth factor (EGF) and 10 ng/ml FGF. An equal number of cells was plated in 48-well plates. Cells were incubated at 37°C in a 5% CO2 incubator for 7 d and added fresh EGF (Peprotec) and FGF (Peprotec) every other day. To test multipotency of neural precursors in vitro, neurospheres were rinsed in 0.1 m phosphate buffer and mechanically dissociated. The same number of cells was plated in 8-well chamber slides in differentiation medium: DMEM/F-12 supplemented with B27, 25 mm glucose, and 1% FBS (Invitrogen). The following day, cells were pretreated with 25 μm cuprizone (Sigma) for 24 h and allowed to differentiate for 5 additional days in differentiation medium in the presence or absence of 20 μm pifithrin-α.
Results
p53 is expressed in the corpus callosum of mice treated with cuprizone, and its ablation and pharmacological inhibition decrease the extent of demyelination
Elevated levels of the transcription factor p53 were previously described in the brain of MS patients characterized by demyelination consequent to oligodendroglial cell death and by microglial infiltration (Kuhlmann et al., 2002; Wosik et al., 2003). The animal model that best recapitulates these neuropathological findings is the cuprizone model of demyelination that is characterized by loss of myelin in the dorsal corpus callosum because of oligodendrocytic death and microglial activation (Matsushima and Morell, 2001). To define whether p53 levels were also upregulated in the corpus callosum of cuprizone-fed mice, we performed real-time PCR on samples isolated at multiple time points during cuprizone treatment (Fig. 1A). In agreement with the previous findings in the brain of MS patients, in this animal model we also detected a twofold increase in p53 transcripts during the first 2–3 weeks of cuprizone diet that temporally correlated with oligodendrocytic apoptosis and microglial infiltration (Matsushima and Morell, 2001). The increased transcripts were accompanied by enhanced immunoreactivity in the corpus callosum of cuprizone-treated mice in coronal sections stained with a commercially available antibody against p53, the specificity of which was supported by the inability to stain sections from knock-out mice (Fig. 1B). Based on these results, we hypothesized that p53 may play an important role in the cuprizone model of demyelination.
Figure 1.

Upregulation of p53 levels in the corpus callosum of mice on a cuprizone diet. A, Quantitative real-time PCR of p53 mRNA levels in samples isolated from the dorsal corpus callosum of mice treated with cuprizone for the indicated times (2wk, 3wk, 4wk, 6wk) and in mice treated with cuprizone and then allowed to recover for 2 or 3 weeks (2wk post cup, 3wk post cup). The levels were normalized to GAPDH, and the levels detected in mice that were fed a normal diet were arbitrarily set as reference point (untreated). Bar graphs represent the mean of at least four independent experiments, each performed in triplicate. Error bars indicate SEM. *p < 0.05 (one-way ANOVA followed by Bonferroni's multiple comparison test). wk, Week; cup, cuprizone. B, Immunohistochemistry of corpus callosum of untreated wild-type mice and cuprizone-treated wild-type (p53+/+) or p53 knock-out (p53−/−) mice stained with antibodies against p53 (red) and DAPI (blue). Staining of the sections from cuprizone-treated p53−/− mice served as control for antibody specificity. Scale bar, 80 μm.
To test this hypothesis, we compared the extent of myelin loss in the dorsal corpus callosum of p53+/+ (n = 4) and p53−/− (n = 4) mice after 4 weeks of cuprizone treatment (Fig. 2). Myelin in the corpus callosum of p53+/+ and p53−/− treated mice was detected by Weil staining. Loss of myelinated fibers was detected in the dorsal corpus callosum of cuprizone-treated p53+/+ mice (Fig. 2A) but not in the same brain region of cuprizone-treated p53−/− siblings (Fig. 2B). The myelin loss detected in p53+/+ mice (n = 3) after 4 weeks of cuprizone was consistent with the decreased levels of myelin transcripts assessed by real-time PCR (Fig. 3A). The levels of ceramide-galactosyl-transferase (cgt) were 66.6 ± 4%, and the levels of 2′,3′cyclic nucleotide-3′-phosphodiesterase (cnp) transcripts in cuprizone-treated p53+/+ mice were 61.8 ± 5% lower than untreated controls. In contrast, in cuprizone-treated p53−/− mice, the cgt levels were only 31.2 ± 5% lower, and those of cnp were 28 ± 6% lower than untreated controls (Fig. 3A). Additional results supporting the preservation of myelinated fibers in the corpus callosum of p53−/− mice (n = 3) was provided by immunohistochemical analysis of coronal sections stained by antibodies specific for MBP (Fig. 3B). The persistence of MBP staining correlated with the greater number of surviving CC1+ oligodendrocytes in the corpus callosum of cuprizone-treated p53−/− mice compared with wild-type mice (n = 3) (Fig. 3C,D). In agreement with the hypothesized role of p53 as a pro-apoptotic gene, decreased apoptosis was detected in the corpus callosum of cuprizone-treated p53−/− mice compared with p53+/+ mice (Fig. 3E). Together, these data suggested an important role for p53 function in myelin loss consequent to oligodendrogliopathy.
Figure 2.
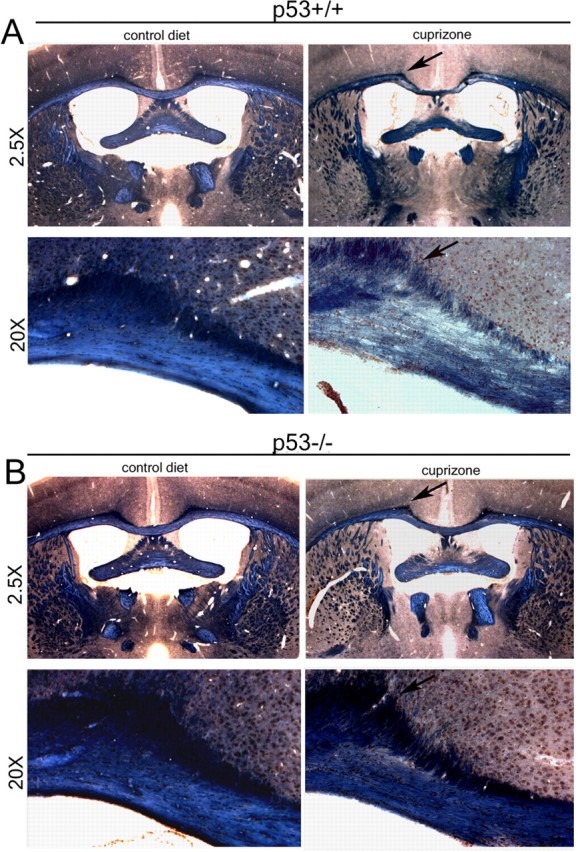
Mice lacking p53 are less sensitive to cuprizone-induced demyelination. A, Bright-field low-power (2.5×) and high-power (20×) micrographs of coronal brain sections of wild-type mice at the level of the dorsal fornix. At 8 weeks of age, wild-type siblings (p53+/+) were fed for 4 weeks with regular food (control diet) or with a modified diet containing 0.2% cuprizone and then killed. Brain sections were stained with Weil's to detect myelinating tracts. Note the reduction of myelinating fibers in the corpus callosum of cuprizone-treated wild-type mice (arrow). B, Low- and high-power micrographs of coronal brain sections from p53-deficient siblings (p53−/−) either untreated (control diet) or treated with cuprizone. Note the sparing of myelin in the corpus callosum of cuprizone-treated mice (arrow).
