

Abstract
Untreatable hereditary macular dystrophy (HMD) presents a major burden to society in terms of the resulting patient disability and the cost to the healthcare provision system. HMD results in central vision loss in humans sufficiently severe for blind registration, and key issues in the development of therapeutic strategies to target these conditions are greater understanding of the causes of photoreceptor loss and the development of restorative procedures. More effective and precise analytical techniques coupled to the development of transgenic models of disease have led to a prolific growth in the identification and our understanding of the genetic mutations that underly HMD. Recent successes in driving differentiation of pluripotent cells towards specific somatic lineages have led to the development of more efficient protocols that can yield enriched populations of a desired phenotype. Retinal pigmented epithelial cells and photoreceptors derived from these are some of the most promising cells that may soon be used in the treatment of specific HMD, especially since rapid developments in the field of induced pluripotency have now set the stage for the production of patient-derived stem cells that overcome the ethical and methodological issues surrounding the use of embryonic derivatives. In this review we highlight a selection of HMD which appear suitable candidates for combinatorial restorative therapy, focusing specifically on where those photoreceptor loss occurs. This technology, along with increased genetic screening, opens up an entirely new pathway to restore vision in patients affected by HMD.
Keywords: Hereditary disease, Stem cells, gene therapy, Macular degeneration, Retinal photoreceptors, Cell therapy, Induced pluripotency, Embryonic stem cells
INTRODUCTION
Numerous hereditary retinal disorders (HRD) that affect central visual function in humans have been identified. The specialized central region of the retina, the macula, is responsible for central visual acuity and has distinct anatomical and physiological properties. The fovea at the epicentre of the macula contains the highest density of retinal cone photoreceptors and receives its blood supply entirely from the choriocapillaris complex of the choroid. Derived from the optic neuroepithelium during eye development, the photoreceptors share a common embryological origin with and are supported by the retinal pigmented epithelium (RPE) [1,2], which is separated from the choriocapillaris by Bruch's membrane, a multilayered basement membrane (Fig. 1). Photoreceptors, RPE, and the choriocapillaris are interdependent on each other; photoreceptor dysfunction and degeneration can occur secondary to RPE pathology (e.g., in Best's macular dystrophy) or be a primary event such as is the case in Stargardt disease (STGD), where RPE pathophysiology resulting from photoreceptor malfunction leads to photoreceptor depletion. Choriocapillaris atrophy and histopathology can be observed after RPE degeneration in various fundus disorders [3,4]. Such features are characteristic of a heterogeneous subgroup of progressive HRD affecting the macula, which can cause profound central visual loss sufficiently severe enough for blind registration. Blindness substantially impairs an individual's quality of life [5–8] and places a large burden on healthcare and support services [9]. There is currently no cure for the underlying causes of any of the macular dystrophies, and available treatments are largely palliative.
Figure 1.
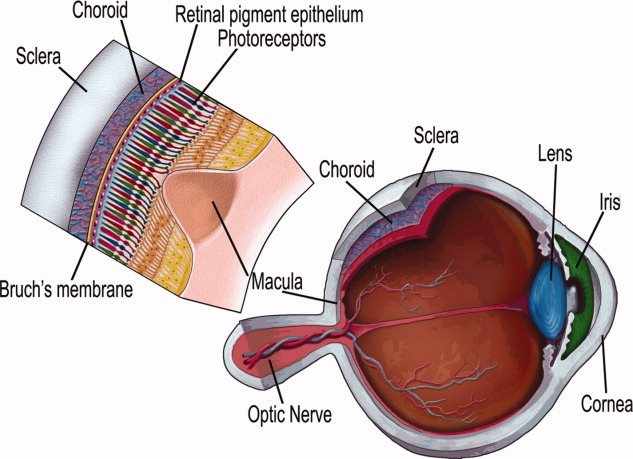
A schematic of the human eye showing the location of the macula, and an enlarged section of the retina at this region showing the organization of the retinal layers and the close relationship of the photoreceptors with the retinal pigment epithelium, Bruch's membrane, and the choriocapillaris of the choroid.
Much effort has been invested in the development of protective and cell-based therapies for HRD. Some success has been achieved in prolonging the survival of photoreceptors by intraocular application of growth or antiapoptotic factors in humans [10]; however, even with repeated application this approach typically slows the progression but fails to arrest the degenerative process [11]. The transfer of genes encoding for these factors into the eye offers additional protection by modifying the physiology of affected cells; however, this approach is ineffective in cells that are dysfunctional due to an underlying genetic mutation. RPE transplantation prevents photoreceptor degeneration in models of RPE dystrophy [12,13], but not in a photoreceptor degeneration model [14], and it is an inappropriate strategy in humans once irreversible loss of photoreceptors has occurred.
A protracted phase of inner neural retinal remodeling occurs after the depletion of the sensory photoreceptors; however, the initial stages largely involve remodeling of the outer nuclear layer [15,16]. Prior to the clinical application of any cell replacement strategy, the progress of global retinal remodeling should first be assessed in order to define the key impediments towards visual reconstitution. A sufficient retinal cyto-architecture must exist for successful graft integration to yield improvements in visual function. Functional cell replacement may be complicated by the retraction of interneuron dendrites and axon terminal fields, Müller glial and horizontal cell hypertrophy, and horizontal neurite sprouting towards the inner plexiform layer [15]. Thus the optimal period for cell engraftment within each disease paradigm should be determined. Nonetheless, a therapeutic window of opportunity for cell replacement may exist prior to significant inner retinal remodeling. Such strategies have proved successful in other degenerative regions of the central nervous system; for example, Parkinsonian symptoms have been reduced for up to six months in monkeys following chemical depletion of dopaminergic cells by transplanting monkey embryonic stem cells (ESCs) or ventral mesencephalon [17,18], and hind limb motor function can be improved in animal models of spinal cord injury following transplantation of olfactory ensheathing cells [19,20]. Cells grafted into the photoreceptor-depleted outer retina prior to the onset of global remodeling could potentially reconstitute the remaining retinal circuitry, partially restoring visual function.
Here we present the clinical manifestations and genetic correlations of a selection of hereditary macular dystrophies (HMDs) that may be suitable candidates for emerging cell- and gene-based therapy. The main focus of this review is the outer retina and diseases affecting central visual acuity where the primary defect results, either directly or indirectly, in the demise of photoreceptors. Here we concentrate on photoreceptor replacement rather than RPE replacement, although in many cases of HMD the replacement of both tissue types is required. We discuss current research successes, the potential implications and limitations of emerging techniques for visual restoration, potential combinatorial approaches, and possible future directions in this highly enigmatic field.
GENETIC BASIS OF RETINAL DISEASE
Hereditary Macular Dystrophy
Although modifiable lifestyle factors such as smoking and a high body mass index increase the risk of retinal disease, new evidence is emerging that stresses the importance of familial influence and the underlying molecular causes of retinal disease; indeed, few clinical conditions are totally without some genetic influence [21]. Below we describe three major types of HMD and discuss the genetic and environmental contribution to the onset and progression of retinal disease. A summary of other HRDs is presented in Table 1.
Table 1.
A summary of the occurrence of cone and macular dystrophies and associated the identified underlying molecular causes
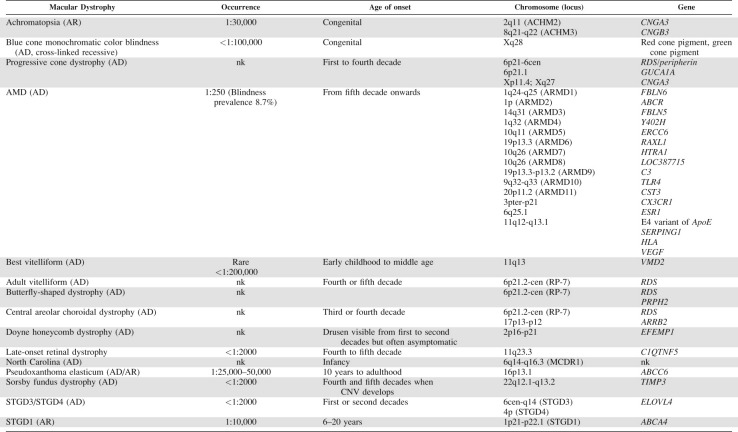 |
Abbreviations: AD, autosomal dominant; AMD, Age-Related Macular Degeneration; AR, autosomal recessive; CNV, choroidal neovascularization; nk, not known.
A feature of many HMDs, although typically characteristic of Age-Related Macular Degeneration (AMD), is the presence of sub-RPE deposits, or drusen [22–24]. This contributes to disease pathogenesis by cleaving RPE cell attachment to Bruch's membrane, which causes disruption of membrane transport and subsequent RPE demise [25]. Concomitant local oxidative stress, chronic inflammation, choriocapillaris changes, and neurosensory degeneration occur [26]. Various underlying molecular causes alter individual susceptibility for drusen accumulation and subsequent pathology in HMD; however, drusen and histopathological changes in RPE can also be detected during normal physiological aging [27]. Photoreceptors are continuously renewed by shedding outer segment (OS) distal disks in a circadian fashion [28]. With aging, the heavy metabolic burden of OS phagocytosis and breakdown on the RPE results in incomplete digestion of internalized material and accumulation of autofluorescent lipofuscin, a component of drusen, within RPE lysosomes and endosomes. A major fluorophore of RPE lipofuscin is A2E [29,30], a minor by-product of the visual cycle [30,31]. Given its high density of photoreceptors, the macula accumulates the greatest levels of A2E in its RPE. The RPE becomes less efficient in coping with increased toxic lysosomal A2E and other visual by-products, thus contributing to AMD [31]. Although fundoscopically similar, the age of onset, pattern of drusen deposits, and visual course separate normal aging from patients with HMD. AMD, once considered effectively an environmentally exacerbated form of aging, can now be considered another form of HMD with onset later in life.
STGD
STGD is the most prevalent HMD with an estimated incidence of one in 10,000 [32]. It accounts for 7% of all HRD and carries a 25% a priori risk to siblings [21]. The traditional presentation (STGD1) is autosomal recessive with juvenile onset (7-12 years); however, some rarer autosomal dominant forms exist [33–36]. STGD is clinically characterized by RPE and photoreceptor inner segment lipofuscin accumulation, RPE and choroidal vascular atrophy, macular photoreceptor loss, and reactive Müller glial hypertrophy [37,38] with progressive loss of central visual acuity during the first two decades [21]. Fundus flavimaculatus, characterized by subtly later onset, slower progression, and widespread, symmetrical, deeply localized retinal flecks [39,40] is understood to be a different manifestation of the same disease.
STGD dystrophies are mapped to chromosome 1p21-p22.1 and vary in onset, clinical course, and severity. This is likely due to allelic mutations, largely missense, producing a continuum of disease pathology and presentation [21,41,42]. The adenosine triphosphate–binding cassette (ABC) transporters function to aid adenosine triphosphate–dependent translocation of substrates across cell membranes and are implicated in human inherited disorders. Deletion of the ABC subfamily A (ABC1), member four (ABCA4, alternatively ABCR) gene [43,44], which encodes for the rod OS protein Rim (RmP) [45,46], functions in the transmembrane transport of vitamin A derivatives to the RPE and accounts for 60% of STGD cases [47]. Mutations in ABCA4 have been identified in retinitis pigmentosa (RP) and cone-rod dystrophy [48–51]. ABCA4 mutations are also associated with all juvenile HMD, recessive RP, and cone-rod degeneration [47,50].
