

Abstract
The use of functional magnetic resonance imaging (fMRI) in animal models of cocaine addiction is an invaluable tool for investigating the neuroadaptations that lead to this psychiatric disorder. We used blood-oxygen-level-dependent (BOLD) MRI in awake rats to identify the neuronal circuits affected by repeated cocaine administration. Rats were given an injection of cocaine (15 mg/kg, i.p.) or its vehicle for 7 days, abstained from injections for 1 week, and challenged with an intracerebroventricular cocaine injection during functional imaging. Acute cocaine produced robust positive BOLD responses across well-known monoamine-enriched brain regions, such as the prefrontal cortex, nucleus accumbens, dorsal striatum, sensory cortex, hippocampus, thalamus, and midbrain areas. However, repeated cocaine administration resulted in lower BOLD responses in the prefrontal cortex, agranular insular cortex, nucleus accumbens, ventral pallidum, and dorsomedial thalamus, among other brain regions. Reductions in BOLD intensity were not associated with variations in cerebrovascular reactivity between drug naïve rats and those repeatedly exposed to cocaine. Therefore, the lower metabolic activation in response to cocaine could reflect a reduced neuronal and/or synaptic activity upon repeated administration.
Keywords: cocaine, drugs of abuse, BOLD imaging, functional MRI, reward circuitry, awake rat, sensitization
INTRODUCTION
The use of animal models of cocaine addiction along with techniques to assess neuronal activity and function has aided the identification of the underlying neuropathology of this brain disorder (Bozarth et al, 1980). The behavioral sensitization model assesses the progressive enhancement of locomotor behavior upon repeated cocaine administration in rats (Post and Rose, 1976). The increased locomotor behavior produced by cocaine can persist after prolonged abstinence periods (Pierce and Kalivas, 1997; Robinson and Berridge, 1993; Vanderschuren and Kalivas, 2000). The neuroadaptations that result in behavioral sensitization in rats have been postulated to involve cocaine-induced changes in neuronal activity that lead to psychostimulant-induced psychosis, as well as pathological craving for cocaine in human addicts (Kalivas et al, 1998; Post and Rose, 1976; Robinson and Berridge, 1993). The brain circuitry mediating cocaine’s initial locomotor and reinforcing action is the mesocorticolimbic dopamine system (Goeders and Smith, 1983; Vanderschuren and Kalivas, 2000). However, this circuit is anatomically and physiologically embedded within other limbic and cortical brain regions (Pierce and Kalivas, 1997). Cocaine-induced alterations within mesocorticolimbic system, such as changes in gene expression (Graybiel et al, 1990), synaptic monoamine levels (Kalivas and Duffy, 1993), dendritic spine formation (Robinson and Kolb, 1999), and the biophysical properties of neurons (Ungless et al, 2001) could also lead to physiological alterations in other brain areas. Therefore, in order to fully identify, and understand, the functional alterations in the neural substrates targeted by cocaine, noninvasive in vivo techniques must be employed.
Cocaine’s effects on neuronal activity within the brain can be assessed in vivo using the blood-oxygen-level-dependent (BOLD) functional magnetic resonance imaging (fMRI) technique. The BOLD fMRI signal is associated with a reduction in paramagnetic deoxyhemoglobin (dHb) concentrations near sites of increased neuronal activity (Ogawa et al, 1990). Increased oxygenation of these areas contributes to the elevations in BOLD signal intensity (Thompson et al, 2003). An acute cocaine injection increases cerebral metabolic activity (Porrino, 1993) and blood flow (Stein and Fuller, 1992); effects that are likely to contribute to changes in BOLD signal intensity (Febo et al, 2004). These alterations could possibly involve effects of cocaine on synaptic monoamine levels throughout cortical and subcortical structures (Goeders and Smith, 1983; Kalivas and Duffy, 1993). In the present experiment, we used BOLD imaging to assess the effects of cocaine in fully awake rats that were drug naïve or had been previously exposed to the drug.
