


Abstract
We report here a new directional cDNA library construction method using an in vitro site-specific recombination reaction, based on the integrase–excisionase system of bacteriophage λ. Preliminary experiments revealed that in vitro recombinational cloning (RC) provided important advantages over conventional ligation-assisted cloning: it eliminated restriction digestion for directional cloning, generated low levels of chimeric clones, reduced size bias and, in our hands, gave a higher cloning efficiency than conventional ligation reactions. In a cDNA cloning experiment using an in vitro synthesized long poly(A)+ RNA (7.8 kb), the RC gave a higher full-length cDNA clone content and about 10 times more transformants than conventional ligation-assisted cloning. Furthermore, characterization of rat brain cDNA clones yielded by the RC method showed that the frequency of cDNA clones >2 kb having internal NotI sites was ∼6%, whereas these cDNAs could not be cloned at all or could be isolated only in a truncated form by conventional methods. Taken together, these results indicate that the RC method makes it possible to prepare cDNA libraries better representing the entire population of cDNAs, without sacrificing the simplicity of current conventional ligation-assisted methods.
INTRODUCTION
Comprehensive cDNA analysis plays a critical role in complementing genomic information, and construction of high-quality cDNA libraries is essential for meaningful cDNA analysis. The information obtained from mammalian cDNA analysis, for example, is often indispensable in reliably predicting protein-coding sequence from genome sequence. Given the importance of comprehensive analysis of human cDNAs, for the past six years we have implemented a cDNA sequencing project that aims to accumulate predicted protein sequences encoded by cDNAs (1,2). The total number of nucleotide residues determined by our project exceeds 7 Mb to date (2). Our project is distinct from others in that we focus our sequencing efforts on large cDNA inserts (>4 kb) (3) and the results of the analysis of large cDNA clones are publicly accessible at http://www.kazusa.or.jp/huge (4). However, through this experience of cDNA analysis, we realized that many technical problems in cDNA library construction still remain, especially for analysis of large cDNAs. For instance, conventional cDNA cloning protocols typically include the use of a restriction enzyme cleavage step for directional cloning (5), and thus risk overlooking cDNA clones that happen to have an internal restriction site that is also cleaved by the same restriction enzyme. Because the frequency of large cDNAs being digested with a particular restriction enzyme should be higher than that of shorter cDNAs, this risk becomes greater as the cDNA size increases. Further, chimeric clones, which harbor intermolecularly ligated cDNA inserts, were found at the rate of ∼3% or more in human large cDNA clones and were almost impossible to identify by sequencing of cDNA ends only (insert sizes between 5 and 7 kb; 6), whereas they are a negligible component in a pool of small cDNA clones (7,8). Also a concern in analysis of large cDNAs is that conventional cDNA libraries under-represent large cDNA clones due to severe size bias in the ligation reaction, as well as during propagation of the cDNA plasmid population in Escherichia coli. Although these concerns may not be serious in conventional cDNA cloning where multiple clones are isolated for a single gene, they are highly problematic for large-scale sequencing of cDNAs in which clones are randomly sampled on a single-clone-for-single-gene basis.
We considered that nearly all of these cloning problems resulted from using ligation-assisted cloning (LC) to construct the cDNA library. Very recently, a new cloning method which exploits the λ-att recombination system of bacteriophage λ in E.coli has been reported (9,10). Briefly, the recombination-assisted cloning (RC) method transfers DNA segments into vectors using site-specific recombination, instead of restriction enzymes and ligase. In one application, PCR products that are flanked by attB1 and attB2 sequences are efficiently transferred by recombination into plasmid vectors containing attP1 and attP2 sequences. Recombination between attB and attP sequences (B×P) is highly specific (11), and mediated by recombination proteins Int and IHF. The product of the B×P reaction is a properly oriented subclone of the PCR product within the vector donated by the attP plasmid. The subcloned PCR product is now flanked by attL sequences, and in this form it can be efficiently transferred to other vectors, if desired, using the reverse reaction (L×R) of the λ-att recombination system. Additional details about the RC method can be found at http://www2.lifetech.com/content/world/gatewayman.pdf.
We thought that use of the same B×P recombination reaction could produce cDNA libraries (from double-stranded cDNA prepared with flanking attB sequences), and that this approach could solve many of the drawbacks associated with conventional cDNA library construction. In particular, RC of cDNA should eliminate the need for restriction enzyme treatment for directional cloning. It should also substantially lower the frequency of chimeric inserts, since terminal attB recombination sequences on the cDNA react only with their respective attP sequences, and not with each other (11).
In the present study, we first characterized the RC method for cDNA cloning with respect to chimerization, size bias and cloning efficiency. Then we applied RC to directional cDNA library construction and analyzed the resultant libraries. The results indicated that cDNA library construction based on RC significantly reduces the major drawbacks of conventional cDNA cloning methods. In addition, this approach provides a new, simple route to prepare cDNA clones that are readily transferred to other vectors, using RC, for further studies.
MATERIALS AND METHODS
Materials
The reagents required for the RC system were obtained from Life Technologies as a field test kit. Comparable reagents are now available commercially from Gibco BRL. Oligonucleotides purified on polyacrylamide gels were purchased from Amersham Pharmacia Biotech, Inc., and used as adapters and an adapter-primer for cDNA synthesis.