Abcr−/− mice display biochemical, physiological, and ultrastructural changes with delayed rod dark adaptation, delayed clearance of all-trans-retinaldehyde (all-trans-RAL), increased levels of PE in rod OS, accelerated A2E accumulation in the RPE, thickening of Bruch's membrane, and visual loss [52,53]. When raised in total darkness, A2E accumulation in Abcr−/− mice is completely inhibited, indicating that the rate of STGD progression in humans might be slowed by limiting light exposure [54].
The two dominant forms are genetically distinct from recessive STGD, mapped to chromosome 6cen-q14 (STGD3) and 4p (STGD4). These affect the elongation of very long-chain fatty acid-like gene four (ELOVL4), expressed abundantly in photoreceptors [55]. A knock-in mouse model of a five base pair deletion in ELOVL4 was recently reported [56]. Mice heterozygous for this mutation show photoreceptor degeneration, while the homozygous variety have abnormally compacted outer epithelium, lack key hydrophobic components of the stratum corneum affecting permeability barrier function, and die shortly after birth.
Sorsby Pseudoinflammatory Fundus Dystrophy
Sorsby pseudoinflammatory fundus dystrophy (SFD) is a highly penetrant, rare, autosomal dominant condition characterized by loss of central vision due to extracellular matrix abnormalities in Bruch's membrane and bilateral choroidal neovascularization (CNV). This disrupts choroidal nutrient and metabolite transport leading to atrophy of the neural retina [57,58]. Onset is typically 40-50 years of age [59,60]. Fine drusen-like deposits, atrophic lesions at the macula [21], and lipid deposits at the RPE/Bruch's membrane interface are observed [22]. This is usually complicated by CNV and associated hemorrhage leading to a disciform macular scar [61]. The peripheral retina is also affected, resulting in night blindness (nyctalopia).
SFD shares several features with late-stage AMD, resulting in SFD becoming an accepted AMD genetic model [62]. Whilst AMD has highly complex and largely unknown etiology, SFD is a single-gene disorder that occurs due to mutations in the TIMP3 gene on chromosome 22 [63–65]. Its product is an RPE enzyme important in extracellular matrix regulation [66,67], and the presence of a mutant form may affect retinal protein turnover [61]. Eight mutations are described, seven affecting the coding sequence in exon 5; six are missense, introducing a novel unpaired cysteine residue into the C-terminal domain [61,62,66,68–70]. Other mutations introduce a stop codon at position 139 [71] or a single base (A) in the splice acceptor site between exons 4 and 5 (CAG to CAAG) [72]. A region of the TIMP3 C-terminus appears particularly vulnerable; three mutations are found between residues 166-168 with two others (172 and 181) in close proximity [61]. The serine to cysteine substitution of residue 181 [Ser181Cys] in exon five is the causal mutation in the majority of Sorsby's families tested in the United Kingdom.
In SFD, mutant TIMP3 accumulates in the RPE and Bruch's membrane, prompting the disease process [65,73,74]; TIMP3 overexpression can induce apoptosis in several cell types including RPE [75]. Unlike other TIMP family members, TIMP3 binds to the extracellular matrix via its C-terminal, the site of all known SFD mutations [61,76–78]. Whereas mice with targeted deletion of TIMP1 show CNV [79], homozygous TIMP3-null mice do not exhibit an obvious retinal phenotype [80]. A knock-in mouse carrying a Ser156Cys mutation in the orthologous murine TIMP3 gene shows clinical features of human SFD, including abnormalities and elevated TIMP3 in Bruch's membrane and RPE [68,81], providing an experimental system in which to investigate SFD pathophysiology. The range of mutations is limited in SFD, meaning that genetic analysis makes diagnosis quick and reliable, although the underlying disease process remains untreatable.
Age-Related Macular Degeneration
AMD is the leading cause of blindness in the developed world over 60 years of age and accounts for 50% of blind registration [82–87]. Incidence of AMD rises exponentially with age [88]. While its prevalence is increasing in the United Kingdom, blind registration from cataract, glaucoma, and optic atrophy has declined [89]. There are two main forms of AMD: an early form characterized by degenerative changes in the RPE and accumulation of drusen, and a late form that manifests with geographic atrophy alongside sub-RPE and subretinal CNV [90]. Therapies to prevent and treat AMD are limited. High-dose antioxidant vitamins and metabolic therapies can reduce the progressive visual loss in patients with early AMD and, in the case of metabolic therapy, the regression of drusen; however, the absolute reduction in new cases is small [91–93]. Although environmental factors are known to play a role in disease pathology and progression, epidemiological and family-based studies provide convincing evidence for a genetic basis for AMD, with inheritance thought to be polygenic [94–100]. Despite its prevalence, the etiology and pathogenesis of AMD remains poorly understood. Susceptibility loci (chromosome 1, 5, 9, 10, and 17) [101–103] and candidate genes that appear to play a role have been identified [104–108], some correlating with AMD pathology more strongly than others; more than 100 different proteins are associated with AMD deposits [108].
The complement system, involved in our immune defense against foreign antigens, is implicated in AMD [109–111]. Complement factor H (CFH) inhibits the complement cascade that ensures the system is directed against pathogens and not the body's own tissues, and is a major susceptibility gene in AMD. Complement components are found alongside drusen and the RPE-choroid interface [110], and abnormal macular dysregulation of the complement cascade in RPE and Bruch's membrane appears to cause unrestrained complement activation and drusen formation [112]. The CFH Y402H haplotype on chromosome 1q32 confers the greatest risk of AMD [113]; however, multiple polymorphism variants in caucasian AMD patients have been determined [104,114–118]. Y402H is not a primary indicator in Japanese patients, however [119], nor in the Chinese population in which this polymorphism is associated specifically with neovascular AMD [120]. The classical complement pathway is largely regulated by the SERPING1 gene product, and a common polymorphism in SERPING1 was recently identified as a causal factor in AMD [121]. Interestingly, a CFH haplotype with a deletion in CFHR1 and CFHR3 elicits an independent decreased risk of AMD [113].
Oxidative stress pathways are also causally involved [122]. Schmidt and colleagues identified a coding change [Ala69Ser] in the LOC387715/ARMS2 gene on chromosome 10q26 as the second major identified susceptibility allele [123]. They report that genetic susceptibility in combination with a modifiable lifestyle factor conveys a significantly higher risk than either factor alone. The population-attributable risk is 36% for LOC387715/ARMS2 and 43% for Y402H, however combining LOC387715 and cigarette smoking increases risk to 61% [123]. The LOC387715/ARMS2 protein product colocalizes with the mitochondrial outer membrane in mammals [106]; thus, the Ala69Ser mutation presumably contributes to AMD pathogenesis by affecting mitochondrial function. Polymorphisms of other immune response genes, such as human leukocyte antigen, are also implicated after observation of strong human leukocyte antigen immunoreactivity in drusen [109,124]. Variants of complement cascade component factor B and complement component two are protective against AMD [105].
Several genes implicated in the etiology of various HMD have been examined in AMD. Carrier relatives of STGD are more likely to develop AMD [125], and genetic similarities may exist between the two [43]; however, it remains controversial as to whether the ABCA4 gene plays a pathogenic role [43,44]. The high level of ABCA4 polymorphism across individuals makes it difficult to determine any responsibility for ABCA4 in AMD [21]; however, its involvement is possible in a small number of cases [107]. A2E, which accumulates in the STGD eye, is also found in the RPE in AMD and perturbs efflux of cholesterol from RPE endosomes/lysosomes, causing cholesterol and cholesteryl ester deposit accumulation [61,126,127]. Initially identified as a causal factor in Doyne's disease, a mutation in the extracellular matrix protein Fibulin 3 gene [128] led to the correlation of missense mutations in Fibulin 5 in 1.7% of 402 AMD cases in the United States [129]. These contribute to AMD by causing reduced Fibulin five and elastin production—a key Bruch's membrane component—but were not causal [129]. The TIMP3 mutation in SFD has not correlated with AMD [66,130], although distribution of the TIMP3 enzyme product in the AMD Bruch's membrane is significantly higher than age-matched controls, remains in active form, and is associated with drusen patches [76].
Late-onset retinal degeneration (LORD) is a rare autosomal dominant disorder that has striking parallels with AMD. Histopathology reveals thick sub-RPE deposits that result in RPE dysfunction and photoreceptor loss [24,131–133]. A Ser163Arg mutation in the CTRP5/C1QTNF5 gene is reported to cause approximately 50% of documented LORD cases [134]. As yet it is unknown whether mutations in CTRP5 influence susceptibility to AMD. Apolipoprotein E (ApoE) and β-amyloid aggregates, associated with Alzheimer's disease, have been found in AMD-related macular drusen [135,136]. Some studies implicate the ApoE ɛ4 allele as a protective factor which delays disease onset, while the ɛ2 allele accelerates disease onset and progression [137,138]. However, although there is currently no significant association between Alzheimer's disease and AMD [139], a recent study supports some relationship between cognitive function and dementia with early AMD in older individuals [140]. Mutations in the Bestrophin gene, responsible for vitelliform macular dystrophy (Best's disease) show a small, nonsignificant correlation with AMD [141,142].
The identification of vascular endothelial growth factor (VEGF) polymorphisms in late-stage AMD has led to the use of optical coherence tomography-guided anti-VEGF (OCTVEGF) treatments which slow disease progression by treating CNV [143]. They do not, however, alter the underlying pathophysiology, and atrophic macular changes can still progress despite successful CNV treatment. A naturally occurring mouse model with autosomal recessive late-onset severe retinal degeneration mapped to the Mdm1 gene on mouse chromosome 10 was recently reported, but is not associated with the human Mdm1 ortholog or AMD [144]. The Sod1−/− and ApoE knock-in mice show the most similarities to the human clinical manifestation of AMD thus serve as good disease models [145,146]. In conclusion, the most prominent genetic factors identified to date in the etiology of AMD are the Y402H variant of CFH, LOC387715, and SERPING1, which are found in more than 60% of cases [121].
Cell Replacement Strategies
While modifiable lifestyle factors present an increased risk of retinal disease to an individual already expressing high-risk alleles, there is clear evidence that genetics plays an important role in the occurrence and development of HMD. Understanding the inheritance and genetic basis of these retinal diseases will undoubtedly provide new treatment platforms for restorative therapy. Gene therapy, whether corrective [147–150] or to overexpress various neuroprotective substances [135] either on its own or combined with stem cell therapy, has enormous potential in these diseases, although it may be less applicable to those cases where the dystrophy is advanced at birth or where perturbed retinal and cortical development has compromised visual function [151]. Stem cell therapy could be used in two main ways, namely by enhancing endogenous repair using stem cells that secrete growth factors or deliver drugs utilizing the innate attraction of diseased structures to various stem cell populations, or by exogenous cell replacement strategies. In the case of HMD, the latter strategy could be a useful therapeutic option depending on integrity of the inner retina, Bruch's membrane, and choriocapillaris, and provided that problems associated with the synaptic integration between transplanted photoreceptors and the host retina can be overcome.