Figure 3.
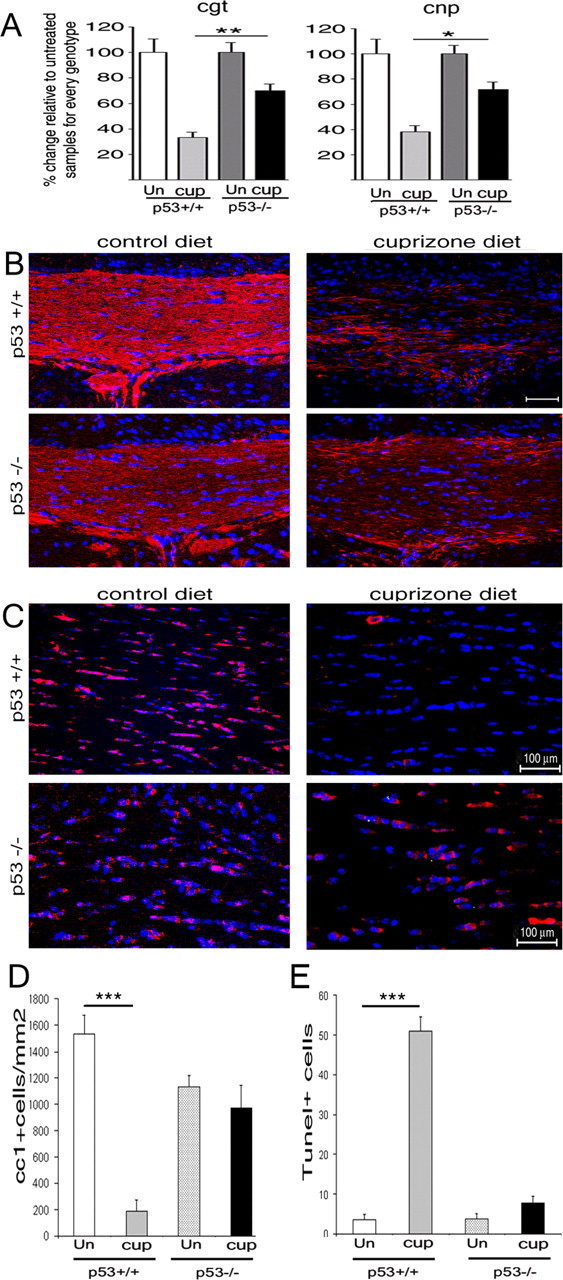
Increased oligodendrocyte survival and myelin sparing in mice lacking p53. A, Relative RNA levels of the myelin gene transcripts for cgt and cnp detected in the dorsal corpus callosum of p53+/+ and p53−/− mice after 4 weeks of feeding on a control or 0.2% cuprizone diet. The RNA levels in each sample were referred to the GAPDH levels, and the levels detected in the untreated animals were chosen as a reference point. Note the higher levels of myelin gene transcripts detected in cuprizone-treated p53−/− mice compared with treated wild-type siblings. *p < 0.05; **p < 0.01. Error bars indicate SEM. B, Confocal image of coronal brain sections of wild-type (p53+/+) and knock-out (p53−/−) siblings fed a control or cuprizone diet for 4 weeks and then stained with antibodies specific for MBP to identify myelinated fibers (red) and DAPI (blue) as nuclear counterstain. Scale bar, 50 μm. C, Confocal image of coronal brain sections of wild-type (p53+/+) and knock-out (p53−/−) siblings fed a control or a 0.2% cuprizone diet for 4 weeks and then stained with the CC1 antibody to identify oligodendrocytes (red) and DAPI (blue) as nuclear counterstain. D, Bar graphs indicate the relative number of CC1+ cells detected in the dorsal corpus callosum of p53+/+ and p53−/− mice receiving either control or 0.2% cuprizone diet. n = 3 animals per condition; ***p < 0.001. E, Bar graphs indicate the relative number of TUNEL+ cells detected in the dorsal corpus callosum of p53+/+ and p53−/− mice after 3 weeks of control or 0.2% cuprizone diet. n = 3 animals per condition; ***p < 0.001. Note that in p53−/− mice the difference in the number of CC1+ and TUNEL+ cells between the untreated and cuprizone-treated groups is not statistically significant. Un, Untreated (control); cup, cuprizone; TUNEL, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling.
To further explore the potential therapeutic value of these findings, we asked whether pharmacological inhibition of p53 with pifithrin-α (Culmsee et al., 2001) would similarly prevent myelin loss in the cuprizone model of demyelination. We adopted two distinct protocols of pifithrin-α administration. In a first set of experiments, wild-type C57BL/6 mice (n = 8) received systemic administration of pifithrin-α (2 mg/kg body weight; n = 4) or vehicle control (n = 4) during the second and third week of cuprizone diet, at a time coincident with the detection of elevated p53 levels and peak of oligodendrocytic apoptosis in the corpus callosum (Fig. 4A). In a second set of experiments, pifithrin-α (n = 4) or vehicle control (n = 4) was administered after the peak of oligodendrocytic death, during the fourth and fifth week of cuprizone treatment (Fig. 4D). Early administration of pifithrin-α significantly increased oligodendrocyte survival (Fig. 4B) while decreasing the number of apoptotic cells (Fig. 4C) detected in the corpus callosum. In contrast, late administration of pifithrin-α had a less prominent effect on oligodendrocyte survival (Fig. 4F), although it still protected the existing myelinated fibers (Fig. 4E). These results suggested that, besides the role in oligodendrocyte apoptosis, p53 might affect the integrity of myelinated fibers also by a second mechanism, possibly involving the activation of microglial cells recruited to the corpus callosum.
Figure 4.