Donor (attP) plasmids carrying an ampicillin-resistance gene were constructed from pSP73 (Promega) and pSPORT-1 (Life Technologies). In brief, an attP cassette containing a ccdB gene and chloramphenicol-resistance gene was prepared from pDONR201 plasmid by PCR. The amplified region corresponds to the nucleotide sequence from 10 to 2643 in pDONR201 (accessible via the World Wide Web at http://www.lifetech.com/content/world/gatewayman.pdf), and flanked by two restriction sites included in the primers, HindIII (attP1 side) and SacI (attP2 side). The attP cassette (2.7 kb) was first subcloned into the HindIII–SacI site of pSP73. The attP cassette subcloned into pSP73 was excised by digestion with XhoI and BglII and then subcloned into the SalI–BamHI site of pSPORT-1. The resultant donor plasmids, which confer ampicillin resistance to E.coli cells, were named attP-pSP73 (5.1 kb) and attP-pSPORT-1 (6.8 kb), and were prepared from DB5 cells [DH10B: gyrA462 endA Δ(srl-recA)1398], provided in the field test kit from Life Technologies. Similarly, to create a plasmid for cloning with LC, an attR cassette containing the ccdB and chloramphenicol-resistance genes was prepared from the plasmid pDEST1 (supplied with the Life Technologies Field Test Kit) by PCR using 5′-CATCGCGAGGTACCAAGCTTTC-3′ and 5′-CCGCCAAAACAGCCGAGCTCTC-3′. Since the amplified attR cassette was also flanked by two artificially generated restriction sites for HindIII and SacI, it was subcloned into the HindIII–SacI site of pSP73, as in the case of the attP cassette described above. The resultant plasmid was called attR-pSP73 (4.8 kb).
Rat β-spectrin III (βSp3) poly(A)+ RNA (7.8 kb) used in a model cDNA cloning experiment was prepared from a NotI-linearized cDNA clone (12) by in vitro RNA synthesis (MEGAscriptTM, Ambion). Rat brain poly(A)+ RNA was obtained from Clontech or prepared from total rat brain RNA using a µMACS mRNA isolation kit (Miltenyi Biotech).
Examination of size bias and chimerization by LC and RC
A part of the multiple cloning site in pBluescript II SK (Stratagene) was amplified by PCR using the following two primers: 5′-TGGGTACCACAAGTTTGTACAAAAAAGCAGGCTCGACGGTATCGAT-3′ and 5′-CTGGAGCTCACCACTTTGTACAAGAAAGCTGGGTGGCGGCCGCTC-3′, where the attB1 and attB2 sequences of the forward and reverse primers, respectively, are underlined. After digestion with KpnI and SacI, the resultant PCR product was subcloned into the KpnI–SacI site of pBluescript II SK. The resultant plasmid, attB-pBluescript, was used as a template for preparation of a DNA fragment containing multiple cloning sites (MCS). MCS (128 bp) was obtained by PCR using attB-pBluescript and two primers (5′-GGGGACAAGTTTGTACAAAAAAGCAGGC-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGG-3′). DNA fragments containing the tetracycline-resistance gene (Tet) and beta-galactosidase gene (βGal) were amplified from control plasmids included in the field test kit provided by Life Technologies using the same set of PCR primers. MCS, Tet and βGal thus obtained were digested with BsrGI (New England BioLabs), which cleaves at the TGTACA sequence within the common 15 bp core region of each att sequence, leaving four-base 5′ overhangs. The products with or without BsrGI digestion were finally purified using the Concert Rapid PCR Purification System (Life Technologies) and then used for LC or RC, respectively. To prepare a vector for LC, attR-pSP73 plasmid was digested with BsrGI and dephosphorylated with bacterial alkaline phosphatase. The linearized vector portion of attR-pSP73 was purified on an agarose gel, recovered using CONCERT Gel Extraction System (Life Technologies) and quantified by absorption at 260 nm. The quantities of MCS, Tet and βGal DNA were estimated from fluorescent band intensities on an agarose gel with a FluoroImager 595 (Molecular Dynamics). Ligation reactions were carried out in 20 µl containing 1× T4 DNA ligase buffer (Life Technologies), 45 ng of dephosphorylated, BsrGI-digested vector portion of attR-pSP73, 90 ng of BsrGI-digested βGal, 45 ng of BsrGI-digested Tet and 5 ng of BsrGI-digested MCS with T4 DNA ligase (1 U) at 25°C for 3 h. Recombination reactions were performed in 20 µl, containing 500 ng of attP-pSP73, 90 ng of βGal, 45 ng of Tet and 5 ng of MCS, at 25°C for 3 h as instructed in the field test kit, and then quenched by the incubation of proteinase K (0.2 µg/µl) at 37°C for 10 min. The reaction products were purified by phenol extraction followed by ethanol precipitation and introduced into E.coli cells by electroporation (ElectroMAX DH10BTM cells, Life Technologies). The transformation efficiency of ElectroMAX DH10B cells was 1.0–1.5 × 1010 c.f.u./µg of pUC19. Electroporated E.coli cells were plated on LB agar plates containing ampicillin (100 µg/ml). Plasmids were recovered from transformants (>105) on ampicillin-containing agar plates after growth in 100 ml of 2× YT medium for 6 h at 37°C. Plasmid preparation was carried out using a Concert High purity Plasmid Maxiprep System (Life Technologies). The obtained plasmids were digested with BsrGI, run on an agarose gel and stained with SYBR-Green I fluorescent dye (Molecular Probes). The fluorescent gel image was scanned with a FluoroImager 599 and analyzed with ImageQuant software (Amersham Pharmacia Biotech). For the chimerization test, transformants on ampicillin-containing plates (∼104) were cultured in 100 ml 2× YT medium in the presence of ampicillin (100 µg/ml) and tetracycline (25 µg/ml) at 37°C for 6 h. After the culture, plasmids were prepared and analyzed as described above.