Various sources of cells and methods of transplantation have been used in the attempt to replenish the retina with functional photoreceptors or healthy RPE following the degeneration of host cells in assorted animal models of retinal degeneration with various degrees of success [152–157]. In the best case, subretinal transplantation of embryonic and postnatal day 1 (P1) mouse retinal cells into murine models of retinal degeneration showed greatest integration, differentiation, and synaptic connectivity within host tissue when already committed to a photoreceptor fate while still morphologically immature. Increases in pupil sensitivity in this study correlated with the number of incorporated Nrl+ donor cells, a result that was not achieved with transplanted proliferating or stem cells [157].
Recent reports describe the successful generation of photoreceptor precursors from ESCs using defined protocols incorporating factors that are known to promote forebrain development, retinal progenitor specification and photoreceptor induction [158–161]. Similarly, protocols for RPE production from ESCs are being refined, and functional studies on human ESC-derived RPE demonstrate their physiological viability [162]. Small numbers of opsin- and rhodopsin-positive cells can be observed after subretinal transplantation of spontaneously differentiated human ESCs (hESCs) into neonatal and adult rat eyes with no tumor formation observed up to 18 weeks post-transplant [163]. More recently, retinal progenitors derived from hESC were subretinally transplanted into Crx−/− mice, a model of Leber congenital amaurosis (LCA) [164]. Grafted cells integrated within the outer nuclear layer; displayed layer-specific expression of opsin, rhodopsin, and recoverin; and restored the light response in previously unresponsive recipients, even though grafted cells expressing photoreceptor-specific markers did not develop OSs. These results highlight the value of hESC as a potentially unlimited source of specialized cell types for transplantation; however, issues do remain regarding their use, including efficient direction of differentiation towards a required lineage and elimination of undifferentiated ESCs from the cell population intended for transplantation in order to minimize the risk of tumorigenesis.
The ETDRS chart, originally used in the Early Treatment of Diabetic Retinopathy Study, has become the standard distance visual acuity as measurement in clinical research [165]. More recently, modest improvements in visual acuity as measured using the ETDRS chart was reported after transplantation of fetal neural retina with its RPE in a patient with dominant RP [166]. These authors recently published their results from an additional fetal retina-RPE transplant clinical trial conducted on six RP and four AMD patients [167]. In this latest trial, 70% of transplant recipients showed improvements in visual acuity, and postoperative loss of RPE pigmentation was not associated with changes in visual outcome [167]. In one patient, improvement in visual acuity and light sensitivity was maintained for 6 years. It is difficult to discern, however, whether observed improvements are due to graft functionality, graft-derived factors, or surgical manipulation eliciting improvements in visual function [168,169]. The development of an artificial Bruch's membrane substitute or the use of scaffolds to maximize the adhesion and safe surgical delivery of the RPE sheets or tissue under the retina may enhance cell-replacement strategies [158,170] provided there is access to functional choriocapillaris. The absence of just one of these vital elements will cause a sequence of events affecting retinal viability and hinder graft integration and function.
In summary, although cell transplantation results are encouraging, only a small proportion of grafted cells (in the best case 0.4% of sorted P1 Nrl+ photoreceptor precursors) integrated within the correct lamina and differentiated appropriately in a model of retinal degeneration [158]; therefore, the efficiency of retinal reconstitution via cell replacement remains a challenge. Encouraging incorporation of grafted photoreceptor precursors is ineffective without a functional RPE/Bruch's membrane/choriocapillaris complex. Studies over the past 10 years have shown that allogeneic RPE largely resists attachment to aged Bruch's membrane in vitro and where successful attachment and proliferation has been achieved, long-term survival is poor [171–173]. Early studies in humans demonstrated that long-term survival of transplanted fetal retina with its RPE (up to 6 months) could be achieved in RP patients, but with no improvement in visual function [174]. Similar studies in AMD patients yielded mixed results; transplantation of fetal or adult RPE [175–178] implied that late-AMD is less conducive for graft survival with patients showing signs of rejection within 3 months [174]. RPE transplanted alongside aggregate retinal transplants or photoreceptor sheets into RP and AMD patients proved the long-term safety and survival of grafts but again patients showed no improvement in visual function [179–181].
Revolutionary Developments in the Stem Cell Field: Induced Pluripotency
As highlighted in the above section, hESCs have been shown to differentiate along photoreceptor and RPE lineages using growth factors that have roles in forebrain development and early and late retinal fate specification. In one very recent study, hESC-derived photoreceptor cells were shown to settle into the appropriate layers and express markers of differentiated rod and cone cells upon intraocular injection into animal models of retinal disease [164]. These are undoubtedly promising results, but many substantial shortcomings of hESC differentiation still await resolution. Among these are the ethical issues associated with the use of embryonic tissue, the apparently embryonic/fetal phenotype of the cells derived during hESC differentiation [183], and the potential of tumorigenesis arising from the presence of undifferentiated progenitors remaining in culture; however, the principal problem is one of immune rejection of differentiated cells after transplantation into the patient. Pioneering work carried out in the last 3 years suggests that immune rejection issues may be overcome by creating embryonic-like stem cells from somatic cells of adult individuals through a process called induced pluripotency [184–186]. This process, which is held as one of the most seminal discoveries in the stem cell field, was first reported by Yamanaka's group [187] and involves overexpression of four key genes (Oct4, Sox2, Klf4, and c-Myc) in murine fibroblasts, resulting in their conversion into cells that resemble ESCs in terms of morphology, gene expression, growth, and differentiation capabilities and are now named induced pluripotent stem cells (iPSCs). This work was soon supplemented by the generation of human iPSCs by two groups, one of whom [186] showed that adult human dermal fibroblasts can also be reprogrammed by overexpression of OCT4, SOX2, KLF4, and c-MYC, while another [188] made use of a slightly different set of factors (OCT4, SOX2, LIN28, and NANOG) to reprogram both fetal and adult human fibroblasts. Since these initial papers there has been remarkable progress in the field, aimed largely at increasing the efficiency of the iPSC generation protocol and replacing the initial retroviral vectors that were used to transfect fibroblasts with OCT4, SOX2, KLF4, and c-MYC. Two approaches to this latter problem have been used with varying degrees of success. The use of nonintegrating retroviruses that maintain transient expression of the reprogramming factor genes from episomes that do not integrate into the host cell genome has shown some potential to generate iPSCs, albeit with lower efficiency than is achievable with integrating retroviruses [189]. Very recently, a protocol using recombinant proteins for induction of pluripotency has been published, which suggests that future methods for the production of iPSCs may be much simpler than those of the initial publications in this area [190].
Together these developments suggest that it is now possible to derive iPSCs free of transgenes and with perhaps reduced risk of tumorigenesis, which was observed in the initial animal-based studies (20–30%) associated with reactivation of c-MYC in adult tissues [191]. Caution is required, however, for the small molecules and chemicals that are required for reprogramming because they may promote global epigenetic and genetic modifications, which may compromise the safety aspect of these cells.
The ability to generate iPSCs readily allows us to progress to one of the principal applications envisaged for this technology, which is the generation of human disease models and potential correction of genetic disease. For example, iPSC technology was combined with gene therapy to correct the mutant human sickle cell anemia allele [192]. The corrected iPSC generated in this study were induced to differentiate to hematopoietic progenitors that were subsequently transplanted into the murine model of sickle cell anemia, leading to functional recovery and providing a “proof of principle” demonstration for future human therapeutic application. Similarly, transplantation of murine iPSC-derived neural progenitors into animal models of Parkinson's disease led to improved behavior 4 weeks after transplantation [193]. Furthermore, iPSCs have been generated from other species, including the rhesus macaque, which is by far one of the most relevant primate models for human disease [194].
To date, a large number of iPSC lines from various diseases such as amyotrophic lateral sclerosis [195]; Parkinson's disease [196]; type I diabetes [197]; spinal muscular atrophy [198]; adenosine deaminase deficiency-related severe combined immunodeficiency (ADA-SCID); Shwachman-Bodian-Diamond syndrome; Gaucher disease; and type III, Duchenne, and Becker muscular dystrophy [199] have been reported.
Creation of iPSC lines on its own is not sufficient and needs to be combined with techniques that have and will be developed in human ESC/iPSC for efficient and directed differentiation towards the desired functional cell type. In view of this, it is important to determine whether iPSCs differentiate into various phenotypes in a manner similar to ESCs, and, although a couple of studies have suggested that this may be the case under in vitro conditions [200–202], in vivo functional studies in animal models need to be performed to fully investigate the potential of iPSC lines for both disease modeling and cell transplantation.
Potential Applications and Limitations of iPSCs for Understanding and Treatment of Retinal Disease
Derivation of iPSCs from patients with diseases causing outer retinal degeneration, alongside in vitro gene correction with a robust differentiation method for producing photoreceptor and RPE cells from these cells, could provide a source of autologous cells for transplantation. A very recent report has shown that human iPSC can be differentiated to photoreceptor cells with efficiency similar to that of human ESCs [203], suggesting that already established protocols for human ESCs can be transferred directly to iPSCs. Of equal importance is the derivation of iPSCs from patients with retinal disease for creation of disease models, which provides an invaluable opportunity to investigate disease pathogenesis and treatment that has not been possible before. Although there are successful animal models of retinal degeneration, most of these models mimic modulation of one or at most two genes [204]. Using iPSCs isolated from affected patients provides new opportunities to complete the current gaps in our understanding [205], especially for disorders such as AMD where multiple genes are likely to play a role in disease initiation and progression as highlighted above (see Age-Related Macular Degeneration) and where animal models that mimic the disease perfectly are difficult to create due the polygenic nature of the disease. Such cells can be used to devise genetic tests for risk prediction and diagnosis as well as testing and design of new drugs that may have an impact on photoreceptor and RPE cell degeneration.
Generation of photoreceptor and RPE cells from patient-specific iPSC lines overcomes the need for immunosuppression and will likely reduce the risk of rejection of grafted tissue within the neurally depleted region. However, in diseases where a single underlying genetic mutation is causing cellular dysfunction or death, one caveat in generating iPSCs from such patients will be the generation of populations of cells that carry the same mutation. Targeting host photoreceptors with gene therapy to supply growth factors or antiapoptotic genes has already been tested in animal models of degenerative retinal disease in addition to antiangiogenic factors in animal models of neovascular retinal disease, with no adverse morphological, inflammatory, or vascular effects [11]. It is therefore possible that the experience gained from gene therapy trials in adult host photoreceptors can be applied to iPSC-derived photoreceptor or RPE cells. Another possibility is the correction of the gene defect at the iPSC stage prior to differentiation to photoreceptors or RPE, although the polygenic nature of many HMD cases means that such an approach currently remains a challenge. Nonetheless, although homologous recombination is still very difficult in hESCs and presumably in iPSCs, inducing the expression of the wild-type gene in mutant iPSCs prior to their differentiation is likely to be achieved using viral vectors, which fits with recent reports of gene correction [148–150]. For example, a recent report has shown that subretinal lentiviral vector delivery of the human ABCA4 gene in a mouse model of STGD was shown to transduce up to 20% of photoreceptors in the injected region and substantially reduce disease-associated lipofuscin accumulation [206]. It can be therefore envisaged that expression of wild-type ABCA4 in iPSCs derived from well-characterized STGD patients with ABCA4 mutations and their further differentiation to RPE and photoreceptor cells could potentially elicit some clinical improvement. Conversely, in cases where differing clinical manifestations of HRD arise from the same gene mutation (for example, adult vitelliform, central areolar choroidal, and butterfly-shaped dystrophies all arise from a mutation in the RDS gene on the short arm of chromosome six (Table 1)), the same corrective gene therapy might be applied to patients spanning more than one dystrophy, eliciting wider clinical impact. Care should be exercised with this approach, however, because overexpression of particular genes may carry the additional risk of unwanted and unpredicted physiological effects. In addition, applications of this approach for other single-gene disorders will depend very much on the size of the gene of interest because vectors have limited packing capacity, which may convey limitations in terms of the size of the defect that could be fixed. This approach will not work in genetic diseases associated with dominant negative effect such as STGD patients with EL0VL4 mutations or SFD patients with TIMP3 [207,208]. In these cases, gene correction at the iPSC stage is likely to be the only means of gene correction. There are already developments in this field mainly applied to hESCs using zinc finger nucleases [209]; however, this method has to be established and tested in human iPSCs and in multiple loci with high efficiency before this approach can be envisaged as a tool in cell and gene therapy.