Systemic administration of pharmacological inhibitors of p53 is most effective in protecting oligodendrocytes and myelin when started during the first 3 weeks of cuprizone treatment. A, Schematic representation of the early administration protocol of pifithrin-α treatment (red arrows) during the second and third week of cuprizone diet (black arrows). The asterisk denotes the time point of analysis when the animals were killed and the brains were processed for immunohistochemistry. B, Bar graphs indicate the number of CC1+ oligodendrocytes in the dorsal corpus callosum of animals that received either a control diet or a 0.2% cuprizone diet for 3.5 weeks in the absence (cup) or presence of systemic pifithrin-α administration (cup+pif). n = 3 for each treatment; ***p < 0.001; **p < 0.01. C, TUNEL+ cell counts in the corpus callosum of animals that received either a control diet or a 0.2% cuprizone diet for 3.5 weeks in the absence (cup) or presence of systemic pifithrin-α administration (cup+pif). n = 3 for each treatment; ***p < 0.001; *p < 0.05. D, Schematic representation of the late administration protocol of pifithrin-α treatment (red arrows) started during the fourth week of cuprizone diet (black arrows). The asterisk denotes the time point of analysis when the animals were killed and the brains were processed for immunohistochemistry. E, Confocal image of coronal brain sections of wild-type mice on a regular diet (control) or fed a cuprizone diet for 5.5 weeks in the absence (cuprizone 5.5 wk) or presence of systemic pifithrin-α administration (cup + pif 5.5 wk). Sections were stained with MBP antibody to identify myelinated fibers (red) and DAPI (blue) as nuclear counterstain. F, Bar graph quantification of CC1+ oligodendrocytes in the dorsal corpus callosum of animals that received either a control diet or a 0.2% cuprizone diet for 5.5 weeks in the absence (cup) or presence of systemic pifithrin-α (cup+pif) administration during the fourth week. n = 3 for each treatment; ***p < 0.001; **p < 0.01. Error bars indicate SEM. pif, Pifithrin-α; cup, cuprizone.
Genetic and pharmacological inhibition of p53 function decreases microglial recruitment and gene expression
In agreement with the protective role of p53 inhibition, gene expression profiling of samples isolated from the corpus callosum of p53+/+ and p53−/− mice using Affymetrix gene microarrays revealed greater levels of myelin gene transcripts (supplemental Table 1, available at www.jneurosci.org as supplemental material) and lower levels of activated microglial gene transcripts (supplemental Table 2, available at www.jneurosci.org as supplemental material), in cuprizone-treated p53−/− compared with p53+/+ mice. As expected, cuprizone-treated p53−/− mice did not exhibit the characteristic upregulation of p53-dependent target genes (i.e., p21, gadd45) detected in treated p53+/+ mice (Fig. 5A). Previous studies had reported the characteristic upregulation of microglial genes, including lysozyme, β2-microglobulin, and several cathepsins in the corpus callosum of cuprizone-fed mice (Morell et al., 1998; Jurevics et al., 2002). In agreement with the hypothesis that p53 function interferes with the program of microglial gene activation, we detected lower levels of lysozyme, β2-microglobulin, and tnfα in the corpus callosum of cuprizone-treated p53−/− mice compared with p53+/+ mice (Fig. 5B). Similar results were obtained in mice receiving systemic administration of pifithrin-α because systemic administration of this inhibitor significantly impaired the cuprizone-induced upregulation of p21 and gadd45 transcripts in cuprizone-fed mice (Fig. 5C) and lowered the transcriptional activation of microglia in response to cuprizone (Fig. 5D).
Figure 5.
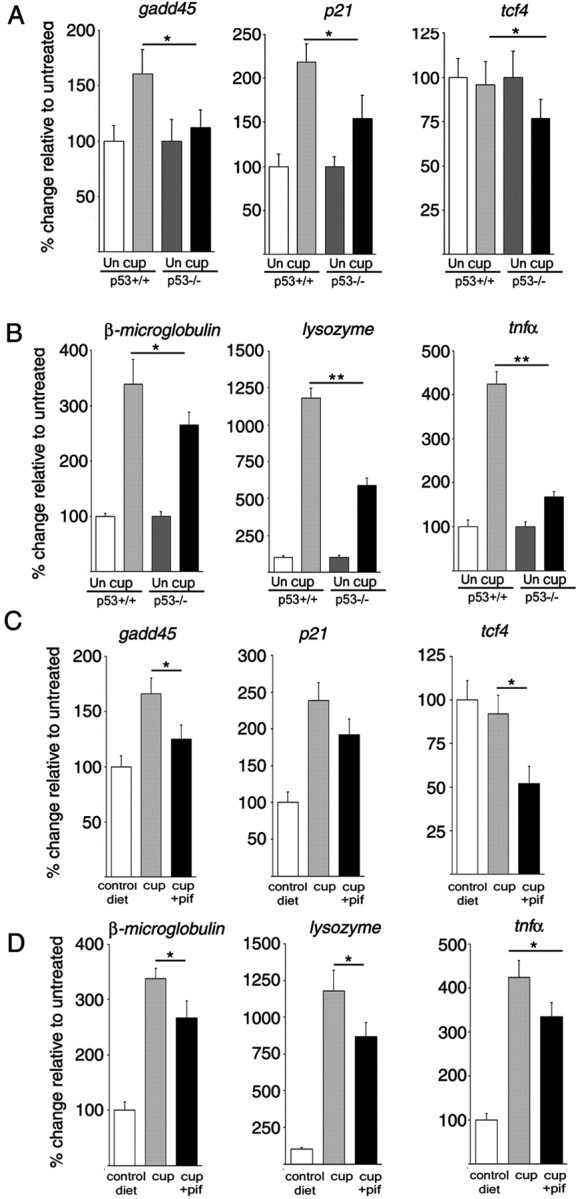
Decreased expression of microglial genes in mice with genetic deletion or pharmacological inhibition of p53. A, B, Quantitative real-time PCR of the mRNA levels for the p53 target genes gadd45, p21, and tcf4 (A) and of β2-microglobulin, lysozyme, and tnf-α (B) in samples isolated from the corpus callosum of untreated (un) and cuprizone-treated (cup) p53+/+ and p53−/− mice. C, D, Quantitative real-time PCR of the mRNA levels for the p53 target genes gadd45, p21, and tcf4 (C) and of β2-microglobulin, lysozyme, and tnf-α (D) in samples isolated from the corpus callosum of untreated controls (control diet), cuprizone-fed mice (cup), and cuprizone-fed mice receiving pifithrin-α treatment (cup+pif). The RNA levels were referred to the GAPDH levels, and the values detected in cuprizone-treated mice in the absence and presence of pifithrin treatment were referred in terms of percentage value relative to the levels detected in untreated controls. *p < 0.05; **p < 0.01. Error bars indicate SEM.