Cloning of β-spectrin III cDNA by RC and LC
cDNA synthesis from in vitro synthesized β-spectrin III (βSp3) mRNA was carried out according to the supplier’s instructions in the SuperScriptTM Plasmid System for cDNA synthesis (Life Technologies). The first-strand synthesis was primed with an oligonucleotide containing dT20, the attB2 site (underlined) and a NotI-recognition site [attB2 dT primer, 5′-GCGAAGCCCACCACTTTGTACAAGAAAGCTGGGCGGCCGC(T)20-3′]. The other adapters included an attB1 site (underlined) and a protruding 5′-end for annealing to the SalI-digested end of the cloning vector (upper strand of attB1 adapter: 5′-TCGACGCGTACAAGTTTGTACAAAAAAGCAGGCTCTTC-3′; lower strand of attB1 adapter: 5′-pGAAGAGCCTGCTTTTTTGTACAAACTTGTACGCG-3′). After adapter ligation, the cDNAs were separated from unligated attB1 adapters by precipitation with polyethylene glycol (PEG), as follows. After precipitation with ethanol, the reaction products were dissolved in 50 µl of 10 mM Tris–HCl pH 8.0 and 1 mM EDTA. After addition of 30 µl PEG solution (20% PEG6000 and 2.5 M NaCl), the reaction products were incubated on ice for 15 min and then collected by centrifugation at 18 000 g for 10 min. The adapter-ligated cDNAs were further size-fractionated by gel electrophoresis using low melting temperature agarose (13). The recovered βSp3 cDNAs (>4 kb) were subjected to the recombination reaction with BP Clonase (Int + IHF, Life Technologies) at room temperature overnight. In the recombination reactions (20 µl), ∼20 ng cDNA and 570 ng attP-pSPORT-1 were included. After the recombination reaction, the reaction was quenched by incubation with proteinase K (0.2 µg/µl) at 37°C for 10 min. As a control reaction, βSp3 cDNA ligated with phosphorylated attB1 adapter was digested with NotI just before agarose gel electrophoresis, the recovered cDNAs >4 kb from an agarose gel (∼20 ng) were subjected to LC with NotI–SalI-digested pSPORT-1 vector (50 ng) and T4 DNA ligase (1 U) in 20 µl at 16°C for 16 h. Both of the reaction products were extracted with phenol, precipitated once with ethanol and then introduced into E.coli cells by electroporation (ElectroMAX DH10BTM cells, Life Technologies).
To characterize the resultant cDNA clones, the recovered plasmids were digested with NotI alone or NotI plus EcoRI, run on an agarose gel and stained with SYBR-Green I (Molecular Probes). After the gel image was captured with FluoroImager 599 (Amersham Pharmacia Biotech), the digested plasmids were subjected to hybridization analysis by the method of Church and Gilbert (14) using a 32P-labeled oligonucleotide probe. An oligonucleotide probe specific to the 3′ extreme end of βSp3 cDNA (5′-GATTAAACAGCTCGGCTTCTC-3′) was 5′-labeled with 32P-ATP and T4 polynucleotide kinase. After hybridization at 45°C overnight, the membrane on which the digested plasmids were immobilized was extensively washed with 2× SSC at 42°C. The radioactive hybridization signals remaining on the membrane were quantitatively detected along the lanes with a BAS-2000 image analyzer (Fuji Film).
Characterization of rat brain cDNA clones generated by RC
After size fractionation on agarose gel to remove cDNAs <1 kb, rat brain cDNAs were used for cDNA library construction by RC, essentially as described for βSp3 cDNA. attP-pSP73 was used as a donor plasmid in this experiment. Rat brain cDNA clones obtained by RC (∼106 colonies) were grown on agar plates containing ampicillin (50 µg/ml) and the plasmids were recovered from these colonies. The plasmids were then size-fractionated on agarose gels and those with insert sizes between 3.5 and 4.5 kb were retrieved from the gels. The recovered plasmids were then reintroduced into E.coli cells by electroporation, and transformants were randomly picked and inoculated in 0.8 ml of 2× YT medium. After growth at 37°C overnight with vigorous shaking, plasmids were prepared from 192 transformants using 96-well plates (as described in Tech Note Lit. No., TN004, Millipore). The resultant plasmids were digested with NotI and run on agarose gels. The gel images were captured and analyzed on a FluoroImager 595.
For a direct comparison of LC- and RC-assisted cDNA library construction methods, rat brain cDNA libraries were prepared by LC and RC from the same preparation of double-stranded cDNAs generated using an attB2 dT primer. The cDNAs subjected to LC were ligated with a SalI adapter provided in the SuperScriptTM Plasmid System for cDNA synthesis (Life Technologies) while the cDNAs for RC were ligated with an attB1 adapter as described above. The rat brain cDNAs used for LC were digested with NotI and then size-fractionated on an agarose gel to remove cDNAs <1 kb, while those used for RC were directly size-fractionated in the same way. The amounts of the recovered cDNAs were estimated from fluorescent intensity of DNA bands on agarose gels. LC and RC were done using ∼20 ng of rat brain cDNAs in 20 µl reaction mixture as described for βSp3 cDNA cloning. To characterize the resultant cDNA clones, a mixture of plasmids was recovered from more than 9 × 104 colonies in each cDNA library. The recovered plasmids were digested with NotI, run on an agarose gel and stained with SYBR-Green I (Molecular Probes). Gel images were captured with a FluoroImager 599 and analyzed with ImageQuant software.
Sequencing of clones generated by RC
βSp3 cDNA clones and rat brain cDNA clones were sequenced with attL1 sequencing primer (5′-CTGAAGCTTGGATCTCGGGC-3′) and attL2 sequencing primer (5′-GCCAGAGCTGCAGCTGGATG-3′) using a BigDye terminator cycle sequencing kit (v1.0, Applied Biosystems) with an ABI373 or ABI377 DNA sequencer (Applied Biosystems). The quality of the sequencing data was enough for characterization of cDNA clones, but not high. This is most likely due to the fact that cDNAs are flanked by highly homologous 100 base attL sites, which is known as the ‘PCR suppression effect’ (15).