Correction of gene defects in other retinal disorders such as AMD already associated with multiple genes, polymorphisms, and environmental factors, however, is unlikely using the iPSC approach, and in this case the usefulness of iPSC technology will mainly rely on the development of screening tools that will enable identification of individuals with high-risk of advanced AMD prior to retinal degeneration.
Translation: Challenges Ahead?
Efficient tissue delivery, graft integration, and synaptic connection with host circuitry remain as issues for the successful clinical translation of cell-based restorative therapies [210]. In patient terms, those affected by severe macular degeneration may be more willing to consider the option of translational human surgery. There is rarely any HRD-associated cognitive impairment, which means that informed consent could be performed with the patients prior to any translational research. Exploiting HMD with well characterized causal mutations is an excellent starting point for the translation of gene therapy combined with iPSC cell-replacement strategies from bench to bedside. For example, STGD is relatively well characterized and quite common, making it important and clinically relevant, and it primarily affects the macula, thus allowing potentially simple surgical replacement of macular cells with genetically-corrected iPSCs.
Cell replacement requires the stable and appropriate nondisruptive integration of functional cells within the correct lamina, the formation of synapses with appropriate interneurons, and the correct electrophysiological and transmitter responses to light to enable the transmission of discernable visual responses to the cortex. It also requires the long-term survival and viability of grafted cells. Variables in the transplantation procedure in humans include the source of cells, age of the donor, transplantation method, and stage of host disease progression [211,212]. The presence of xenogeneic molecules in protocols used to direct derivation and differentiation in vitro will limit clinical use. Similar to the use of hESCs, driving patient-specific stem cells towards a desired lineage in appropriate numbers remains a challenge. Recent advances in differentiating stem cells to photoreceptors have been achieved, meaning there are already some methodologies in place for generating the cells of interest [160,161,213], and the results of the study by MacLaren et al. provides information about the developmental time point to which we should drive cellular differentiation to encourage graft integration [158]. Although much work needs to be done to enhance the incorporation of grafted cells, it may be that in some patients only a small number of integrated cells are needed for functional restoration. In one study, the onset of obvious visual symptoms in glaucoma patients was reported only after loss of 25-35% of ganglion cells [214]. It should be noted here that the possibility of correcting the underlying etiology in the end stage of disease by iPSCs, gene correction, and transplantation is attractive, but the comprehensiveness of this approach is dependent on the viability of other vital supportive retinal components. Patients affected by perturbed retinal cytoarchitecture will be less amenable to this therapy, making clinical assessment of these parameters by in vivo imaging and electrophysiology vital.
The continued identification of candidate genes and suitable delivery vehicles is key. Research into the molecular genetics of HRD is at an early stage, making disease classification on the basis of molecular pathology difficult. Patients previously have been divided by phenotype; however, the importance of accurately genotyping individual cases must be realized. This is likely currently complicated by expense or the lack of a well identified gene defect with an available test for exploitation.
Additionally, HRDs are inherited due to numerous types of mutations (loss of function, gain of function) and are clinically and genetically heterogeneous [215]. Mutation screening in STGD patients has led to the identification of 400 sequence variations in the ABCA4 gene [216–219]. In early-onset HRD, genetic tests are able to be conducted more readily; however, this becomes problematic in the case of AMD where, due to the late onset, parents of affected individuals are often deceased and their offspring yet to be affected [220]. In representative animal models of HRD, the efficiency of gene correction techniques can be tested [53,81], and this will be further enhanced by the availability of patient-derived iPSCs.
One concern might be whether inheritance of HRD will affect gene therapy potential; for example, STGD is recessive and the mutations give variable penetration and expressivity. However, recessive models have been more amenable to gene therapy by restoration of the wild-type form of mutant genes. Photoreceptor rescue was achieved in 1996 by Bennett and colleagues who used adenovirus to introduce wild-type PDE6β in the rd mouse [221], an effect that was improved by the use of second-generation adenovirus and then lentivirus [222,223]. The results of three independent human retinal RPE65-replacement trials, published recently, demonstrate the safety of subretinal vector delivery in humans and show that gene therapy in advanced cases of human LCA could induce modest improvements in vision [148–150,224]. Two of the studies [148,149] used the ETDRS chart and an obstacle course combined with various other tests; however, only one reported electroretinography results [149]. Each study utilized recombinant AAV-2 vector delivered subretinally. In the study by Maguire et al. in Philadelphia [148], human RPE65 carrying a chicken β-actin promoter was introduced into three patients, two with homozygous missense and one with null mutation in RPE65. Patients reported improvements in vision at 2 weeks and measured improvements in pupillometry and nystagmus frequency over several weeks, concomitant with improvements in confidence and time taken to complete an obstacle course in all subjects [148]. The London study reported by Bainbridge et al., [149] delivered the human RPE65 gene under a human RPE65 promoter in three LCA patients with RPE65 missense mutations. Unlike the first study, only one patient showed visual improvement as measured by ETDRS alongside significantly improved retinal sensitivity, dark-adapted perimetry, and dramatic improvements in mobility through an obstacle course (from 77 to 14 seconds). Electroretinography results however, were not improved [149]. The third study reported treatment of three young patients with LCA. All three patients showed significantly increased visual sensitivity in vector treated retinal regions, yet the kinetics of the newly restored retinoid cycle in rod and cone photoreceptors was slow [150]. In all studies there were no adverse systemic effects. The safety of the technique has thus been demonstrated, though visual improvements were modest, which suggests that perhaps treatment needs to occur at an earlier stage in the disease course or in combination with cell-replacement therapy.
It is important to show that iPSCs can be generated from older patients and whether patients at later stages of disease are amenable to combinatorial therapy. Bainbridge et al. proposed that the patient who benefited from gene therapy did so due to being treated while at a less advanced stage than their counterparts [149], highlighting the importance of understanding the optimal window of opportunity for genetic intervention. It cannot be ruled out, however, that observed differences between the studies may have occurred due to the use of different promoters, the subretinal delivery of varying amounts (150 μl vs. 1,000 μl), or slight variations in methodology.
Furthermore, although the eye and subretinal space in particular is relatively immunologically privileged, the immune response may be exacerbated and affect graft survival in HRD cases where the blood retinal barrier is compromised [11,211]. There is evidence that AMD has an inflammatory element, and avoiding any element of immune rejection would be vital in terms of disease recurrence. Use of iPSCs would avoid an immune response to the donor cells themselves, but genetically engineered modifications are being made to viral vectors to limit the immune response using this approach [225].
Questions and Future Directions
Whilst developing the potential of iPSCs as a therapeutic strategy overcomes many of the issues associated with the use of hESCs (ethics, immunorejection), there are some very important control experiments that must be performed before translation of iPSC-based cell therapies to the clinic can become a reality. Fundamental questions include: Are these cells identical to hESCs? Do they have a normal karyotype? Can we efficiently differentiate iPSCs to photoreceptors and RPE? Given recent successes in generating photoreceptor precursors and functional RPE from hESCs, one would imagine similar results could be achieved with iPSCs. There is already evidence that iPSCs can differentiate into derivatives of the three germ layers [186]; however, it remains unknown whether retroviral transduction has any long lasting effects or whether cells might be reactivated under stress or with a lack in instructive cues, leading to the presence of potentially oncogenic cells. In light of this last comment, virus-free, nonintegrating plasmid reprogramming has been demonstrated in embryonic mouse fibroblasts [226], albeit at low efficiency compared with retroviral methods. These nonviral iPSCs were free from transgene integration in host chromosomes, demonstrated a capacity to differentiate towards all three germ layers, and could produce chimeric mice when injected into blastocysts. It is clear that while HMD with clearly identified etiology and a common causal mutation are the best candidates for combinatorial iPSC/gene/transplantation therapy, much work remains to be done to transform the current course of action after diagnosis of HMD for such therapy to become a reality in the near future. Improved knowledge of the mechanisms and molecular basis of HMD alongside continued safety evaluation of the long-term action and consequences of gene therapy and cell restoration will allow us to move towards tailoring safe, more specific and efficient restorative therapies for patients affected by this heterogeneous group of currently incurable diseases.
Acknowledgments
The authors are grateful to Simon Foster for help with the graphics and Prof. John Finlay, Newcastle Health Charity, Sunderland Eye Infirmary, Newcastle University, UK NIHR Biomedical Research Centre for Ageing and Age-related disease and the Newcastle upon Tyne NHS Hospitals Trust, for funding this work.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors indicate no potential conflicts of interest.