MATERIALS AND METHODS
Subjects
In total, 24 adult male Sprague–Dawley rats weighing 350–450 g were obtained from Charles River Laboratories (Charles River, MA). Animals were housed in pairs, maintained on 12 : 12 light : dark cycle (lights on at 0900) and provided food and water ad libitum. All animals were acquired and cared for in accordance with the guidelines published in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publications No. 85-23, Revised 1985).
Cocaine Administration
Cocaine hydrochloride was purchased from Sigma-Aldrich (St Louis, MO) and dissolved in sterile saline (0.9%). Rats were given a daily cocaine (15 mg kg−1, intraperitoneal) or saline vehicle (0.1 ml/100 g) injection for 7 consecutive days, and allowed 1 week free of injections before brain imaging experiments (Figure 1a). The i.p. cocaine pretreatment dose and injection regime has been previously reported to produce robust locomotor sensitization in rats that lasts up to 3 weeks of withdrawal (Uslaner et al, 2003). Cocaine-induced brain activation was then assessed during functional imaging following 1 week of abstinence from repeated injections. This resulted into three groups: (1) rats that received vehicle injections on all days (vehicle control; n = 4); (2) those that were given cocaine only on the day of functional imaging (acute cocaine; n = 8); and (3) those that received cocaine injections on all days (repeated cocaine; n = 8 per group) (Figure 1a).
Figure 1.
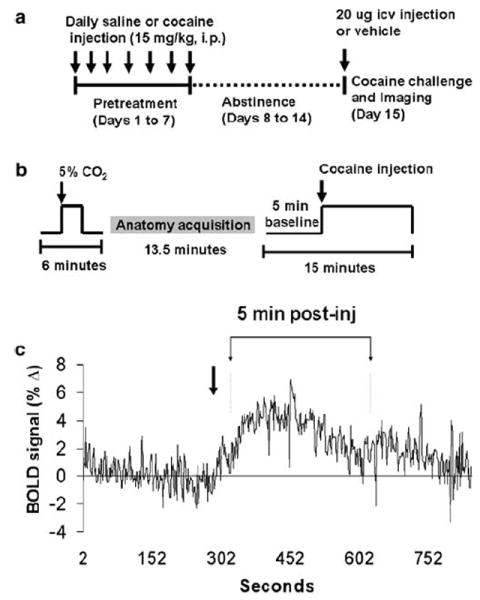
Study design. (a) Cocaine injection dose and injection paradigm used. (b) Functional imaging experimental design. (c) BOLD signal response to cocaine, as reported in Febo et al (2004), showing the timepoint at which the peak signal intensity is attained during the timeseries.
Functional Imaging
Studies were performed with a multiconcentric dual-coil, small animal restrainer developed by Insight NeuroImaging Systems, LLC, (Worcester, MA) (Ludwig et al, 2004). Prior to imaging studies, animals were acclimated to the restrainer and the imaging protocol as described previously by Febo et al (2004). Briefly, animals were lightly anesthetized with 2% isoflurane and secured into the dual-coil restrainer. When fully conscious, the restraining unit was placed into a black opaque tube that served as a ‘mock scanner’ and a tape-recording of an MRI pulse sequence played for 90 min to simulate the bore of the magnet and an imaging protocol. This procedure was repeated on 3 consecutive days. Experiments were conducted in a Bruker Biospec 4.7-T/40-cm horizontal magnet (Oxford Instrument, Oxford, UK) equipped with a Biospec Bruker console (Bruker, Billerica, MA, USA) and a 20-G/cm magnetic field gradient insert (ID = 12 cm, 120-μs rise time).