RESULTS
Size bias and chimerization in the RC system
To compare size dependence of cloning efficiency and chimerization rate between LC and RC, we carried out a preliminary cloning experiment using three different DNA fragments (0.13, 1.4 and 3.0 kb) generated by PCR: the 0.13 kb fragment was prepared from a MCS of pBluescript vector; the 1.4 kb fragment contained a tetracycline-resistance gene (Tet); the 3.0 kb fragment encoded beta-galactosidase (βGal). Each fragment was prepared with terminal attB sequences, as substrates for RC. Each attB sequence also comprised a recognition site for the restriction enzyme BsrGI, which leaves four-base cohesive termini suitable for cloning by LC. Vectors of similar size and derivation were used for the cloning, with the main difference being that the LC vector was digested first with BsrGI. Equimolar amounts of the three DNA fragments were mixed with the corresponding vector in a single tube and subjected to either the RC or the LC reaction. When the reaction products were introduced into E.coli cells by electroporation using ElectroMax DH10B cells (∼1010 c.f.u./µg of pUC19), LC and RC yielded 3.0 × 108 and 1.4 × 109 c.f.u./µg of insert DNA, respectively. In several similar experiments, the absolute cloning efficiencies varied depending on transformation efficiency of competent cells used, but RC was always more efficient than LC. These results indicate that RC is nearly five times more efficient than LC under these cloning conditions. Since these cloning efficiencies are higher (by at least three orders of magnitude) than the background transformation efficiencies of LC and RC, virtually all the clones harbored inserts. Both cloning reactions contained the same molar amounts of DNA fragment, so the size distribution of cloned inserts should reflect the respective cloning size bias. The recovered plasmids from >105 colonies were linearized by digestion with BglII and then run on an agarose gel (Fig. 1A). From fluorescent signal intensities of DNA bands, we estimated the relative molar contents of plasmids carrying MCS, Tet and βGal to be 100:11:0.5 for RC and 100:4:<0.1 for LC. Thus, although both methods produced size bias, these results demonstrate that RC caused considerably less size bias than LC. If we take the difference in cloning efficiency between LC and RC into account, the yield of clones carrying large DNA inserts produced by RC is clearly better than that by LC.
Figure 1.
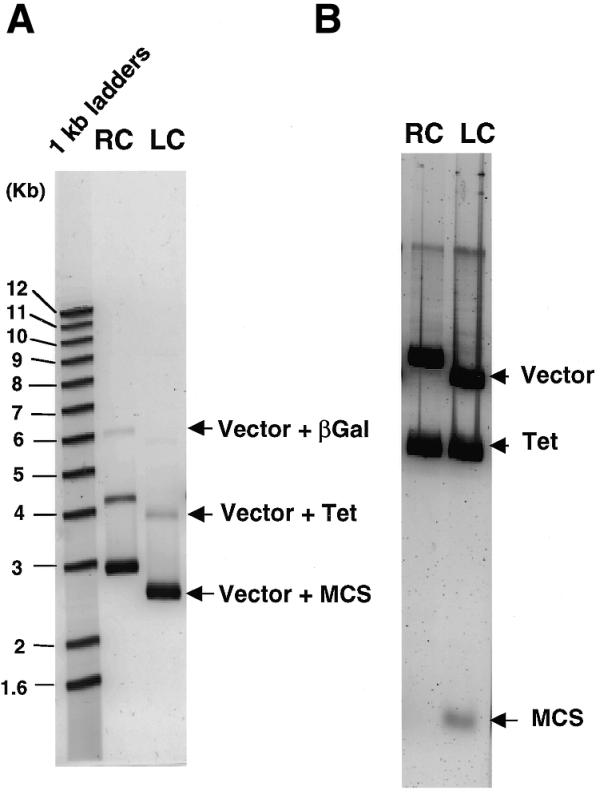
Comparison of cloned plasmids produced from an equimolar mixture of three DNA fragments of different sizes by RC and LC. Plasmids retrieved from transformants generated by RC and LC were digested with BglII (A) or BsrGI (B), and then run on 0.7 or 1.2% agarose gels, respectively. Plasmids were linearized by BglII digestion, and inserts were excised from the vector (pSP73) by BsrGI digestion. Because the resultant clones by RC contained recombination site sequences in the vector portion, the size of the RC plasmid became slightly larger than that of the LC plasmid even when it carried the same insert. In (B), contaminating E.coli chromosomal DNA appeared as the uppermost band in both lanes and the excised βGal fragment, which should run slightly slower than the vectors, was not seen in either lane.
In the next experiment, we examined occurrence of chimeric (i.e., intermolecularly ligated) clones. Ampicillin-resistant clones (∼104 colonies) obtained by LC or RC were cultured in liquid medium containing ampicillin and tetracycline, and the plasmids were then recovered from the E.coli cells. If no intermolecularly ligated molecules are cloned, the resultant plasmids should harbor only Tet since neither MCS nor βGal conferred tetracycline resistance on E.coli cells. The gel images of the recovered plasmids after digestion with BsrGI reveal that this was the case for RC, whereas the plasmids recovered from clones yielded by LC obviously contain intermolecularly ligated products, where MCS is fused with Tet through the BsrGI site (Fig. 1B). These results demonstrate that RC produces virtually no chimeric clones, as expected.
cDNA library construction using an in vitro synthesized RNA by the RC system
For directional cDNA cloning by RC, slight modifications to conventional cDNA library construction methods are required. Since cDNA library construction based on the method of Gubler and Hoffman (5) is widely used, we modified this method to replace the ligation step with RC for insertion of cDNAs into a vector. Figure 2 shows the overall scheme of the RC-assisted cDNA library construction method. In this scheme, we used an attB1 adapter with 5′-end phosphorylated lower strand, as in the case of conventional ligation-assisted cloning. Although ligation of phosphorylated attB1 adapter was anticipated to yield a considerable number of products flanked by two attB1 adapters, as shown in the box in Figure 2, only the attB1 adapter at the 5′-end should serve as a productive recombination site for cDNA cloning. This was confirmed by the actual cloning experiments described below.
Figure 2.