REFERENCES
- 1.Mund ML, Rodrigues MM. Embryology of the human retinal pigment epithelium. In: Zinn RM, Marmor MF, editors. The Retinal Pigment Epithelium. London: Harvard University Press; 1979. pp. 45–52. [Google Scholar]
- 2.Moshiri A, Close J, Reh TA. Retinal stem cells and regeneration. Int J Dev Biol. 2004;48:1003–1014. doi: 10.1387/ijdb.041870am. [DOI] [PubMed] [Google Scholar]
- 3.Korte GE, Reppucci V, Henkind P. RPE destruction causes choriocapillary atrophy. Invest Ophthalmol Vis Sci. 1984;25:1135–1145. [PubMed] [Google Scholar]
- 4.Armstrong JD, Meyer D, Xu S, Elfervig JL. Long-term follow-up of Stargardt's disease and fundus flavimaculatus. Ophthalmology. 1998;105:448–457. doi: 10.1016/S0161-6420(98)93026-3. [DOI] [PubMed] [Google Scholar]
- 5.Rovner BW, Zisselman PM, Shmuely-Dulitzki Y. Depression and disability in older people with impaired vision: a follow-up study. J Am Geriatr Soc. 1996;44:181–184. doi: 10.1111/j.1532-5415.1996.tb02436.x. [DOI] [PubMed] [Google Scholar]
- 6.Stuck AE, Walthert JM, Nikolaus T, et al. Risk factors for functional status decline in community-living elderly people: a systematic literature review. Soc Sci Med. 1999;48:445–469. doi: 10.1016/s0277-9536(98)00370-0. [DOI] [PubMed] [Google Scholar]
- 7.Grisso JA, Kelsey JL, Strom BL, et al. Risk factors for falls as a cause of hip fracture in women. The Northeast Hip Fracture Study Group. N Eng J Med. 1991;324:1326–1331. doi: 10.1056/NEJM199105093241905. [DOI] [PubMed] [Google Scholar]
- 8.Williams RA, Brody BL, Thomas RG, et al. The psychosocial impact of macular degeneration. Arch Ophthalmol. 1998;116:514–520. doi: 10.1001/archopht.116.4.514. [DOI] [PubMed] [Google Scholar]
- 9.Lanchoney DM, Maguire MG, Fine SL. A model of the incidence and consequences of choroidal neovascularization secondary to age-related macular degeneration. Comparative effects of current treatment and potential prophylaxis on visual outcomes in high-risk patients. Arch Ophthalmol. 1998;116:1045–1052. doi: 10.1001/archopht.116.8.1045. [DOI] [PubMed] [Google Scholar]
- 10.Sieving PA, Caruso RC, Tao W, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci USA. 2006;103:3896–3901. doi: 10.1073/pnas.0600236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harvey AR, Hu Y, Leaver SG, et al. Gene therapy and transplantation in CNS repair: the visual system. Prog Ret Eye Res. 2006;25:449–489. doi: 10.1016/j.preteyeres.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Turner JE. Inherited retinal dystrophy in the RCS rat: prevention of photoreceptor degeneration by pigment epithelial cell transplantation. Exp Eye Res. 1988;47:911–917. doi: 10.1016/0014-4835(88)90073-5. [DOI] [PubMed] [Google Scholar]
- 13.Lopez R, Gouras P, Kjeldbye H, et al. Transplanted retinal pigment epithelium modifies the retinal degeneration in the RCS rat. Invest Ophthalmol Vis Sci. 1989;30:586–588. [PubMed] [Google Scholar]
- 14.Li L, Sheedlo HJ, Turner JE. Retinal pigment epithelial cell transplants in retinal degeneration slow mice do not rescue photoreceptor cells. Invest Ophthalmol Vis Sci. 1993;34:2141–2145. [PubMed] [Google Scholar]
- 15.Marc RE, Jones BW, Watt CB, et al. Neural remodeling in retinal degeneration. Prog Ret Eye Res. 2003;22:607–655. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- 16.Li ZY, Kljavin IJ, Milam AH. Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci. 1995;15:5429–5438. doi: 10.1523/JNEUROSCI.15-08-05429.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collier TJ, Sortwell CE, Elsworth JD, et al. Embryonic ventral mesencephalic grafts to the substantia nigra of MPTP-treated monkeys: feasibility relevant to multiple-target grafting as a therapy for Parkinson's disease. J Comp Neurol. 2002;442:320–330. doi: 10.1002/cne.10108. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi J, Takagi Y, Saiki H. Transplantation of embryonic stem cell-derived dopaminergic neurons in MPTP-treated monkeys. Methods Mol Biol. 2009;482:199–212. doi: 10.1007/978-1-59745-060-7_13. [DOI] [PubMed] [Google Scholar]
- 19.Plant GW, Christensen CL, Oudega M, Bunge MB. Delayed transplantation of olfactory ensheathing glia promotes sparing/regeneration of supraspinal axons in the contused adult rat spinal cord. J Neurotrauma. 2003;20:1–16. doi: 10.1089/08977150360517146. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki M, Li B, Lankford KL, et al. Remyelination of the injured spinal cord. Prog Brain Res. 2007;161:419–433. doi: 10.1016/S0079-6123(06)61030-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Black GCM. In: Genetics for Ophthalmologists: The Molecular Genetic Basis of Ophthalmic Disorders. Hatchwell E, editor. London, UK: Remedica Publishing; 2002. [Google Scholar]
- 22.Capon MR, Marshall H, Krafft JI, et al. Sorsby's fundus dystrophy: a light and electron microscopic study. Ophthalmology. 1989;96:1769–1777. doi: 10.1016/s0161-6420(89)32664-9. [DOI] [PubMed] [Google Scholar]
- 23.Green WR, Enger C. Age-related macular degeneration histopathologic studies: the 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100:1519–1535. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- 24.Kuntz CA, Jacobson SG, Cideciyan AV, et al. Subretinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 1996;37:1772–1782. [PubMed] [Google Scholar]
- 25.Kunze C, Elsner AE, Beausencourt E, et al. Spatial extent of pigment epithelial detachments in age-related macular degeneration. Ophthalmology. 1999;106:1830–1840. doi: 10.1016/S0161-6420(99)90364-0. [DOI] [PubMed] [Google Scholar]
- 26.Enzmann V, Yolcu E, Kaplan HJ, et al. Stem cells as tools in regenerative therapy for retinal degeneration. Arch Ophthalmol. 2009;127:563–571. doi: 10.1001/archophthalmol.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sippy BD, Hinton DR. Aging of retina and retinal pigment epithelium. In: Lim JI, editor. Age-Related Macular Degeneration. New York: Marcel Dekker, Inc; 2002. pp. 1–14. [Google Scholar]
- 28.Bok D. The retinal pigment epithelium: a versatile partner in vision. J Cell Sci Suppl. 1993;17:189–195. doi: 10.1242/jcs.1993.supplement_17.27. [DOI] [PubMed] [Google Scholar]
- 29.Sparrow JR, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res. 2005;80:595–606. doi: 10.1016/j.exer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 30.Travis GH, Golczak M, Moise AR, et al. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rattner A, Nathans J. Macular degeneration: recent advances and therapeutic opportunities. Nat Rev Neurosci. 2006;7:860–872. doi: 10.1038/nrn2007. [DOI] [PubMed] [Google Scholar]
- 32.Blacharski PA. Fundus flavimaculatus. In: Newsome DA, editor. Retinal Dystrophies and Degenerations. New York: Raven Press; 1988. pp. 135–159. [Google Scholar]
- 33.Bither PP, Berns LA. Dominant inheritance of Stargardt's disease. J Am Optom Assoc. 1988;59:112–117. [PubMed] [Google Scholar]
- 34.Puech B, Hache JC, Turut P, et al. X-shaped macular dystrophy with flavimaculatus flecks. Ophthalmologica. 1989;199:146–157. doi: 10.1159/000310033. [DOI] [PubMed] [Google Scholar]
- 35.Lopez PF, Maumenee IH, de la Cruz Z, et al. Autosomal-dominant fundus flavimaculatus. Ophthalmology. 1990;97:798–809. doi: 10.1016/s0161-6420(90)32508-3. [DOI] [PubMed] [Google Scholar]
- 36.Mansour AM. Long-term follow-up of dominant macular dystrophy with flecks (Stargardt) Ophthalmologica. 1992;205:138–143. doi: 10.1159/000310329. [DOI] [PubMed] [Google Scholar]
- 37.Stargardt K. Über familiare, progressive Degeneration in der Makulagegend des Auges. Albrecht von Graefes Arch Klin Exp Ophthal. 1909;71:534–549. [Google Scholar]
- 38.Birnbach CD, Järveläinen M, Possin DE, et al. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994;101:1211–1219. doi: 10.1016/s0161-6420(13)31725-4. [DOI] [PubMed] [Google Scholar]
- 39.Franceschetti A. Über tapeto-retinale Degeneration im Kindesalter. In: Sautter H, editor. Entwicklung und Fortschritt in der Augenheilkunde. Enke, Stuttgart: 1963. pp. 107–120. [Google Scholar]
- 40.Franceschetti A, Francois J. Fundus flavimaculatus. Arch Ophtalmol Rev Gen Ophtalmol. 1965;25:505–530. [PubMed] [Google Scholar]
- 41.Kaplan J, Gerber S, Larget-Piet D, et al. A gene for Stargardt's disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet. 1993;5:308–311. doi: 10.1038/ng1193-308. [DOI] [PubMed] [Google Scholar]
- 42.Gerber S, Rozet JM, Bonneau D, et al. A gene for late-onset fundus flavimaculatus with macular dystrophy maps to chromosome 1p13. Am J Hum Genet. 1995;56:396–399. [PMC free article] [PubMed] [Google Scholar]
- 43.Allikmets R. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;17:122. doi: 10.1038/ng0997-122a. [DOI] [PubMed] [Google Scholar]
- 44.Stone EM, Webster AR, Vandenburgh K, et al. Allelic variation in ABCR associated with Stargardt disease but not age-related macular degeneration. Nat Genet. 1998;20:328–329. doi: 10.1038/3798. [DOI] [PubMed] [Google Scholar]
- 45.Illing M, Molday LL, Molday RS. The 220-kDa rim protein of retinal rod OSs is a member of the ABC transporter superfamily. J Biol Chem. 1997;272:10303–10310. doi: 10.1074/jbc.272.15.10303. [DOI] [PubMed] [Google Scholar]
- 46.Azarian SM, Travis GH. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardts-disease (ABCR) FEBS Lett. 1997;409:247–252. doi: 10.1016/s0014-5793(97)00517-6. [DOI] [PubMed] [Google Scholar]
- 47.Koenekoop RK. The gene for Stargardt disease, ABCA4, is a major retinal gene: a mini-review. Ophthalmic Genet. 2003;24:75–80. doi: 10.1076/opge.24.2.75.13996. [DOI] [PubMed] [Google Scholar]
- 48.Martinez-Mir A, Bayes M, Vilageliu L, et al. A new locus for autosomal recessive retinitis pigmentosa (RP19) maps to 1p13–1p21. Genomics. 1997;40:142–146. doi: 10.1006/geno.1996.4528. [DOI] [PubMed] [Google Scholar]
- 49.Martinez-Mir A, Paloma E, Allikmets R, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18:11–12. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 50.Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt disease gene ABCR. Hum Mol Genet. 1998;7:355–362. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 51.Maugeri A, Klevering BJ, Rohrschneider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000;67:960–966. doi: 10.1086/303079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weng J, Mata NL, Azarian SM, et al. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 53.Radu RA, Mata NL, Nusinowitz S, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt's macular degeneration. Proc Natl Acad Sci USA. 2003;100:4742–4747. doi: 10.1073/pnas.0737855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA. 2000;97:7154–7159. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Donoso LA, Frost AT, Stone EM, et al. Autosomal dominant Stargardt-like macular dystrophy: founder effect and reassessment of genetic heterogeneity. Arch Ophthalmol. 2001;119:564–570. doi: 10.1001/archopht.119.4.564. [DOI] [PubMed] [Google Scholar]