Challenge cocaine injections were made via the intracerebroventricular (ICV) route while the animal was inside the magnet. The dose of cocaine used was 20 μg dissolved in 10 μl of artificial cerebrospinal fluid. The injection route and dose have been shown to reduce cardiac and respiratory complications associated with peripheral psychostimulant administration (Febo et al, 2004). Immediately prior to imaging, rats were anesthetized under 2% isoflurane, the skull surface was exposed and the landmark suture Bregma located. A 26-gauge cannula of polyethylene tubing (PE-10: inner diameter 0.28 mm, outer diameter 0.61 mm) was implanted into the lateral cerebral ventricle (1mm caudal to Bregma, 2mm lateral to the mid-sagittal sinus, and 4mm ventral to dura) and secured to the skull with surgical glue. Cocaine injections were made via a plastic syringe connected at the end of the tubing. Cannula placement was verified prior to cocaine imaging with a short anatomical MR scan. Only animals with correct placement were included in the study.
Functional images were obtained with a spin echo EPI pulse sequence (64 × 64 data matrix, TE = 55 ms, TR = 2000 ms). Images were continuously acquired during 15 min that were divided between a 5-min baseline and a 10-min period following ICV cocaine injection (Figure 1b). High-resolution anatomical scans were collected using a fast spin echo pulse sequence (TE = 48 ms, TR = 2500 ms, FOV = 3 cm, 1.2mm slice thickness, 256 × 256 data matrix, 16 RareFactor).
In total, 20–25 min prior ICV cocaine injections, drug naïve, and cocaine pretreated rats were exposed to a 2-min 5% CO2 pulse during a short functional scan (6 min) (Figure 1b). Global cerebral blood flow can be modulated by several physiological parameters; among these is CO2 (Cohen et al, 2002; Sicard et al, 2003). In general, both cerebral blood flow and BOLD intensity increase in the presence of hypercapnic conditions (Cohen et al, 2002; Sicard et al, 2003). Thus, we used the hypercapnia stimulus as a way of assessing differences in cerebrovascular reactivity between the two treatment groups.
Data Analysis
Selection of regions of interest (ROI) and statistical analysis were performed using the Stimulate software (Strupp, 1996). Movies of functional scans were generated and carefully examined to detect gross movements (ie frequent voxel displacements during time series) and the raw data time series were analyzed for course spikes. Data from two animals in the repeated cocaine group and two in the acute group were discarded due to excess motion artifact. ROI were drawn according to the Atlas of the Rat Brain (Paxinos and Watson, 1997) and are shown in Figure 2a. Before tracing the ROI, functional scans were aligned to a reference anatomical scan using the image analysis software ImageJ (Wayne Rasband, http://rsb.info.nih.gov/ij/; Karl Schmidt http://www.quickvol.com). Each slice of the functional maps and anatomical images from each subject were manually coregistered to a reference anatomical scan using custom software and the following degrees of freedom: x and y translation, independent x and y scale, and rotation about the z-axis. The original images were then re-sampled without interpolation in the transformation to the atlas coordinate system, and a new set of images was created.
Figure 2.