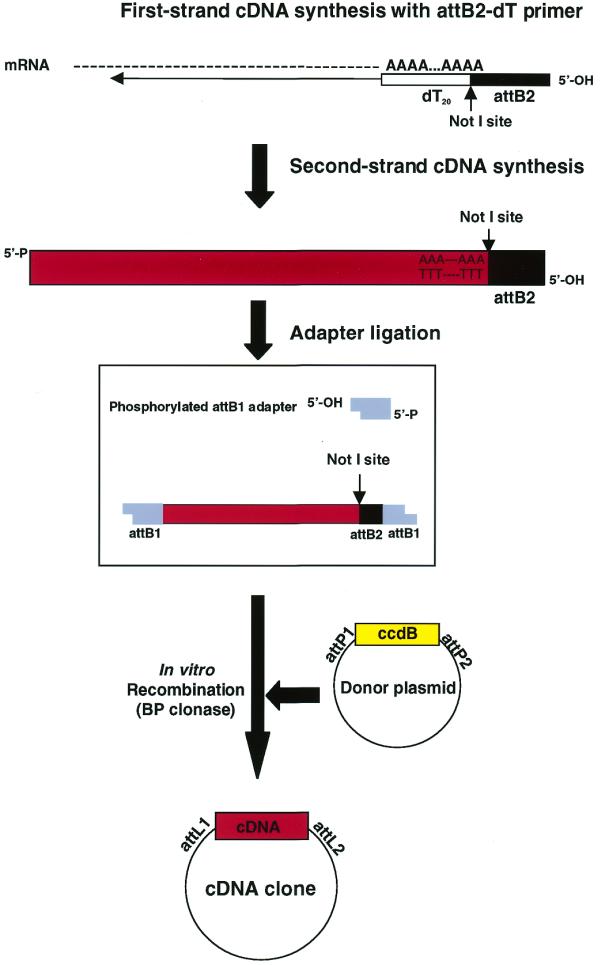
A scheme of cDNA library construction based on RC. Steps involved in RC-based cDNA library construction are schematically shown and were almost identical to those described in the instructions of SuperScript Plasmid System (Life Technologies) except for the use of the attB2-dT adapter primer, attB1 adapter and inclusion of an in vitro recombination reaction. Oligonucleotide sequences used for the attB2-dT primer adapter, upper and lower strands of attB1 adapter are as follows: attB2-dT primer, 5′-GCGAAGCCCACCACTTTGTACAAGAAAGCTGGGCGGCCGC(T)20-3′; upper and lower strands of attB1 adapter, 5′-TCGACGCGTACAAGTTTGTACAAAAAAGCAGGCTCTTC-3′ and 5′-GAAGAGCCTGCTTTTTTGTACAAACTTGTACGCG-3′, where the attB1 and attB2 sequences are underlined. An expected reaction product using phosphorylated attB1 adapter is illustrated. In the phosphorylated attB1 adapter, the 5′-end of only the lower strand of the attB1 adapter was chemically phosphorylated.
In the first experiment, we applied the overall scheme depicted in Figure 2 for cloning of long mRNA to compare the cloning efficiency and the size bias of RC with those of LC. In this case, long poly(A)-tailed RNA (7.8 kb) was synthesized from rat βSp3 cDNA (12) and its integrity was confirmed on a formaldehyde-denaturing agarose gel (data not shown). Double-stranded cDNAs were prepared using the in vitro synthesized RNA as a template and then separate cloning reactions were performed using two different βSp3 cDNA preparations to compare the resultant cDNA libraries: double-stranded cDNAs ligated with phosphorylated attB1 adapter with and without NotI digestion. Before cloning, both of the reaction products were size-fractionated on an agarose gel to enrich for cDNAs >4 kb. The cDNA preparation without NotI digestion was subjected to RC, while the cDNA with NotI digestion was used for conventional LC cDNA library construction, by ligation into NotI–SalI-digested pSPORT-1. When the reaction products of RC and LC were introduced into E.coli cells by electroporation, the cloning efficiencies by RC and LC were 5.0 × 108 c.f.u./µg and 3.6 × 107 c.f.u./µg of the cDNA insert, respectively.
To analyze the size distribution of the mixture of βSp3 cDNA inserts thus obtained, we prepared cDNA plasmids from >2 × 104 colonies of these two libraries. These plasmid mixtures were digested with restriction enzymes that do not cut internally in βSp3: NotI to linearize the plasmids, or NotI plus EcoRI to release the inserts. A NotI site is located between the 3′-end of cDNA and attL2 site, and two EcoRI sites are present, one 145 bp upstream of the 5′-end of cDNA (in the multiple cloning site of pSPROT-1) and the other 330 bp downstream of the 3′-end of cDNA. Figure 3 displays the staining patterns and the βSp3-specific hybridization patterns of the cDNA samples digested with NotI (Fig. 3A) and NotI plus EcoRI (Fig. 3B). Although all these plasmids were derivatives of pSPORT-1 vector, the vector portion of RC clones is ∼450 bp larger than that of LC clones due to the presence of attL sites and their flanking sequences. In addition, because attL1 sites are present in the excised inserts from RC clones, actual cDNA insert sizes of RC clones are ∼100 bp smaller than those estimated from migration rates of NotI–EcoRI fragments on an agarose gel. For RC clones, a small fragment carrying the attL2 site (∼330 bp) was also excised from the vector, which is seen as a faint band in the DNA staining pattern of the RC sample in Figure 3B. Taking these differences in plasmid structure into account, we carefully examined the staining and the hybridization patterns of RC and LC clones. It is notable that the clones yielded by LC and RC are clearly different: cloning of βSp3 cDNA (>4 kb) by LC generated considerable amounts of spurious clones, because plasmids smaller than pSPORT-1 vector (which would migrate at the position indicated by the star in Fig. 3A and B) and without hybridization signals are apparent in Figure 3A (indicated by a brace). In contrast, the lanes of RC products show little or no evidence of such spurious clones. In Figure 3A and B, βSp3 cDNA plasmids yielded by LC and RC were loaded on the gel so as to give approximately the same signal intensities of full-length cDNA (7.8 kb). Thus, the stronger bands of the LC sample in Figure 3A and B indicate that the full-length content of LC clones was lower than that of RC clones. The size distribution of cloned βSp3 cDNA was quantitatively examined by monitoring the radioactive hybridization signals along the lanes in (B) (Fig. 3C). From the quantitation of the hybridization signals, the contents of the full-length βSp3 cDNA were estimated to be 7% in the RC system and 4% in the LC system, while the insert DNA before cloning contained 13% of the full-length βSp3 cDNA. These results confirm that RC caused less size bias than LC in actual cDNA cloning. Nine clones randomly sampled from the RC library were sequenced from both the attL sites, and all were found to carry inserts in the expected orientation (data not shown).