- 56.Vasireddy V, Uchida Y, Salem N, et al. Loss of functional ELOVL4 depletes very long-chain fatty acids (≥C28) and the unique ω-O-acylceramides in skin leading to neonatal death. Hum Mol Genet. 2007;16:471–482. doi: 10.1093/hmg/ddl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sorsby A, Mason MEJ, Gardener N. A fundus dystrophy with unusual features. Br J Ophthalmol. 1949;33:67–97. doi: 10.1136/bjo.33.2.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gregory-Evans K. What is Sorsby's fundus dystrophy? Br J Ophthalmol. 2000;84:679–680. doi: 10.1136/bjo.84.7.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamilton WK, Ewing CC, Ives EJ, et al. Sorsby's fundus dystrophy. Ophthalmology. 1989;96:1755–1762. doi: 10.1016/s0161-6420(89)32647-9. [DOI] [PubMed] [Google Scholar]
- 60.Weisinger HS, Pesudovs K. Sorsby's fundus dystrophy. Optometry. 2001;72:435–440. [PubMed] [Google Scholar]
- 61.Li CM, Chung BH, Presley JB, et al. Lipoprotein-like particles and cholesteryl esters in human Bruch's membrane: initial characterization. Invest Ophthalmol Vis Sci. 2005;46:2576–2586. doi: 10.1167/iovs.05-0034. [DOI] [PubMed] [Google Scholar]
- 62.Tymms MJ. Sorsby's fundus dystrophy: what does TIMP3 tell us about general mechanisms underlying macular degeneration? Clin Exp Optom. 1999;82:124–129. doi: 10.1111/j.1444-0938.1999.tb06789.x. [DOI] [PubMed] [Google Scholar]
- 63.Weber BH, Vogt G, Pruett RC. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby's fundus dystrophy. Nat Genet. 1994;8:352–356. doi: 10.1038/ng1294-352. [DOI] [PubMed] [Google Scholar]
- 64.Weber BH, Vogt G, Wolz W, et al. Sorsby's fundus dystrophy is genetically linked to chromosome 22q13-qter. Nat Genet. 1994;7:158–161. doi: 10.1038/ng0694-158. [DOI] [PubMed] [Google Scholar]
- 65.Della NG, Campochiaro PA, Zack DJ. Localization of TIMP-3 mRNA expression to the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1996;37:1921–1924. [PubMed] [Google Scholar]
- 66.Felbor U, Doepner D, Schneider U, et al. Evaluation of the gene encoding the tissue inhibitor of metalloproteinases-3 in various maculopathies. Invest Ophthalmol Vis Sci. 1997;38:1054–1059. [PubMed] [Google Scholar]
- 67.Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000;1477:267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- 68.Felbor U, Stöhr H, Amann T, et al. A novel Ser156Cys mutation in the tissue inhibitor of metalloproteinases-3 (TIMP3) in Sorsby's fundus dystrophy with unusual clinical features. Hum Mol Genet. 1995;4:2415–2416. doi: 10.1093/hmg/4.12.2415. [DOI] [PubMed] [Google Scholar]
- 69.Jacobson SG, Cideciyan AV, Regunath G, et al. Night blindness in Sorsby's fundus dystrophy reversed by vitamin A. Nat Genet. 1995;11:27–32. doi: 10.1038/ng0995-27. [DOI] [PubMed] [Google Scholar]
- 70.Jacobson SG, Cideciyan AV, Bennett J, et al. Novel mutation in the TIMP3 gene causes Sorsby fundus dystrophy. Arch Ophthalmol. 2002;120:376–379. doi: 10.1001/archopht.120.3.376. [DOI] [PubMed] [Google Scholar]
- 71.Langton KP, McKie N, Curtis A, et al. A novel tissue inhibitor of metalloproteinases-3 mutation reveals a common molecular phenotype in Sorsby's fundus dystrophy. J Biol Chem. 2000;275:27027–27031. doi: 10.1074/jbc.M909677199. [DOI] [PubMed] [Google Scholar]
- 72.Tabata Y, Isashiki Y, Kamimura K, et al. A novel splice site mutation in the tissue inhibitor of the metalloproteinases-3 gene in Sorsby's fundus dystrophy with unusual clinical features. Hum Genet. 1998;103:179–182. doi: 10.1007/pl00008707. [DOI] [PubMed] [Google Scholar]
- 73.Chong NH, Alexander RA, Gin T. TIMP-3, collagen, and elastin immunohistochemistry and histopathology of Sorsby's fundus dystrophy. Invest Ophthalmol Vis Sci. 2000;41:898–902. [PubMed] [Google Scholar]
- 74.Fariss RN, Apte SS, Olsen BR. Tissue inhibitor of metalloproteinases-3 is a component of Bruch's membrane of the eye. Am J Pathol. 1997;150:323–328. [PMC free article] [PubMed] [Google Scholar]
- 75.Majid MA, Smith VA, Easty DL. Sorsby's fundus dystrophy mutant tissue inhibitors of metalloproteinase-3 induce apoptosis of retinal pigment epithelial and MCF-7 cells. FEBS Lett. 2002;529:281–285. doi: 10.1016/s0014-5793(02)03359-8. [DOI] [PubMed] [Google Scholar]
- 76.Kamei M, Hollyfield JG. TIMP-3 in Bruch's membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999;40:2367–2375. [PubMed] [Google Scholar]
- 77.Langton KP, Barker MD, McKie N. Localization of the functional domains of human tissue inhibitor of metalloproteinases-3 and the effects of a Sorsby's fundus dystrophy mutation. J Biol Chem. 1998;273:16778–16781. doi: 10.1074/jbc.273.27.16778. [DOI] [PubMed] [Google Scholar]
- 78.Lee MH, Dodds P, Verma V, et al. Tailoring tissue inhibitor of metalloproteinases-3 to overcome the weakening effects of the cysteine-rich domains of tumour necrosis factor-alpha converting enzyme. Biochem J. 2003;371:369–376. doi: 10.1042/BJ20021538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamada E, Tobe T, Yamada H, et al. TIMP-1 promotes VEGF-induced neovascularization in the retina. Histol Histopathol. 2001;16:87–97. doi: 10.14670/HH-16.87. [DOI] [PubMed] [Google Scholar]
- 80.Leco KJ, Waterhouse P, Sanchez OH, et al. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3) J Clin Invest. 2001;108:817–829. doi: 10.1172/JCI12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weber BH, Lin B, White K, et al. A mouse model for Sorsby fundus dystrophy. Invest Ophthalmol Vis Sci. 2002;43:2732–2740. [PubMed] [Google Scholar]
- 82.Leibowitz HM, Krueger DE, Maunder LR, et al. The Framingham Eye Study monograph: an ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973–1975. Surv Ophthalmol. 1980;24:335–610. [PubMed] [Google Scholar]
- 83.Rosenberg T, Klie F. The incidence of registered blindness caused by age-related macular degeneration. Acta Ophthalmol Scand. 1996;74:399–402. doi: 10.1111/j.1600-0420.1996.tb00717.x. [DOI] [PubMed] [Google Scholar]
- 84.Evans J. Causes of blindness and partial sight in England and Wales 1990–1991: studies on medical and population subjects. No 57. London: HMSO; 1995. [Google Scholar]
- 85.Balatsoukas DD, Sioulis C, Parisi A, et al. Visual handicap in south-east Scotland. J R Coll Surg Edinb. 1995;40:49–51. [PubMed] [Google Scholar]
- 86.Owen CG, Fletcher AE, Donoghue M, et al. How big is the burden of visual loss caused by age related macular degeneration in the United Kingdom? Br J Ophthalmol. 2003;87:312–317. doi: 10.1136/bjo.87.3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.la Cour M, Kiilgaard JF, Nissen MH. Age-related macular degeneration: epidemiology and optimal treatment. Drugs Aging. 2002;19:101–133. doi: 10.2165/00002512-200219020-00003. [DOI] [PubMed] [Google Scholar]
- 88.Lotery A, Trump D. Progress in defining the molecular biology of age related macular degeneration. Hum Genet. 2007;122:219–236. doi: 10.1007/s00439-007-0406-3. [DOI] [PubMed] [Google Scholar]
- 89.Evans J, Wormald R. Is the incidence of registrable age-related macular degeneration increasing? Br J Ophthalmol. 1996;80:9–14. doi: 10.1136/bjo.80.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arnold JJ, Sarks SH. Extracts from “clinical evidence”: age related macular degeneration. BMJ. 2000;321:741–744. doi: 10.1136/bmj.321.7263.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feher J, Kovacs B, Kovacs I, et al. Improvement of visual functions and fundus alterations in early age-related macular degeneration treated with a combination of acetyl-L-carnitine, n-3 fatty acids, and coenzyme Q10. Ophthalmologica. 2005;219:154–166. doi: 10.1159/000085248. [DOI] [PubMed] [Google Scholar]
- 93.Fehér J, Kovács B, Kovács I, et al. Metabolic therapy for early treatment of age-related macular degeneration. Orv Hetil. 2007;148:2259–2268. doi: 10.1556/OH.2007.28250. [DOI] [PubMed] [Google Scholar]
- 94.Smith W, Mitchell P. Family history and age-related maculopathy: the Blue Mountains Eye Study. Aust Nz J Ophthalmol. 1998;26:203–206. doi: 10.1111/j.1442-9071.1998.tb01311.x. [DOI] [PubMed] [Google Scholar]
- 95.Hyman L, Neborsky R. Risk factors for age-related macular degeneration: an update. Curr Opin Ophthalmol. 2002;13:171–175. doi: 10.1097/00055735-200206000-00007. [DOI] [PubMed] [Google Scholar]
- 96.Meyers SM. A twin study on age-related macular degeneration. Trans Am Ophthalmol Soc. 1994;92:775–843. [PMC free article] [PubMed] [Google Scholar]
- 97.Klein ML, Francis PJ. Genetics of age-related macular degeneration. Ophthalmol Clin North Am. 2003;16:567–574. doi: 10.1016/s0896-1549(03)00063-4. [DOI] [PubMed] [Google Scholar]
- 98.Seddon JM, Santangelo SL, Book K, et al. A genomewide scan for age-related macular degeneration provides evidence for linkage to several chromosomal regions. Am J Hum Genet. 2003;73:780–790. doi: 10.1086/378505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seddon JM, Cote J, Page WF, et al. The US twin study of age-related macular degeneration: relative roles of genetic and environmental influences. Arch Ophthalmol. 2005;123:321–327. doi: 10.1001/archopht.123.3.321. [DOI] [PubMed] [Google Scholar]
- 100.Hammond CJ, Webster AR, Snieder H, et al. Genetic influence on early age-related maculopathy: a twin study. Ophthalmology. 2002;109:730–736. doi: 10.1016/s0161-6420(01)01049-1. [DOI] [PubMed] [Google Scholar]
- 101.Klein ML, Schultz DW, Edwards A, et al. Age-related macular degeneration. Clinical features in a large family and linkage to chromosome 1q. Arch Ophthalmol. 1998;116:1082–1088. doi: 10.1001/archopht.116.8.1082. [DOI] [PubMed] [Google Scholar]
- 102.Weeks DE, Conley YP, Tsai HJ, et al. Age-related maculopathy: an expanded genome-wide scan with evidence of susceptibility loci within the 1q31 and 17q25 regions. Am J Ophthalmol. 2001;132:682–692. doi: 10.1016/s0002-9394(01)01214-4. [DOI] [PubMed] [Google Scholar]
- 103.Weeks DE, Conley YP, Mah TS, et al. A full genome scan for age-related maculopathy. Hum Mol Genet. 2000;9:1329–1349. doi: 10.1093/hmg/9.9.1329. [DOI] [PubMed] [Google Scholar]
- 104.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kanda A, Chen W, Othman M, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci USA. 2007;104:16227–16232. doi: 10.1073/pnas.0703933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Patel N, Adewoyin T, Chong NV. Age-related macular degeneration: a perspective on genetic studies. Eye. 2008;22:768–776. doi: 10.1038/sj.eye.6702844. [DOI] [PubMed] [Google Scholar]
- 108.Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mullins RF, Russell SR, Anderson DH, et al. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–846. [PubMed] [Google Scholar]
- 110.Hageman GS, Luthert PJ, Victor Chong NH, et al. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 111.Johnson LV, Leitner WP, Staples MK, et al. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- 112.Sivaprasad S, Chong NV. The complement system and age-related macular degeneration. Eye. 2006;20:867–872. doi: 10.1038/sj.eye.6702176. [DOI] [PubMed] [Google Scholar]