Decreased expression of genes characteristic of activated microglia in p53−/− mice could result from impaired recruitment of microglial cells to the corpus callosum of cuprizone-fed mice and/or from a transcriptional effect of p53 in the recruited microglial cells. The presence of microglial cells was first assessed in the corpus callosum of cuprizone-fed p53−/− mice using immunohistochemistry and antibodies specific for the antigen Mac1. Fewer Mac1+ cells were detected in the corpus callosum of cuprizone-treated p53−/− mice compared with p53+/+ mice (Fig. 6A,B). Similar results were detected in mice receiving pifithrin-α treatment either before (Fig. 6C) or after the occurrence of oligodendroglial death (Fig. 6D,E). When pifithrin-α treatment was started during the early stages of cuprizone diet, we observed a decline in oligodendrocytic apoptosis and the nearly complete absence of Mac1+ cells in the corpus callosum of treated animals (Fig. 6C). This effect could be attributable to the fact that fewer microglial cells were recruited to the corpus callosum if fewer oligodendrocytes died. In a second set of experiments, pifithrin-α treatment was initiated after the period of massive oligodendrocytic death had already occurred. Also in this case, however, pifithrin-α-treated mice showed a significantly reduced number of Mac1+-immunoreactive cells in the corpus callosum (Fig. 6D). Together, these results suggest that modulation of p53 function affects the behavior of microglial cells in multiple ways.
Figure 6.
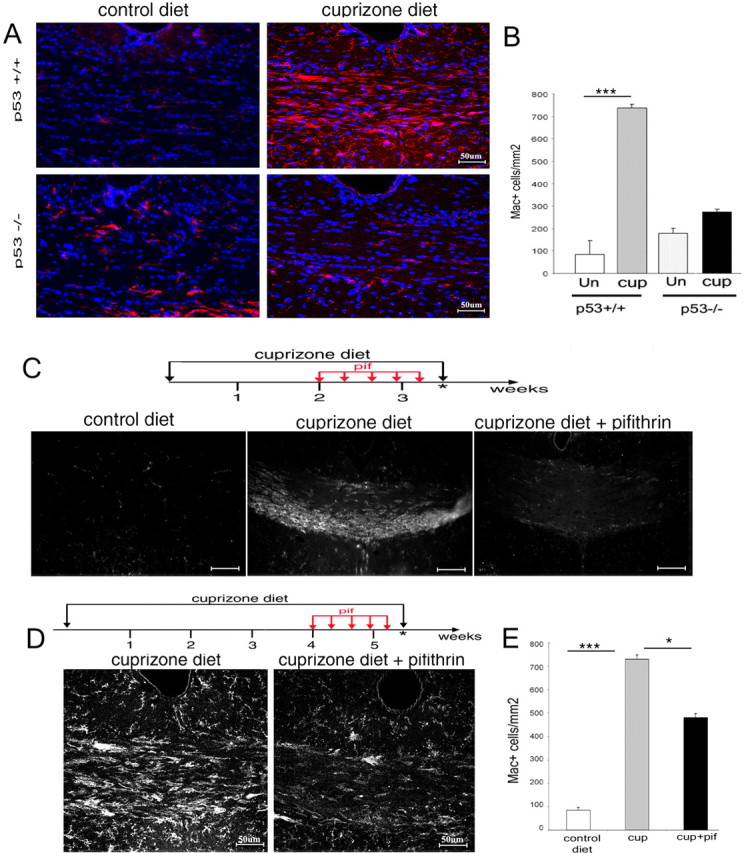
Pifithrin-α treatment of cuprizone-fed mice reduces recruitment of microglial cells to the corpus callosum even if initiated after oligodendrocyte apoptosis has occurred. A, Confocal image of coronal brain sections of wild-type (p53+/+) and knock-out (p53−/−) siblings fed either a control diet or a 0.2% cuprizone diet for 4 weeks and stained with Mac1 antibody (red) to identify microglial cells and with DAPI (blue) as nuclear counterstain. B, Bar graphs indicate the relative number of Mac1+ cells detected in the dorsal corpus callosum of p53+/+ and p53−/− mice receiving either control (Un) or 0.2% cuprizone (cup) diet. n = 3 animals per condition; ***p < 0.001. C, Schematic representation of the duration of the early pifithrin-α treatment (red arrows) in mice fed a cuprizone diet for 3.5 weeks (black arrows). The asterisk denotes the time point of analysis when the animals were killed and the brains were processed for immunohistochemistry (top). Immunohistochemistry of coronal brain sections of the medial corpus callosum at the level of the dorsal fornix, stained with antibodies specific for the microglial marker Mac1 (bottom), is shown. Note the presence of immunoreactive cells only in the cuprizone-fed mice but not in mice on a control diet or in mice treated with pifithrin-α during the first 3 weeks of cuprizone diet. Scale bars, 80 μm. D, Schematic representation of the duration of the late pifithrin-α treatment (red arrows) in mice fed a cuprizone diet for 5.5 weeks (black arrows). The asterisk denotes the time point of analysis when the animals were killed and the brains were processed for immunohistochemistry. Immunohistochemistry of coronal brain sections of the medial corpus callosum at the level of the dorsal fornix, of mice treated as described in C, is shown. Sections were stained with antibodies specific for the microglial marker Mac1. E, Bar graph representation of Mac1+ cells in the dorsal corpus callosum of animals that received either a control diet or a 0.2% cuprizone diet for 5.5 weeks in the absence (cup) or presence of systemic pifithrin-α (cup+pif) administration during the fourth week. n = 3 for each treatment; ***p < 0.001; *p < 0.05. Error bars indicate SEM. pif, Pifithrin-α.
To further define whether the effect of p53 inhibition on microglial cells was cell intrinsic and independent of cross talks with the oligodendrocytic population, we adopted a reductionist approach and used either the BV2 microglial cell line or primary microglial cultures isolated from the cortices of p53−/− and p53+/+ mice. The advantage of the in vitro experimental system is the possibility to evaluate the effect of p53 on microglial gene expression, using a homogeneous and controlled experimental setting. Consistent with the possibility that p53 modulated the expression of genes characteristic of activated microglia, we detected increased p53 levels in cultured microglial BV2 cells after LPS (100 ng/ml) stimulation (Fig. 7A). LPS also induced the expression of the p53 gene target p21, and this increase was blocked by cotreatment with pifithrin-α (20 μm) (Fig. 7B). The LPS-dependent elevation of p21 transcript levels was also detected in p53+/+ primary cultures of microglial cells from mice, but not in cells isolated from p53−/− mice (Fig. 7C). Treatment of BV2 cells with pifithrin-α effectively reduced the LPS-dependent activation of the microglial genes lysozyme and β2-microglobulin, although it had no effect on the levels of cathepsin Z (Fig. 7D). Similarly, LPS stimulation or cuprizone treatment (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) of primary microglial cultures elicited a greater increase in the transcript levels of β2-microglobulin and tnfα in p53+/+ cells compared with p53−/− mice (Fig. 7E). The fact that pifithrin or genetic ablation of p53 did not completely abolish the transcriptional response of microglia to LPS is consistent with the importance of nuclear factor-κb (NF-κb) regulation of this transcriptional program (Lu et al., 2007). Together, these data support a role for p53 as one of the modulators of gene expression in activated microglial cells.
Figure 7.