Figure 3.
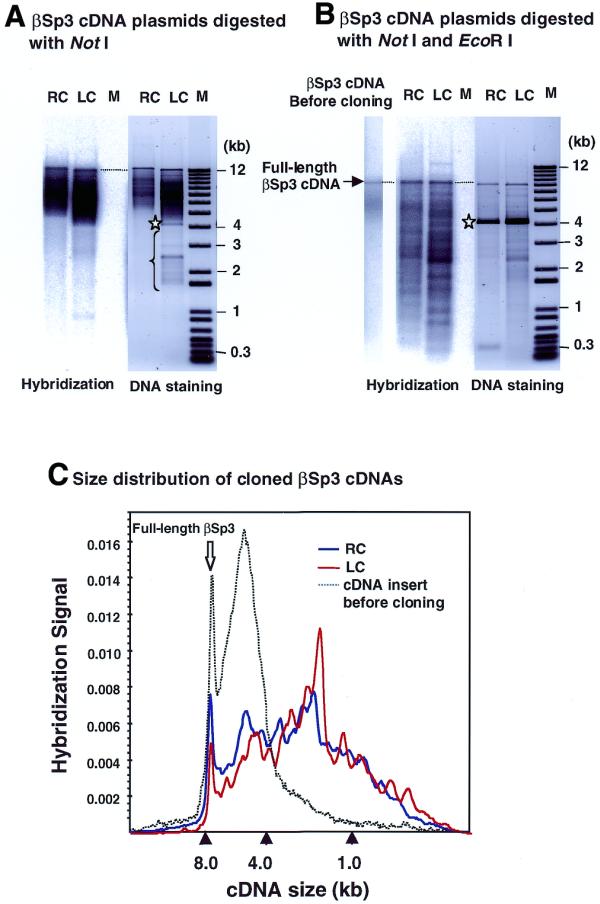
Comparison of size bias effects imposed on cloning of a 7.8 kb βSp3 cDNA by LC and RC. RC and LC shown above lanes in (A) and (B) indicate recovered cDNA plasmids produced by RC and by LC, respectively. DNAs were separated on a 0.7% agarose gel, stained with SYBR-Green I, transferred onto a nylon membrane and hybridized with a 32P-labeled oligonucleotide probe specific to the 3′-end sequence of βSp3. (A) and (B) show the DNA staining patterns and the hybridization pattern of βSp3 cDNA plasmids digested with NotI and NotI plus EcoRI, respectively. In (B) the hybridization pattern of the gel-recovered βSp3 cDNA before cloning is also shown. Size marker DNAs (1 kb Plus DNA Ladder, Life Technologies) were run in lane M. (C) Distribution of hybridization signals along each lane of the gel-recovered βSp3 cDNA and cDNA plasmids in (B). Stars in (A) and (B) indicate the position of linearized pSPORT-1.
Construction and characterization of rat brain cDNA library prepared by the RC system
The results described above show that RC offers several advantages over conventional LC, particularly for large cDNAs. In the next step, we applied RC to the construction of an actual cDNA library, prepared from rat brain poly(A)+ RNA, and analyzed the resultant cDNA clones. Double-stranded rat brain cDNAs after attB1 adapter ligation were size-fractionated on an agarose gel and the cDNAs >1 kb were subjected to RC. The average cloning efficiency of rat brain cDNAs (>1 kb) was 6.0 × 108 c.f.u./µg of cDNA insert in three independent experiments (ranging from 2.3 × 108 c.f.u./µg to 8.6 × 108 c.f.u./µg). The transformation with attP pSPORT-1 plasmid alone gave 104 c.f.u./µg of vector. When we characterized ten randomly sampled cDNA clones by DNA sequencing from both attL sites, all the clones carried cDNA inserts flanked by the recombination sites in the expected orientation (i.e., attL1 and attL2 sites were located at the 5′- and 3′-ends, respectively). Although our preliminary experiments had suggested that phosphorylated attB1 adapters were ligated with most cDNAs at both ends before the recombination reaction (data not shown), these results thus indicate that the BxP reaction can use DNAs bearing two attB1 sites and one attB2 site as substrates to faithfully generate expected recombination products.
To compare the RC method to the conventional LC method in actual cDNA cloning, we prepared two cDNA libraries, one by the RC- and the other by the LC-assisted method, using the same lot of rat brain cDNA in each case. For LC, the cDNA was digested first with NotI then size fractionated on an agarose gel to remove cDNAs smaller than 1 kb, prior to ligation with vector; for RC, the cDNA was directly size fractionated, prior to use in the BxP recombination reaction. Once again, the cloning efficiency obtained with the RC method was ∼6 × 108 c.f.u./µg of cDNA insert, whereas the cloning efficiency by LC with the same cDNAs after NotI digestion was 1.5 × 108 c.f.u./µg. In this LC experiment, we used the SalI adapter provided in the kit in place of the attB1 adapter as described in Materials and Methods because the SalI adapter gave a higher LC efficiency than the attB1 adapter.
We next compared the size distribution of cDNA inserts cloned by RC and LC. For this, cDNA plasmids were digested with NotI, run on an agarose gel and the fluorescence intensity of the linearized plasmid was monitored along each lane. In this comparison, we adjusted the calculated insert sizes to account for the 500 bp larger size of the RC vector. Overall, a similar distribution of insert sizes was obtained with RC and LC. Because most of rat brain cDNAs were between 1 and 2 kb, the difference in size bias between RC and LC was difficult to see clearly using these small cDNAs. In recent experiments (O.Ohara and T.Watanabe, unpublished), we have found that a lower size bias of RC is more apparent when cloning cDNA populations >3 kb, consistent with the results of our model experiment cloning 7.8 kb rat βSp3 cDNA by LC and RC, described above.