- 113.Hughes AE, Orr N, Esfandiary H, et al. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–1177. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 114.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Edwards AO, Ritter Iii R, Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 116.Zareparsi S, Branham KE, Li M, et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet. 2005;77:149–153. doi: 10.1086/431426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Conley YP, Thalamuthu A, Jakobsdottir J, et al. Candidate gene analysis suggests a role for fatty acid biosynthesis and regulation of the complement system in the etiology of age-related maculopathy. Hum Mol Genet. 2005;14:1991–2002. doi: 10.1093/hmg/ddi204. [DOI] [PubMed] [Google Scholar]
- 118.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 119.Gotoh N, Yamada R, Hiratani H, et al. No association between complement factor H gene polymorphism and exudative age-related macular degeneration in Japanese. Hum Genet. 2006;120:1–5. doi: 10.1007/s00439-006-0187-0. [DOI] [PubMed] [Google Scholar]
- 120.Lau LI, Chen SJ, Cheng CY, et al. Association of the Y402H polymorphism in Complement factor H Gene and neovascular age-related macular degeneration in Chinese patients. Invest Ophthalmol Vis Sci. 2006;47:3242–3246. doi: 10.1167/iovs.05-1532. [DOI] [PubMed] [Google Scholar]
- 121.Ennis S, Jomary C, Mullins R, et al. Association between the SERPING1 gene and age-related macular degeneration: a two-stage case–control study. Lancet. 2008;372:1828–1834. doi: 10.1016/S0140-6736(08)61348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Conley YP, Jakobsdottir J, Mah T, et al. CFH, ELOVL4, PLEKHA1, and LOC387715 genes and susceptibility to age-related maculopathy: AREDS and CHS cohorts and meta-analyses. Hum Mol Genet. 2006;15:3206–3218. doi: 10.1093/hmg/ddl396. [DOI] [PubMed] [Google Scholar]
- 123.Schmidt S, Hauser MA, Scott WK, et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006;78:852–864. doi: 10.1086/503822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Penfold P, Liew S, Madigan M, et al. Modulation of major histocompatibility complex class II expression in retinas with age-related macular degeneration. Invest Ophthalmol Vis Sci. 1997;38:2125–2133. [PubMed] [Google Scholar]
- 125.Shroyer NF, Lewis RA, Lupski JR. Analysis of the ABCR (ABCA4) gene in 4-aminoquinoline retinopathy: is retinal toxicity by chloroquine and hydroxychloroquine related to Stargardt disease. Am J Ophthal. 2001;131:761–766. doi: 10.1016/s0002-9394(01)00838-8. [DOI] [PubMed] [Google Scholar]
- 126.Lakkaraju A, Finnemann SC, Rodriguez-Boulan E. The lipofuscin fluorophore A2E perturbs cholesterol metabolism in retinal pigment epithelial cells. Proc Natl Acad Sci USA. 2007;104:11026–11031. doi: 10.1073/pnas.0702504104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Curcio CA, Presley JB, Malek G, et al. Esterified and unesterified cholesterol in drusen and basal deposits of eyes with age-related maculopathy. Exp Eye Res. 2005;81:731–741. doi: 10.1016/j.exer.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 128.Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- 129.Stone EM, Braun TA, Russell SR, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346–353. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- 130.de la Paz MA, Pericak-Vance MA, Lennon F, et al. Exclusion of TIMP3 as a candidate locus in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1997;38:1060–1065. [PubMed] [Google Scholar]
- 131.Duvall J, McKechnie NM, Lee WR, et al. Extensive subretinal pigment epithelial deposit in two brothers suffering from dominant retinitis pigmentosa. A histopathological study. Graefes Arch Clin Exp Ophthal. 1986;224:299–309. doi: 10.1007/BF02143075. [DOI] [PubMed] [Google Scholar]
- 132.Milam AH, Curcio CA, Cideciyan AV, et al. Dominant late-onset retinal degeneration with regional variation of sub-retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology. 2000;107:2256–2266. doi: 10.1016/s0161-6420(00)00419-x. [DOI] [PubMed] [Google Scholar]
- 133.Jacobson SG, Cideciyan AV, Wright E, et al. Phenotypic marker for early disease detection in dominant late-onset retinal degeneration. Invest Ophthal Vis Sci. 2001;42:1882–1890. [PubMed] [Google Scholar]
- 134.Hayward C, Shu X, Cideciyan AV, et al. Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: a genetic model for age-related macular degeneration. Hum Mol Genet. 2003;12:2657–2667. doi: 10.1093/hmg/ddg289. [DOI] [PubMed] [Google Scholar]
- 135.Klaver CC, Kliffen M, van Duijn CM, et al. Genetic association of apolipoprotein E with age-related macular degeneration. Am J Hum Genet. 1998;63:200–206. doi: 10.1086/301901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Anderson DH, Talaga KC, Rivest AJ, et al. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004;78:243–256. doi: 10.1016/j.exer.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 137.Baird PN, Guida E, Chu DT, et al. The epsilon2 and epsilon4 alleles of the apolipoprotein gene are associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:1311–1315. doi: 10.1167/iovs.03-1121. [DOI] [PubMed] [Google Scholar]
- 138.Zareparsi S, Reddick AC, Branham KE, et al. Association of apolipoprotein E alleles with susceptibility to age-related macular degeneration in a large cohort from a single center. Invest Ophthalmol Vis Sci. 2004;45:1306–1310. doi: 10.1167/iovs.03-1253. [DOI] [PubMed] [Google Scholar]
- 139.Roca-Santiago HM, Lago-Bouza JR, Millan-Calenti JC, et al. Alzheimer's disease and age-related macular degeneration. Arch Soc Esp Oftalmol. 2006;81:73–78. doi: 10.4321/s0365-66912006000200006. [DOI] [PubMed] [Google Scholar]
- 140.Baker ML, Wang JJ, Rogers S, et al. Early age-related macular degeneration, cognitive function, and dementia: the Cardiovascular Health Study. Arch Ophthalmol. 2009;127:667–673. doi: 10.1001/archophthalmol.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Allikmets R, Seddon JM, Bernstein PS, et al. Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies. Hum Genet. 1999;104:449–453. doi: 10.1007/s004390050986. [DOI] [PubMed] [Google Scholar]
- 142.Lotery AJ, Munier FL, Fishman GA, et al. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41:1291–1296. [PubMed] [Google Scholar]
- 143.Haines JL, Schnetz-Boutaud N, Schmidt S, et al. Functional candidate genes in age-related macular degeneration: significant association with VEGF, VLDLR, and LRP6. Invest Ophthalmol Vis Sci. 2006;47:329–335. doi: 10.1167/iovs.05-0116. [DOI] [PubMed] [Google Scholar]
- 144.Chang B, Mandal MN, Chavali VR, et al. Age-related retinal degeneration (arrd2) in a novel mouse model due to a nonsense mutation in the Mdm1 gene. Hum Mol Genet. 2008;17:3929–3941. doi: 10.1093/hmg/ddn295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Imamura Y, Noda S, Hashizume K, et al. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc Natl Acad Sci USA. 2006;103:11282–11287. doi: 10.1073/pnas.0602131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Malek G, Johnson LV, Mace BE, et al. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci USA. 2005;102:11900–11905. doi: 10.1073/pnas.0503015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vollrath D, Feng W, Duncan JL, et al. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Natl Acad Sci USA. 2001;98:12584–12589. doi: 10.1073/pnas.221364198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Eng J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 150.Cideciyan AV, Aleman TS, Boye SL, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Buch PK, MacLaren RE, Ali RR. Neuroprotective gene therapy for the treatment of inherited retinal degeneration. Curr Gene Ther. 2007;7:434–445. doi: 10.2174/156652307782793531. [DOI] [PubMed] [Google Scholar]
- 152.Seiler M, Aramant RB, Ehinger B, et al. Transplantation of embryonic retina to adult retina in rabbits. Exp Eye Res. 1990;51:225–228. doi: 10.1016/0014-4835(90)90076-7. [DOI] [PubMed] [Google Scholar]
- 153.Gouras P, Du J, Kjeldbye H, et al. Long-term photoreceptor transplants in dystrophic and normal mouse retina. Invest Ophthalmol Vis Sci. 1994;35:3145–3153. [PubMed] [Google Scholar]
- 154.Young MJ, Ray J, Whiteley SJ, et al. Neuronal differentiation and morphological integration of hippocampal progenitor cells transplanted to the retina of immature and mature dystrophic rats. Mol Cell Neurosci. 2000;16:197–205. doi: 10.1006/mcne.2000.0869. [DOI] [PubMed] [Google Scholar]
- 155.Chacko DM, Rogers JA, Turner JE, et al. Survival and differentiation of cultured retinal progenitors transplanted in the subretinal space of the rat. Biochem Biophys Res Commun. 2000;268:842–846. doi: 10.1006/bbrc.2000.2153. [DOI] [PubMed] [Google Scholar]
- 156.Radtke ND, Aramant RB, Petry HM, et al. Vision improvement in retinal degeneration patients by implantation of retina together with retinal pigment epithelium. Am J Ophthalmol. 2008;146:172–182. doi: 10.1016/j.ajo.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 157.Vugler A, Lawrence J, Walsh J, et al. Embryonic stem cells and retinal repair. Mech Dev. 2007;124:807–829. doi: 10.1016/j.mod.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 158.MacLaren RE, Pearson RA, MacNeil A, et al. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006;444:203–207. doi: 10.1038/nature05161. [DOI] [PubMed] [Google Scholar]
- 159.Ikeda H, Osakada F, Watanabe K, et al. Generation of Rx+/Pax6+ neural retinal precursors from embryonic stem cells. Proc Natl Acad Sci USA. 2005;102:11331–11336. doi: 10.1073/pnas.0500010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Merhi-Soussi F, Angénieux B, Canola K, et al. High yield of cells committed to the photoreceptor fate from expanded mouse retinal stem cells. Stem Cells. 2006;24:2060–2070. doi: 10.1634/stemcells.2005-0311. [DOI] [PubMed] [Google Scholar]
- 161.Osakada F, Ikeda H, Mandai M, et al. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol. 2008;26:215–224. doi: 10.1038/nbt1384. [DOI] [PubMed] [Google Scholar]
- 162.Carr AJ, Vugler A, Lawrence J, et al. Molecular characterization and functional analysis of phagocytosis by human embryonic stem cell-derived RPE cells using a novel human retinal assay. Mol Vis. 2009;15:283–295. [PMC free article] [PubMed] [Google Scholar]
- 163.Banin E, Obolensky A, Idelson M, et al. Retinal incorporation and differentiation of neural precursors derived from human embryonic stem cells. Stem Cells. 2006;24:246–257. doi: 10.1634/stemcells.2005-0009. [DOI] [PubMed] [Google Scholar]
- 164.Lamba DA, Gust J, Reh TA. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell. 2009;9(4):73–79. doi: 10.1016/j.stem.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Humayun MS, de Juan E, Jr., del Cerro M, et al. Human neural retinal transplantation. Invest Ophthalmol Vis Sci. 2000;41:3100–3106. [PubMed] [Google Scholar]
- 166.Kaplan HJ, Tezel TH, Berger AS, et al. Human photoreceptor transplantation in retinitis pigmentosa. A safety study. Arch Ophthalmol. 1997;115:1168–1172. doi: 10.1001/archopht.1997.01100160338012. [DOI] [PubMed] [Google Scholar]
- 167.Kaplan HJ, Tezel TH, Berger AS, et al. Retinal transplantation. Chem Immunol. 1999;73:207–219. doi: 10.1159/000058747. [DOI] [PubMed] [Google Scholar]
- 168.Early Treatment Diabetic Retinopathy Study (ETDRS) Available at http://clinicaltrials.gov/ct2/show/NCT00000151. Accessed December 3, 2008.