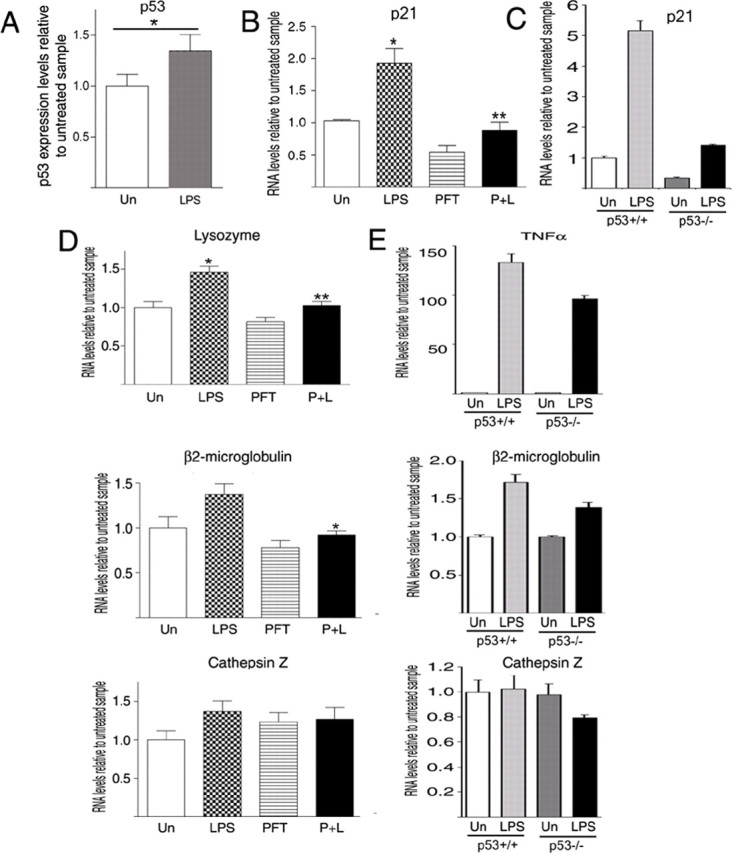
Pharmacological inhibition of p53-dependent transcriptional activity decreases the transcriptional activation induced by LPS in microglial cells. A, Quantitative real-time PCR of p53 mRNA levels in samples isolated from BV2 microglial cells either untreated (Un) or treated with 100 ng/ml LPS. *p < 0.01 versus untreated cells. B, Quantitative real-time PCR of the p21 mRNA levels in samples extracted from BV2 cells either untreated or treated with 100 ng/ml LPS alone (LPS), 20 μm pifithrin-α alone, or cotreated for 6 h. **p < 0.001 versus LPS-treated cells; *p < 0.01 versus untreated cells. C, Real-time PCR of p21 transcript levels measured in RNA samples from primary microglial cultures isolated from p53+/+ and p53−/− mice and stimulated with LPS. D, Quantitative real-time PCR of the mRNA levels in samples isolated from BV2 cells as described in B. Lysozyme, *p < 0.01 versus untreated cells, **p < 0.01 versus LPS; β2-microglobulin, *p < 0.05 versus LPS. The levels of cathepsin Z were not affected by pifithrin-α. Bar graphs represent the mean ± SEM of four independent experiments run in triplicate (ANOVA followed by Bonferroni's multiple comparison test). E, Effect of LPS treatment on transcripts indicative of microglial cell activation. The bar graphs represent the results of quantitative real-time PCR performed on RNA isolated from primary cultures of p53+/+ and p53−/− microglial cells stimulated with LPS. Note the lower level of microglial gene activation detected in p53−/− cells and the similarity with the in vivo data in cuprizone-fed mice, shown in Figure 5. Un, Untreated; PFT, pifithrin-α; P+L, pifithrin-α plus LPS. Error bars indicate SEM.
We have previously mentioned that primary demyelination consequent to oligodendrogliopathy and microglial infiltration is also associated with defective repair. Because p53 affected both oligodendrocyte loss and microglial activation, we asked whether this molecule could also modulate the behavior of cells affecting repair. Previous studies had shown that demyelination of the corpus callosum is repaired by resident oligodendrocyte progenitors and by multipotential cells residing in the adult SVZ (Nait-Oumesmar et al., 1999; Picard-Riera et al., 2002; Murtie et al., 2005; Trebst et al., 2007). Other studies (Billon et al., 2004), including those from our group (Gil-Perotin et al., 2006), had reported an important role for p53 in regulating the pool of oligodendrocyte progenitors in the developing brain and in modulating the behavior of multipotential cells in the adult SVZ. However, the potential role of p53 in repair after demyelination remained to be defined. To address this question, we tested p53 expression in the SVZ of cuprizone-treated mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and compared the responsiveness of multipotential SVZ cells in p53+/+ mice with that observed in p53−/− and also in pifithrin-treated p53+/+ mice after cuprizone-induced demyelination. Immunohistochemical analysis of coronal brain sections using antibodies specific for the neural stem cell marker Sox 2 (Kondo and Raff, 2004) revealed an increase in immunoreactive cells in response to cuprizone-induced demyelination, a response that was enhanced by pifithrin-treated mice (Fig. 8A). In agreement with previous reports on the role of p53 in multipotential progenitors of the adult SVZ (Gil-Perotin et al., 2006), p53−/− mice had greater resting numbers of Sox2+ cells, and cuprizone treatment significantly increased their number (Fig. 8B). Because SVZ cells can be also characterized for the presence of the oligodendrocyte precursor surface molecule polysialic acid-neural cell adhesion molecule (PSA-NCAM) (Ben-Hur et al., 1998; Nait-Oumesmar et al., 1999, 2007; Decker et al., 2000), we conducted a similar analysis on the adult SVZ of cuprizone-treated mice. Consistent with the results obtained for Sox2, cuprizone treatment per se induced a significant increase in PSA-NCAM+ cells in the SVZ of untreated p53+/+ mice (Fig. 8C). This increase was further enhanced by either treatment with pharmacological inhibitors of p53 (Fig. 8C) or in animals with genetic ablation (Fig. 8D). Overall, these results suggest that p53 function is important for regulating the number of multipotential progenitors in the SVZ that are involved in repair.
Figure 8.