Conventional LC for directional cDNA library construction, in contrast to RC, includes restriction digestion of cDNAs with a rare cutter, like NotI, prior to cloning. We sought to demonstrate that cDNAs containing internal NotI sites could be cloned with RC (Table 1). For this analysis, rat brain supercoiled cDNA was fractionated on an agarose gel to enrich for clones harboring inserts of 3–4 kb, because frequencies of clones having internal NotI sites should be higher in large cDNAs than in small cDNAs. Then the size-fractionated DNA was introduced into E.coli, and plasmids were prepared from 192 randomly picked colonies. According to the cDNA construction method used in this study, all the clones should have one artificially generated NotI site at the 3′-end of the cDNA insert. NotI digestion is thus expected to yield single bands of linear cDNA plasmids, unless additional NotI sites are present internally. Therefore the appearance of multiple bands after NotI digestion on agarose gel electrophoresis indicates the presence of internal NotI sites in a cDNA insert.
Table 1. Frequency of occurrence of internal NotI sites in rat brain cDNA clones obtained by RC.
| cDNA Insert size (bp)a | Number of NotI sites per cDNA | cDNA Insert size (bp)a | Number of NotI sites per cDNA | |
| |
1 |
2 |
0 |
Total |
| 500–1000 | 15(2)b | 1 | 0 | 16 |
| 1000–2000 | 76 | 0 | 1 | 77 |
| 2000–3000 | 19 | 2 | 2 | 23 |
| 3000–4000 | 57 | 4(2)c | 0 | 61 |
| 4000–5000 | 13 | 1 | 0 | 14 |
| 5000–6000 | 0 | 1 | 0 | 1 |
| Total | 180 | 9 | 3d | 192 |
aBecause plasmids harboring 3–4 kb cDNA inserts in a supercoiled form co-migrated with those harboring 1–2 kb cDNAs in an open circular form on agarose gel electrophoresis, the size distribution of analyzed cDNA inserts had two peaks, at 1–2 and 3–4 kb, as shown.
bThe number of plasmids found as dimers is shown in parentheses.
cThe number of chimeric clones is shown in parentheses.
dOne plasmid carried a mutation at the position corresponding to a NotI site while two other plasmids were accidentally recovered attP donor plasmids.
Nine of the 192 cDNA clones examined (4.7%) contain an internal NotI site, and eight of the 100 clones (8%) with inserts >2 kb have internal NotI sites (Table 1). DNA sequencing revealed that all nine clones were independent. Two of these clones contain chimeric inserts, which are readily discerned by the presence of an additional attB2-oligo-dT-adapter-primer sequence (harboring the second NotI site) immediately following the expected attL1 sequence. The arrangement of the recombination sites in these chimeras suggests that they arose by 5′-end–5′-end ligation of cDNA-attB2-oligo-dT molecules during ligation of the attB1 adapters, following second strand synthesis (data not shown). Three of the 192 clones have no NotI site. Two of these are unreacted attP-pSP73 donor plasmids, and the remaining clone contains a mutation within the NotI site of the attB2 dT adapter-primer. Taken together, these data (i) confirm RC cloning of cDNAs containing internal NotI sites and (ii) verify a low incidence of chimeric inserts (1%) and empty vector (1%). Finally, we classify two of the 192 clones as plasmid dimers, because NotI digestion halved their sizes, yielding single bands on agarose gels, and the sizes of the inserts excised from the vector were consistent with the sizes estimated in a NotI-digested form rather than those in a covalently closed circular form (data not shown). Overall, the incidence of internal NotI sites is six out of the 97 clones with inserts >2 kb. Additional results demonstrate that the molar content of cDNA >4 kb in a rat brain double-strand cDNA pool was reduced by NotI digestion from 16 to 10% (O.Ohara and T.Watanabe, unpublished).
DISCUSSION
Prior descriptions of RC have emphasized its utility for rapid and efficient cloning of PCR products, together with their parallel subcloning into multiple types of expression vectors (9,10). In the present study, we evaluated several potential advantages for directional cDNA cloning that RC should provide over conventional methods using LC. These include directional cloning without use of restriction enzymes and lower levels of chimeric inserts. These advantages derive from the different cutting and joining mechanisms employed by these two approaches.
The relatively long (∼25 bp) recombination sequences used for RC should occur by chance very rarely, if at all, in any population of cDNA. Consequently, internal cleavage of cDNA insert sequences by the RC reaction should be negligible. In contrast, restriction enzyme cleavage sites, even for rare cutting enzymes, exist much more commonly in a large population of cDNA molecules. Internal cleavage of cDNAs becomes a more serious concern in the cloning and analysis of large cDNAs, since the occurrence of internal rare restriction sites in cDNA increases with increasing cDNA size. Consistent with this, our results (Table 1) demonstrate NotI cleavage sites in ∼6% (6 out of 97) randomly sampled cDNA clones with inserts ≥2 kb, as expected if one NotI site occurs roughly every 48 bp. Although other LC-based cDNA library construction methods may avoid this problem (for example see 16), the simplicity, robustness and high cloning efficiency of the Gubler–Hoffman protocol continue to make this a popular method (5). Our results indicate that RC-based cDNA library construction resolves this problem without sacrificing any of the advantages of conventional cDNA cloning methods.
Another important difference between RC and LC for preparation of cDNA libraries is the level and the structure of chimeric cDNA inserts. The attB recombination sites on cDNA molecules used in RC can recombine only with respective attP sites on cloning vectors, but not with attB sites on other cDNA molecules as demonstrated in a model experiment described in this study. In contrast, the cDNA inserts employed in LC can react not only with the ends of vectors, but with other cDNA molecules as well. Thus LC inevitably promotes some level of random concatenation of both insert cDNAs and vector, leading to significant levels of chimeric inserts. Although RC does not include any step expected to generate chimeric inserts, we observed chimeric clones in rat brain cDNA library constructed by RC-assisted method at a rate of ∼1%. The structure of these chimeric clones suggests that they were generated as by-products of attB1 adapter ligation, not by the B×P reaction. By increasing the amount of attB1 adapter and/or dephosphorylating the cDNA-attB2 molecules prior to ligation to the attB1 adapter, it should prove possible to reduce the frequency of chimeric clones even further. An important point is that chimeric clones arising in cDNA libraries constructed by conventional LC approaches are difficult to detect by end sequencing alone. In contrast, our ability to easily identify these chimeric clones in the RC-assisted library only by end-sequencing of cDNAs is a significant benefit in using this library for comprehensive cDNA analysis.