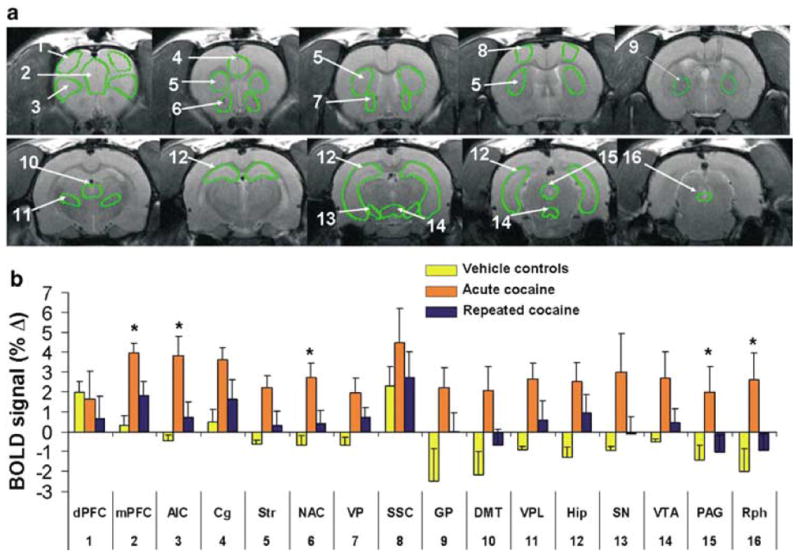
Cocaine-induced BOLD activation in drug naïve and cocaine pretreated rats. (a) Representative high-resolution anatomical images showing regions of interest. Numbers indicate the following regions: (1) dorsal prefrontal cortex (dPFC); (2) medial prefrontal cortex (mPFC); (3) agranular insular cortex (AIC); (4) anterior cingulate cortex (Cg); (5) dorsal striatum (Str); (6) nucleus accumbens (NAC); (7) ventral pallidum (VP); (8) somatosensory cortex (SSC); (9) globus pallidus (GP); (10) dorsomedial thalamus (DMT); (11) ventroposterolateral thalamus (VPL); (12) hippocampus (Hip); (13) substantia nigra (SN); (14) ventral tegmental area (VTA); (15) periaqueductal grey (PAG); (16) raphé (Rph). (b) Average BOLD responses to cocaine in brain areas depicted in (a). Data are averages of the first 5 min after ICV cocaine injections and expressed as mean±SEM. *denotes p <0.05 comparing saline vs cocaine pretreated. Numbers along the x-axis indicate the brain areas depicted in (a).
Raw signal intensity values per ROI of each animal were extracted and normalized to their time series baseline (expressed as percent change from baseline). Data for each animal was then averaged during the first 5-min post-cocaine injection in order to make statistical comparisons (Figure 1c). Statistical analysis was performed using a t-test to compare percent changes in BOLD signal intensity and activated pixels between drug naïve and cocaine pretreated rats.
In order to generate positive BOLD activation maps for visualization, the composite functional maps were subjected to a pixel-by-pixel t-test comparing signal intensity during the 5-min preinjection baseline period and 5 min immediately after cocaine injection (Figure 1c). The selected postinjection period corresponds to the timeframe where the peak BOLD response to ICV cocaine occurs (Febo et al, 2004). Pixels whose BOLD percentage change relative to the baseline period was significantly different at a 95% confidence level were overlaid onto the reference anatomical data set.
RESULTS
BOLD Response to Acute Cocaine
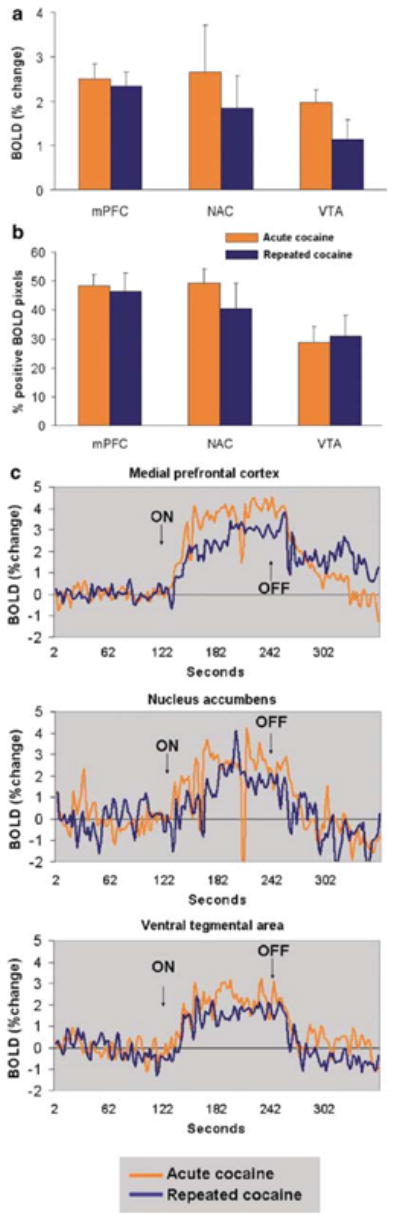
The spatial pattern of positive BOLD activation following an acute dose of cocaine resembled the distribution of monoamine synaptic terminals (Hokfelt et al, 1974; Thierry et al, 1973) and the pattern of cocaine-induced [14C]2-deoxyglucose labeling in the rat brain (Porrino, 1993). As reported previously by our laboratory, immediate and robust positive BOLD activity was observed in several cortical subregions, such as the medial prefrontal, anterior cingulate, agranular insular, primary motor, and somatosensory cortices (Figures 2 and 3) (Febo et al, 2004). Positive BOLD signal changes were also observed in limbic and extrapyramidal structures (Figures 2 and 3). The temporal activation pattern is consistent with our previous findings (Febo et al, 2004). A peak positive BOLD response of 3.2–3.9% in the nucleus accumbens and prefrontal cortex and 1.9% in the ventral tegmental area was attained by 100 s following ICV cocaine administration (Figure 4). The amount of negative BOLD pixels in response to acute cocaine was not as dramatic as with positive BOLD pixels. The percentage of negative BOLD pixels sampled per ROI is shown in Figure 5.