Increased Sox2+ and PSA-NCAM+ cells in the SVZ of mice with genetic or pharmacological inhibition of p53 in vivo after cuprizone-induced demyelination. A, Confocal image of brain sections from adult p53+/+mice receiving a 4 week control diet or a 0.2% cuprizone diet in the absence (cup) or presence of systemic administration of pifithrin-α (cup+pif). Sections were stained with antibodies against Sox2 (green) and DAPI (blue). The bar graphs represent the number of Sox2+ cells counted in coronal sections at the level of the dorsal corpus callosum of at least three mice per group. **p < 0.01; *p < 0.05. Note the wider distribution of the Sox2+ cells in mice treated with cuprizone compared with controls and the greater response detected in the animals treated with pifithrin-α. B, Confocal image of brain sections from untreated (control diet) or cuprizone-treated (cup) p53−/− adult mice stained with antibodies against Sox2 (green) and DAPI (blue). The bar graphs represent the number of Sox2+ cells. Also in this case, cuprizone treatment significantly increased the number of Sox2+ cells in the SVZ of p53−/− mice. *p < 0.05. Note the greater number observed in these mice compared with wild type (A). C, Confocal image of PSA-NCAM+ cells in the adult SVZ of p53+/+ mice receiving a 4 week control diet or a 0.2% cuprizone diet in the absence (cup) or presence of systemic administration of pifithrin-α (cup+pif). Sections were stained with antibodies against PSA-NCAM (red) and DAPI (blue). The bar graphs represent the number of PSA-NCAM+ cells counted in the SVZ at the level of the dorsal corpus callosum of at least three mice per group. **p < 0.01; ***p < 0.001. Note that the combination of cuprizone and pifithrin-α had a stronger effect on the number of PSA-NCAM+ cells than cuprizone alone. D, Confocal image of PSA-NCAM+ (red) cells in the adult SVZ of p53−/− mice untreated (control diet) or after 4 weeks of cuprizone diet (cup). Note that the number of PSA-NCAM+ cells is already elevated in untreated controls and that after cuprizone stimulation it increases to levels that are comparable with those detected in the pifithrin-treated wild-type animals. Scale bar, 50 μm. Error bars indicate SEM. V, Ventricle.
Because p53 is an important modulator of the cell cycle, we asked whether the increased number of Sox2+ cells was attributable to increased proliferation and possibly altered differentiation of SVZ cells. Pifithrin-α treatment increased the number of proliferating cells in vivo, as assessed by the increased number of Ki67+ cells in the SVZ (Fig. 9A), similar to what previously reported for the characterization of the p53-deficient mice (Gil-Perotin et al., 2006). To further define whether the effect on the multipotential progenitors was direct or mediated by other cell types, we dissected cells from the adult SVZ of p53−/− and p53+/+ mice (Fig. 9B), cultured them as neurospheres, and allowed them to differentiate into distinct lineages in cuprizone-containing medium. Consistent with an inhibitory role of p53 in oligodendrogliogenesis, we detected increased generation of oligodendrocytes and reduced generation of astrocytes in p53−/− cultures, as described previously (Gil-Perotin et al., 2006). The experiment was then repeated with SVZ cells isolated from wild-type animals and treated with cuprizone and pifithrin-α, to mimic the in vivo condition (Fig. 9C). Also in this case, p53 inhibition favored oligodendrogliogenesis at the expenses of astrocyte generation (Fig. 9D).
Figure 9.

Pharmacological inhibition of p53-dependent transcriptional activity increases the proliferation and the generation of O4+ cells from multipotential neural progenitors derived from the adult SVZ. A, Quantification of Ki67+ cells in the SVZ of mice that received only a 0.2% cuprizone (cup) diet or also received systemic administration of pifithrin-α (cup+pif) during the second (2wk) or fourth (4wk) week of cuprizone diet. *p < 0.05; **p < 0.01. B, Primary cultures of SVZ cells were obtained from adult p53+/+ and p53−/− mice, cultured as neurospheres, dissociated, exposed to cuprizone (25 μm), and allowed to differentiate. Red, GFAP (astrocytes); green, O4 (oligodendrocytes); blue, DAPI as nuclear counterstain. Scale bar, 20 μm. C, SVZ cells isolated from adult p53+/+ mice were cultured as neurospheres, dissociated, exposed to cuprizone (25 μm), and allowed to differentiate either in the absence (cuprizone) or presence of 20 μm pifithrin-α (cuprizone+pifithrin) or 5 d. Cultures were then stained with antibodies specific for GFAP (red, astrocytes), O4 (green, oligodendrocytes), and DAPI (blue) as nuclear counterstain. D, Bar graphs indicate the percentage of immunoreactive cells relative to total DAPI+ cells. *p < 0.01; **p < 0.001. Error bars indicate SEM.
Based on these cumulative results, we suggest that transient p53 inhibition should be further explored for the development of attractive therapeutic target for demyelinating conditions characterized by primary demyelination.
Discussion
The transcription factor p53 is a well characterized molecule with the ability to induce cell-cycle arrest and apoptosis in several cell types, in response to a wide variety of toxic stimuli (Giaccia and Kastan, 1998). Previous studies had identified p53 gene polymorphism in patients with MS (Kuhlmann et al., 2002) and reported high levels of protein expression in the brain of patients with demyelinated lesions characterized by extensive oligodendrogliopathy and lack of remyelination (Wosik et al., 2003). In this study, we define the functional significance of p53 inhibition using the cuprizone model of demyelination, characterized by oligodendrocytic apoptosis and microglial infiltration, the two neuropathological findings described in the brains of MS patients with elevated p53 levels. We also identify pifithrin-α as a promising novel therapeutic agent that reduces microglial activation and myelin loss, while stimulating the cells responsible for endogenous repair.
In addition to patients with specific forms of MS, p53 was also detected in the brain of patients with progressive multifocal leukoencephalopathy, where it was expressed at high levels in oligodendrocytes infected by the John Cunningham virus and undergoing apoptosis (Yang and Prayson, 2000). Studies in cultured oligodendrocytes further defined this molecule as a downstream effector of pro-apoptotic pathways induced by tumor necrosis factor-α (Ladiwala et al., 1999), oxidative stress (Uberti et al., 1999), or glutamate agonists (Villapol et al., 2007). In agreement with the pro-apoptotic role of p53 in oligodendroglial apoptosis, we show here that p53−/− mice and mice receiving systemic administration of the p53 inhibitor pifithrin-α are less susceptible to cuprizone-induced demyelination and show enhanced oligodendrocytic survival.