RC also addresses the difficulties inherent with the subsequent transfer of large cDNAs from one vector to another, such as for functional analysis. Large cDNAs frequently amplify inefficiently by PCR, and finding appropriate restriction sites with which to excise large cDNA inserts from a vector can be problematic. But when cDNAs are introduced into vectors using RC, their subsequent transfer to other vectors by recombination is fast and efficient, making it possible to subclone large cDNAs as easily as small cDNAs into a variety of vectors (9,10). Therefore, RC is a method highly suitable for both the cloning and analysis of large cDNAs, which have remained elusive targets in conventional cDNA analysis.
In previous studies, open reading frames were introduced into the RC system either by PCR with primers bearing attB (25 bp) recombination sites or by LC into a vector with attL recombination sites (9,10). The present method therefore provides an efficient new route to introduce cDNAs into the RC system, which avoids the need for restriction enzyme digestion and minimizes the incidence of chimeric clones. To date, we have successfully constructed several different cDNA libraries by this method, and used these for comprehensive cDNA analysis.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Chris Gruber, PhD for providing numerous helpful technical suggestions and for making available to us the results of his experiments using a related approach to cDNA cloning based on the RC method. We also thank Mr Takashi Watanabe for his excellent technical assistance, and Jim Hartley, PhD for his insightful suggestions on the manuscript. This study was supported by a grant from the Kazusa DNA Research Institute.
References
- 1.Nomura N., Miyajima,N., Sazuka,T., Tanaka,A., Kawarabayasi,Y., Sato,S., Nagase,T., Seki,N., Ishikawa,K.-I. and Tabata,S. (1994) Prediction of the coding sequences of unidentified human genes. I. The coding sequences of 40 new genes (KIAA0001-KIAA0040) deduced by analysis of randomly sampled cDNA clones from human immature myeloid cell line KG-1. DNA Res., 1, 27–35. [DOI] [PubMed] [Google Scholar]
- 2.Nagase T., Kikuno,R., Ishikawa,K.-I., Hirosawa,M. and Ohara,O. (2000) Prediction of the coding sequences of unidentified human genes. XVI. The complete sequences of 150 new cDNA clones from brain which code for large proteins in vitro. DNA Res., 7, 65–73. [DOI] [PubMed] [Google Scholar]
- 3.Ohara O., Nagase,T., Ishikawa,K.-I., Nakajima,D., Ohira,M., Seki,N. and Nomura,N. (1997) Construction and characterization of human brain cDNA libraries suitable for analysis of cDNA clones encoding relatively large proteins. DNA Res., 4, 53–59. [DOI] [PubMed] [Google Scholar]
- 4.Kikuno R., Nagase,T., Suyama,M., Waki,M., Hirosawa,M. and Ohara,O. (2000) HUGE: a database for human large proteins identified in the Kazusa cDNA sequencing project. Nucleic Acids Res., 28, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gubler U. and Hoffman,B.J. (1983) A simple and very efficient method for generating cDNA libraries. Gene, 25, 263–269. [DOI] [PubMed] [Google Scholar]
- 6.Seki N., Ohira,M., Nagase,T., Ishikawa,K.-I., Miyajima,N., Nakajima,D., Nomura,N. and Ohara,O. (1997) Characterization of cDNA clones in size-fractionated cDNA libaries from human brain. DNA Res., 5, 345–349. [DOI] [PubMed] [Google Scholar]
- 7.Aaronson J.S., Eckman,B., Blevins,R.A., Borkowski,J.A., Myerson,J., Imran,S. and Elliston,K.O. (1996) Toward the development of a gene index to the human genome: an assessment of the nature of high-throughput EST sequence data. Genome Res., 6, 829–845. [DOI] [PubMed] [Google Scholar]
- 8.Hillier L.D., Lennon,G., Becker,M., Bonaldo,M.F., Chiapelli,B., Chissoe,S., Dietrich,N., DuBuque,T., Favello,A., Gish,W. et al. (1996) Generation and analysis of 280,000 human expressed sequence tags. Genome Res., 6, 807–828. [DOI] [PubMed] [Google Scholar]
- 9.Walhout A.J., Sordella,R., Lu,X., Hartley,J.L., Temple,G.F., Brasch,M.A., Thierry-Mieg,N. and Vidal,M. (2000) Protein interaction mapping in C. elegans using proteins involved in vulval development. Science, 287, 116–122. [DOI] [PubMed] [Google Scholar]
- 10.Hartley J.L., Temple,G.F. and Brasch,M.A. (2000) DNA cloning using in vitro site-specific recombination. Genome Res., 10, 1788–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauer C.E., Gardner,J.F. and Gumport,R.I. (1985) Extent of sequence homology required for bacteriophage lambda site-specific recombination. J. Mol. Biol., 181, 187–197. [DOI] [PubMed] [Google Scholar]
- 12.Ohara O., Ohara,R., Yamakawa,H., Nakajima,D. and Nakayama,M. (1998) Characterization of a new beta-spectrin gene which is predominantly expressed in brain. Mol. Brain Res., 57, 181–192. [DOI] [PubMed] [Google Scholar]
- 13.Greene J.R. and Guarente,L. (1987) Subcloning. Methods Enzymol., 152, 512–522. [DOI] [PubMed]
- 14.Church G.M. and Gilbert,W. (1984) Genomic sequencing. Proc. Natl Acad. Sci. USA, 81, 1991–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siebert P.D., Chenchik,A., Kellog,D.E., Lukyanov,K.A. and Lukyanov,S.A. (1995) An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res., 23, 1087–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okayama H. and Berg,P. (1983) A cDNA cloning vector that permits expression of cDNA inserts in mammalian cells. Mol. Cell Biol., 3, 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]