- 169.Radtke ND, Aramant RB, Seiler MJ, et al. Vision change after sheet transplant of foetal retina with retinal pigment epithelium to a patient with retinitis pigmentosa. Arch Ophthalmol. 2004;122:1159–1165. doi: 10.1001/archopht.122.8.1159. [DOI] [PubMed] [Google Scholar]
- 170.Silverman MS, Hughes SE. Photoreceptor rescue in the RCS rat without pigment epithelium transplantation. Curr Eye Res. 1990;9:183–191. doi: 10.3109/02713689008995205. [DOI] [PubMed] [Google Scholar]
- 171.Del Priore LV, Geng L, Tezel TH, et al. Extracellular matrix ligands promote RPE attachment to inner Bruch's membrane. Curr Eye Res. 2002;25:79–89. doi: 10.1076/ceyr.25.2.79.10158. [DOI] [PubMed] [Google Scholar]
- 172.Tezel TH, Kaplan HJ, Del Priore LV. Fate of human retinal pigment epithelial cells seeded onto layers of human Bruch's membrane. Invest Ophthalmol Vis Sci. 1999;40:467–476. [PubMed] [Google Scholar]
- 173.Gullapalli VK, Sugino IK, Van Patten Y, et al. Impaired RPE survival on aged submacular human Bruch's membrane. Exp Eye Res. 2005;80:235–248. doi: 10.1016/j.exer.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 174.Radtke ND, Seiler MJ, Aramant RB, Petry HM, Pidwell DJ. Transplantation of intact sheets of foetal neural retina with its retinal pigment epithelium in retinitis pigmentosa patients. Am J Ophthalmol. 2002;133:544–550. doi: 10.1016/s0002-9394(02)01322-3. [DOI] [PubMed] [Google Scholar]
- 175.Algvere PV, Berglin L, Gouras P, et al. Transplantation of RPE in age-related macular degeneration: observations in disciform lesions and dry RPE atrophy. Graefes Arch Clin Exp Ophthalmol. 1997;235:149–158. doi: 10.1007/BF00941722. [DOI] [PubMed] [Google Scholar]
- 176.Algvere P, Berglin L, Gouras P, et al. Transplantation of foetal retinal pigment epithelium in age-related macular degeneration with subfoveal neovascularization. Graefes Arch Clin Exp Ophthalmol. 1994;232:707–716. doi: 10.1007/BF00184273. [DOI] [PubMed] [Google Scholar]
- 177.Algvere PV, Gouras P, Dafgard Kopp E. Long-term outcome of RPE allografts in non-immunosuppressed patients with AMD. Eur J Ophthalmol. 1999:217–230. doi: 10.1177/112067219900900310. [DOI] [PubMed] [Google Scholar]
- 178.Weisz JM, Humayun MS, De Juan E, Jr, et al. Allogenic foetal retinal pigment epithelial cell transplant in a patient with geographic atrophy. Retina. 1999;19:540–545. doi: 10.1097/00006982-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 179.Das T, del Cerro M, Jalali S, et al. The transplantation of human foetal neuroretinal cells in advanced retinitis pigmentosa patients: results of a long-term safety study. Exp Neurol. 1999;157:58–68. doi: 10.1006/exnr.1998.6992. [DOI] [PubMed] [Google Scholar]
- 180.Zarbin MA. Retinal pigment epithelium-retina transplantation for retinal degenerative disease. Am J Ophthalmol. 2008;146:151–153. doi: 10.1016/j.ajo.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Del Priore LV, Tezel TH, Kaplan HJ. Maculoplasty for age-related macular degeneration: reengineering Bruch's membrane and the human macula. Prog Ret Eye Res. 2006;25:539–562. doi: 10.1016/j.preteyeres.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 182.Tao S, Young C, Redenti S, et al. Survival, migration and differentiation of retinal progenitor cells transplanted on micro-machined poly(methyl methacrylate) scaffolds to the subretinal space. Lab Chip. 2007;7:695–701. doi: 10.1039/b618583e. [DOI] [PubMed] [Google Scholar]
- 183.Lamba DA, Karl MO, Ware CB, et al. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci USA. 2006;22(103):12769–12774. doi: 10.1073/pnas.0601990103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lowry WE, Richter L, Yachechko R, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci USA. 2008;105:2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Park IH, Lerou PH, Zhao R, et al. Generation of human-induced pluripotent stem cells. Nat Protoc. 2008;3:1180–1186. doi: 10.1038/nprot.2008.92. [DOI] [PubMed] [Google Scholar]
- 186.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 187.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 188.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 189.Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009 doi: 10.1126/science.1172482. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhou H, Wu S, Joo JY, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 192.Hanna J, Wernig M, Markoulaki S, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 193.Wernig M, Zhao JP, Pruszak J, et al. Neurons derived from reprogrammed fibroblasts functionally integrate into the foetal brain and improve symptoms of rats with Parkinson's disease. Proc Natl Acad Sci USA. 2008;105:5856–5861. doi: 10.1073/pnas.0801677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liu H, Zhu F, Yong J, et al. Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell. 2008;3:587–590. doi: 10.1016/j.stem.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 195.Dimos JT, Rodolfa KT, Niakan KK, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 196.Soldner F, Hockemeyer D, Beard C, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tateishi K, He J, Taranova O, et al. Generation of insulin-secreting islet-like clusters from human skin fibroblasts. J Biol Chem. 2008;283:31601–31607. doi: 10.1074/jbc.M806597200. [DOI] [PubMed] [Google Scholar]
- 198.Ebert AD, Yu J, Rose FF, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Park IH, Arora N, Huo H, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Choi KD, Yu J, Smuga-Otto K, et al. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells. 2009;27:559–567. doi: 10.1634/stemcells.2008-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Karumbayaram S, Novitch BG, Patterson M, et al. Directed differentiation of human-induced pluripotent stem cells generates active motor neurons. Stem Cells. 2009;27:806–811. doi: 10.1002/stem.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Schenke-Layland K, Rhodes KE, Angelis E, et al. Reprogrammed mouse fibroblasts differentiate into cells of the cardiovascular and hematopoietic lineages. Stem Cells. 2008;26:1537–1546. doi: 10.1634/stemcells.2008-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hirami Y, Osakada F, Takahashi K, et al. Generation of retinal cells from mouse and human induced pluripotent stem cells. Neurosci Lett. 2009;458:126–131. doi: 10.1016/j.neulet.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 204.Tuo J, Bojanowski CM, Zhou M, et al. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–3836. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lensch MW, Daley GQ. Scientific and clinical opportunities for modeling blood disorders with embryonic stem cells. Blood. 2006;107:2605–2612. doi: 10.1182/blood-2005-07-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kong J, Kim SR, Binley K, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15:1311–1320. doi: 10.1038/gt.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Grayson C, Molday RS. Dominant negative mechanism underlies autosomal dominant Stargardt-like macular dystrophy linked to mutations in ELOVL4. J Biol Chem. 2005;280:32521–32530. doi: 10.1074/jbc.M503411200. [DOI] [PubMed] [Google Scholar]
- 208.Li Z, Clarke MP, Barker MD, et al. TIMP3 mutation in Sorsby's fundus dystrophy: molecular insights. Expert Rev Mol Med. 2005;7:1–15. doi: 10.1017/S1462399405010045. [DOI] [PubMed] [Google Scholar]
- 209.Lombardo A, Genovese P, Beausejour CM, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 210.Jones BW, Watt CB, Frederick JM, et al. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- 211.Lund RD, Ono SJ, Keegan DJ, et al. Retinal transplantation: progress and problems in clinical application. J Leukoc Biol. 2003;74:151–160. doi: 10.1189/jlb.0103041. [DOI] [PubMed] [Google Scholar]
- 212.Boulton M, Roanowska M, Wess T. Ageing of the retinal pigment epithelium: implications for transplantation. Graefes Arch Clin Exp Ophthalmol. 2004;242:76–84. doi: 10.1007/s00417-003-0812-8. [DOI] [PubMed] [Google Scholar]
- 213.Vugler A, Carr AJ, Lawrence J, et al. Elucidating the phenomenon of HESC-derived RPE: anatomy of cell genesis expansion and retinal transplantation. Exp Neurol. 2008;214:347–361. doi: 10.1016/j.expneurol.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 214.Kerrigan-Baumrind LA, Quigley HA, Pease ME, et al. Number of ganglion cells in glaucoma eyes compared with threshold visual field tests in the same persons. Invest Ophthalmol Vis Sci. 2000;41:741–748. [PubMed] [Google Scholar]
- 215.Sullivan LS, Daiger SP. Inherited retinal degeneration: exceptional genetic and clinical heterogeneity. Mol Med Today. 1996;2:380–386. doi: 10.1016/s1357-4310(96)10037-x. [DOI] [PubMed] [Google Scholar]
- 216.Rozet JM, Gerber S, Souied E, et al. Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur J Hum Genet. 1998;6:291–295. doi: 10.1038/sj.ejhg.5200221. [DOI] [PubMed] [Google Scholar]
- 217.Maugeri A, van Driel MA, van de Pol DJ, et al. The 2588GC mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet. 1999;64:1024–1035. doi: 10.1086/302323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Rivera A, White K, Stohr H, et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Human Genet. 2000;67:800–813. doi: 10.1086/303090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Simonelli F, Testa F, de Crecchio G, et al. New ABCR mutations and clinical phenotype in Italian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2000;41:892–897. [PubMed] [Google Scholar]
- 220.Michaelides M, Hunt DM, Moore AT. The genetics of inherited macular dystrophies. J Med Genet. 2003;40:641–650. doi: 10.1136/jmg.40.9.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bennett J, Tanabe T, Sun D, et al. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat Med. 1996;2:649–654. doi: 10.1038/nm0696-649. [DOI] [PubMed] [Google Scholar]
- 222.Kumar-Singh R, Farber DB. Encapsidated adenovirus minichromosome-mediated delivery of genes to the retina: application to the rescue of photoreceptor degeneration. Hum Mol Genet. 1998;7:1893–1900. doi: 10.1093/hmg/7.12.1893. [DOI] [PubMed] [Google Scholar]
- 223.Takahashi M, Miyoshi H, Verma IM, et al. Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J Virol. 1999;73:7812–7816. doi: 10.1128/jvi.73.9.7812-7816.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Koenekoop RK. Successful RPE65 gene replacement and improved visual function in humans. Ophthalmic Genet. 2008;29:89–91. doi: 10.1080/13816810802216480. [DOI] [PubMed] [Google Scholar]
- 225.Borras T. Recent developments in ocular gene therapy. Exp Eye Res. 2003;73:643–652. doi: 10.1016/s0014-4835(03)00030-7. [DOI] [PubMed] [Google Scholar]
- 226.Okita K, Nakagawa M, et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]