Figure 3.
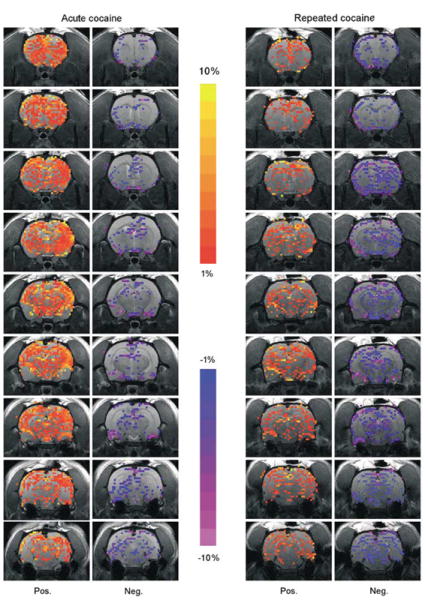
Mesocorticolimbic BOLD response to cocaine in drug naïve and cocaine pretreated rats. (a) Activation maps correspond to composite images of rats given acute cocaine (n = 8) and repeated cocaine injections (n = 8). Colored pixels represent brain areas that showed signal intensity values significantly different from baseline (scale bar hue indicates percent change value). Statistical significance was determined using pixel-by-pixel t-test analysis (Stimulate software, Strupp, 1996) comparing baseline to a cocaine injection period (α = 0.05, uncorrected).
Figure 4.
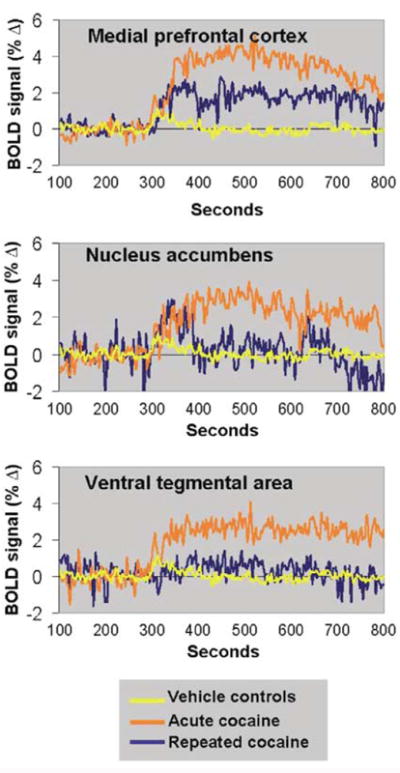
Change in BOLD signal intensity over time following ICV cocaine administration. Data in each treatment group correspond to the average BOLD response across all animals. Arrow indicates the timepoint of cocaine injection. Rats pretreated with cocaine (blue lines) showed lower BOLD responses in the mesocorticolimbic system than rats given an acute cocaine injection (orange lines) or vehicle injections (yellow lines).