We also describe a role for p53 in modulating the behavior of microglial cells, which is independent of the effect on the oligodendrocytes. Indeed, systemic administration of pharmacological inhibitors of p53 (i.e., pifithrin-α) to cuprizone-treated mice positively impacted the course of the disease, by decreasing microglial accumulation and gene expression even if started after the occurrence of oligodendroglial apoptosis. The effect of p53 on gene expression was also suggested by the results of the in vitro studies in LPS-stimulated primary microglial cultures and in the BV2 microglial cell line. In agreement with previous reports on the increased expression of p53 in microglial cells in response to infectious or excitotoxic stimuli (Garden et al., 2004; Villapol et al., 2007), we have detected the upregulation of p53 and of its downstream target gene p21 in cultures stimulated with LPS and suggested that p53 functions as a modulator of the transcriptional response characteristic of activated microglia. Our results indicate lower levels of β2-microglobulin levels in p53−/− microglial cultures compared with controls, in response to the activating stimulus. Treatment of the BV2 cells with pifithrin-α similarly reduced the LPS-dependent activation of the microglial genes lysozyme and β2-microglobulin. The effect of genetic ablation of p53 or pharmacological treatment with pifithrin-α was selective because it did not affect the levels of cathepsin Z. Our results cannot determine whether the effect of p53 on microglial gene expression is direct or indirect, possibly by modulating additional components of the transcriptional network. In peripheral macrophages, for instance, p53 has been shown to inhibit cytokine expression (Komarova et al., 2005), by interfering with NF-κb-dependent gene expression (Dijsselbloem et al., 2007). Although it is becoming increasingly evident that peripheral macrophages and resident microglial cells constitute two distinct populations (Melchior et al., 2006), our data do not address whether p53-dependent regulation of gene expression might represent an additional difference between these two cell types. However, our results support a p53-mediated transcriptional response during microglial activation.
The role of microglia in demyelination is well established (Carson, 2002; Kim and de Vellis, 2005; Sanders and De Keyser, 2007), and the studies in the cuprizone model of demyelination (Hiremath et al., 1998; Pasquini et al., 2007) have provided support to the concept that microglial activation might contribute to oligodendrocytic demise and neuronal damage (Pasquini et al., 2007). These premises have led to the therapeutic use of the tetracycline derivative minocycline, an antibiotic with the ability to inhibit the inflammatory response by modulation of the microglial component (Yrjanheikki et al., 1998, 1999; Yong et al., 2004). Whereas this approach has proven to be effective in limiting the lesion size in response to cuprizone (Pasquini et al., 2007) and in experimental autoimmune encephalomyelitis models (Popovic et al., 2002), it is also clear that it has negatively impacted remyelination and repair (Kotter et al., 2001; Li et al., 2005). The negative effect of minocycline treatment on remyelination could be attributed to several potential reasons. One possibility is that minocycline exerts a dose-dependent inhibition of oligodendrocyte progenitor proliferation, thereby reducing the pool of cells that can form new myelin (Li et al., 2005). An alternative possibility is that minocycline reduces the production of important trophic factors (i.e., BDNF) from microglial cells, thereby affecting oligodendrocyte progenitor cell proliferation and differentiation (Dougherty et al., 2000; Du et al., 2003). Therefore, the general consensus is that it is important to limit microglial activation, without necessarily abolishing it, to provide a source of trophic support for the repair mechanism. Our study, using genetic or pharmacological inhibition of p53, suggests that transient inhibition of this transcription factor might provide multiple beneficial effects in demyelinating disorders consequent to oligodendrogliopathy and massive microglial infiltration. Systemic administration with pifithrin-α, for instance, decreases the extent of myelin loss and microglial accumulation and selectively reduced the levels of cytotoxic microglial gene products (i.e., lysozyme) without affecting the expression of trophic genes (i.e., BDNF). An added beneficial effect of this treatment was the proliferative and differentiative effect on multipotential progenitors in the adult SVZ that resulted in an increased number of Sox2-positive cells and enhanced oligodendrogliogenesis at the expenses of astrogliogenesis. We have previously reported that cells cultured from the SVZ of p53−/− mice have the ability to generate more O4+ oligodendrocytes than cells isolated from p53+/+ siblings (Gil-Perotin et al., 2006). Although we cannot rule out cell-extrinsic mechanisms, our current study shows that treatment with pifithrin-α results in a remarkably similar phenotype.
In conclusion, we propose that pharmacological compounds with transient inhibitory activity on p53, should be further investigated as potential therapeutic agents for primary demyelination. The design of small peptides with the ability to inactivate transiently and possibly in a reversible manner p53 in a cell-specific manner is highly desirable to avoid the complications that sustained inactivation of p53 could have on lymphocyte apoptosis (Okuda et al., 2002) and tumorigenesis (Donehower et al., 1992; Donehower, 1996; Lozano, 2007).
Footnotes
This work was supported by National Institutes of Health (NIH)–National Insitute of Neurological Disorders and Stroke Grant RO1 NS052738, NIH–National Institute of Child Health and Human Development Grant PO1 HD06576 and HD04612 (J.D.V.), National Multiple Sclerosis Society (P.C.-B.), the Christopher and Dana Reeve Foundation, and by the Multiple Sclerosis Research Foundation (P.C.-B.).
References
- Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Bruck W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62:25–33. doi: 10.1093/jnen/62.1.25. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Ben-Hur T, Rogister B, Murray K, Rougon G, Dubois-Dalcq M. Growth and fate of PSA-NCAM+ precursors of the postnatal brain. J Neurosci. 1998;18:5777–5788. doi: 10.1523/JNEUROSCI.18-15-05777.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billon N, Terrinoni A, Jolicoeur C, McCarthy A, Richardson WD, Melino G, Raff M. Roles for p53 and p73 during oligodendrocyte development. Development. 2004;131:1211–1220. doi: 10.1242/dev.01035. [DOI] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Carson MJ. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia. 2002;40:218–231. doi: 10.1002/glia.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- Decker L, Avellana-Adalid V, Nait-Oumesmar B, Durbec P, Baron-Van Evercooren A. Oligodendrocyte precursor migration and differentiation: combined effects of PSA residues, growth factors, and substrates. Mol Cell Neurosci. 2000;16:422–439. doi: 10.1006/mcne.2000.0885. [DOI] [PubMed] [Google Scholar]
- Dijsselbloem N, Goriely S, Albarani V, Gerlo S, Francoz S, Marine JC, Goldman M, Haegeman G, Vanden Berghe W. A critical role for p53 in the control of NF-kappaB-dependent gene expression in TLR4-stimulated dendritic cells exposed to Genistein. J Immunol. 2007;178:5048–5057. doi: 10.4049/jimmunol.178.8.5048. [DOI] [PubMed] [Google Scholar]
- Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol. 1996;7:269–278. doi: 10.1006/scbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Dougherty KD, Dreyfus CF, Black IB. Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiol Dis. 2000;7:574–585. doi: 10.1006/nbdi.2000.0318. [DOI] [PubMed] [Google Scholar]
- Du Y, Fischer TZ, Lee LN, Lercher LD, Dreyfus CF. Regionally specific effects of BDNF on oligodendrocytes. Dev Neurosci. 2003;25:116–126. doi: 10.1159/000072261. [DOI] [PubMed] [Google Scholar]
- Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Moller T, Morrison RS. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004;18:1141–1143. doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, Garcia-Verdugo JM, Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107–1116. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiremath MM, Saito Y, Knapp GW, Ting JP, Suzuki K, Matsushima GK. Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol. 1998;92:38–49. doi: 10.1016/s0165-5728(98)00168-4. [DOI] [PubMed] [Google Scholar]
- Jurevics H, Largent C, Hostettler J, Sammond DW, Matsushima GK, Kleindienst A, Toews AD, Morell P. Alterations in metabolism and gene expression in brain regions during cuprizone-induced demyelination and remyelination. J Neurochem. 2002;82:126–136. doi: 10.1046/j.1471-4159.2002.00954.x. [DOI] [PubMed] [Google Scholar]
- Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, Nedospasov SA, Hazen SL, Feinstein E, Gudkov AV. p53 is a suppressor of inflammatory response in mice. FASEB J. 2005;19:1030–1032. doi: 10.1096/fj.04-3213fje. [DOI] [PubMed] [Google Scholar]
- Kondo T, Raff M. Chromatin remodeling and histone modification in the conversion of oligodendrocyte precursors to neural stem cells. Genes Dev. 2004;18:2963–2972. doi: 10.1101/gad.309404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJ. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia. 2001;35:204–212. doi: 10.1002/glia.1085. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Glas M, zum Bruch C, Mueller W, Weber A, Zipp F, Bruck W. Investigation of bax, bcl-2, bcl-x and p53 gene polymorphisms in multiple sclerosis. J Neuroimmunol. 2002;129:154–160. doi: 10.1016/s0165-5728(02)00167-4. [DOI] [PubMed] [Google Scholar]
- Ladiwala U, Li H, Antel JP, Nalbantoglu J. p53 induction by tumor necrosis factor-alpha and involvement of p53 in cell death of human oligodendrocytes. J Neurochem. 1999;73:605–611. doi: 10.1046/j.1471-4159.1999.0730605.x. [DOI] [PubMed] [Google Scholar]
- Li WW, Setzu A, Zhao C, Franklin RJ. Minocycline-mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non-immune model of demyelination. J Neuroimmunol. 2005;158:58–66. doi: 10.1016/j.jneuroim.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Lozano G. The oncogenic roles of p53 mutants in mouse models. Curr Opin Genet Dev. 2007;17:66–70. doi: 10.1016/j.gde.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Lu DY, Tang CH, Liou HC, Teng CM, Jeng KC, Kuo SC, Lee FY, Fu WM. YC-1 attenuates LPS-induced proinflammatory responses and activation of nuclear factor-kappaB in microglia. Br J Pharmacol. 2007;151:396–405. doi: 10.1038/sj.bjp.0707187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior B, Puntambekar SS, Carson MJ. Microglia and the control of autoreactive T cell responses. Neurochem Int. 2006;49:145–153. doi: 10.1016/j.neuint.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell P, Barrett CV, Mason JL, Toews AD, Hostettler JD, Knapp GW, Matsushima GK. Gene expression in brain during cuprizone-induced demyelination and remyelination. Mol Cell Neurosci. 1998;12:220–227. doi: 10.1006/mcne.1998.0715. [DOI] [PubMed] [Google Scholar]
- Murtie JC, Zhou YX, Le TQ, Vana AC, Armstrong RC. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis. 2005;19:171–182. doi: 10.1016/j.nbd.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Decker L, Lachapelle F, Avellana-Adalid V, Bachelin C, Van Evercooren AB. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur J Neurosci. 1999;11:4357–4366. doi: 10.1046/j.1460-9568.1999.00873.x. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Picard-Riera N, Kerninon C, Decker L, Seilhean D, Hoglinger GU, Hirsch EC, Reynolds R, Baron-Van Evercooren A. Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc Natl Acad Sci USA. 2007;104:4694–4699. doi: 10.1073/pnas.0606835104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda Y, Okuda M, Bernard CC. The suppression of T cell apoptosis influences the severity of disease during the chronic phase but not the recovery from the acute phase of experimental autoimmune encephalomyelitis in mice. J Neuroimmunol. 2002;131:115–125. doi: 10.1016/s0165-5728(02)00267-9. [DOI] [PubMed] [Google Scholar]
- Pasquini LA, Calatayud CA, Bertone Una AL, Millet V, Pasquini JM, Soto EF. The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem Res. 2007;32:279–292. doi: 10.1007/s11064-006-9165-0. [DOI] [PubMed] [Google Scholar]
- Picard-Riera N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B, Liblau R, Pham-Dinh D, Evercooren AB. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci USA. 2002;99:13211–13216. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic N, Schubart A, Goetz BD, Zhang SC, Linington C, Duncan ID. Inhibition of autoimmune encephalomyelitis by a tetracycline. Ann Neurol. 2002;51:215–223. doi: 10.1002/ana.10092. [DOI] [PubMed] [Google Scholar]
- Sanders P, De Keyser J. Janus faces of microglia in multiple sclerosis. Brain Res Rev. 2007;54:274–285. doi: 10.1016/j.brainresrev.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Stadelmann C, Ludwin S, Tabira T, Guseo A, Lucchinetti CF, Leel-Ossy L, Ordinario AT, Bruck W, Lassmann H. Tissue preconditioning may explain concentric lesions in Balo's type of multiple sclerosis. Brain. 2005;128:979–987. doi: 10.1093/brain/awh457. [DOI] [PubMed] [Google Scholar]
- Trebst C, Heine S, Lienenklaus S, Lindner M, Baumgärtner W, Weiss S, Stangel M. Lack of interferon-beta leads to accelerated remyelination in a toxic model of central nervous system demyelination. Acta Neuropathol. 2007;114:587–596. doi: 10.1007/s00401-007-0300-z. [DOI] [PubMed] [Google Scholar]
- Uberti D, Yavin E, Gil S, Ayasola KR, Goldfinger N, Rotter V. Hydrogen peroxide induces nuclear translocation of p53 and apoptosis in cells of oligodendroglia origin. Brain Res Mol Brain Res. 1999;65:167–175. doi: 10.1016/s0169-328x(98)00339-8. [DOI] [PubMed] [Google Scholar]
- Villapol S, Acarin L, Faiz M, Castellano B, Gonzalez B. Distinct spatial and temporal activation of caspase pathways in neurons and glial cells after excitotoxic damage to the immature rat brain. J Neurosci Res. 2007;85:3545–3556. doi: 10.1002/jnr.21450. [DOI] [PubMed] [Google Scholar]
- Wosik K, Antel J, Kuhlmann T, Bruck W, Massie B, Nalbantoglu J. Oligodendrocyte injury in multiple sclerosis: a role for p53. J Neurochem. 2003;85:635–644. doi: 10.1046/j.1471-4159.2003.01674.x. [DOI] [PubMed] [Google Scholar]
- Yang B, Prayson RA. Expression of Bax, Bcl-2, and P53 in progressive multifocal leukoencephalopathy. Mod Pathol. 2000;13:1115–1120. doi: 10.1038/modpathol.3880206. [DOI] [PubMed] [Google Scholar]
- Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3:744–751. doi: 10.1016/S1474-4422(04)00937-8. [DOI] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]