Figure 5.
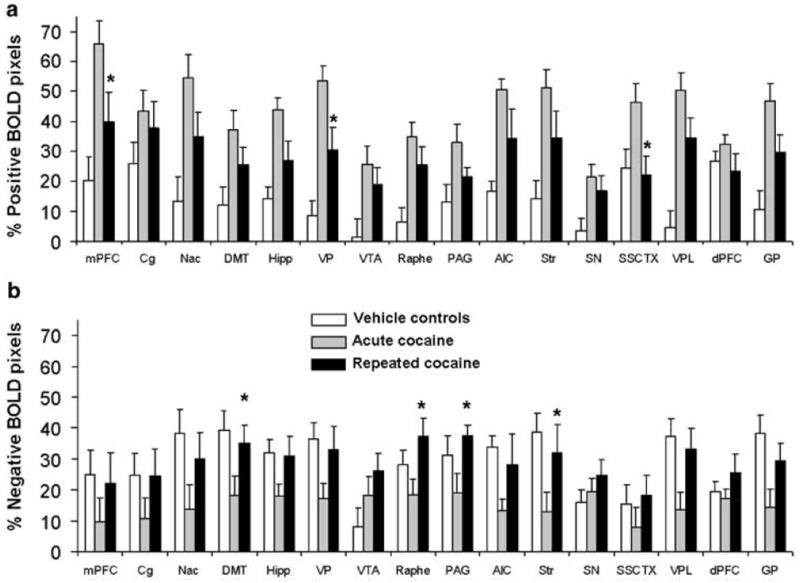
Percent positive and negative BOLD pixels following ICV cocaine injections in drug naïve and cocaine pretreated rats. (a) Positive BOLD pixels. (b) Negative BOLD pixels. Data expressed as mean number of pixels±SEM. * denotes p <0.05 comparing saline vs cocaine pretreated. Abbreviations are the same as indicated in Figure 2.
BOLD Response to Cocaine Following Repeated Administration
Rats pretreated with cocaine for 7 days showed a lower BOLD response to cocaine challenge than saline pretreated animals (Figure 2). This reached statistical significance for the following brain areas: the agranular insular cortex, nucleus accumbens, ventral pallidum, dorsomedial thalamus, and substantia nigra (p <0.05). Other brain structures showed a nonsignificant tendency towards reduced BOLD activity (Figure 2). These included the medial prefrontal cortex, dorsal striatum, globus pallidus, substantia nigra, dorsal raphé, and periaqueductal grey (p <0.1). Several other brain areas showed no difference in the BOLD response to cocaine among cocaine and saline pretreated animals (Figure 2). These included the somatosensory cortex, dorsal prefrontal cortex, ventral pallidum, ventroposterolateral thalamus, hippocampus, ventral tegmental area, and the cingulate cortex.
The relative amount of positive and negative BOLD pixels within each region of interest was also quantified (Figure 5). The nucleus accumbens, dorsomedial thalamus, ventral pallidum, dorsal striatum, and the ventroposterolateral thalamus of cocaine pretreated rats showed a lower percentage of positive BOLD pixels than cocaine naïve rats (Figure 5). Cocaine pretreated rats also showed a greater amount of negative BOLD pixels in the cingulate, accumbens, dorsal striatum, raphe, VPL, and globus pallidus (Figure 5).
BOLD Response to 5% CO2
No significant differences in hypercapnia-induced BOLD signal changes were observed either globally (data not shown) or within specific brain structures of the mesocorticolimbic system (Figure 6). Although naïve rats exhibited a tendency towards greater BOLD signal changes in the prefrontal cortex than cocaine pretreated animals (Figure 6), this effect did not reach statistical significance (p = 0.08). Thus, the lower BOLD signal changes in response to cocaine in pretreated animals were not associated with lower cerebrovascular reactivity (Figure 6).
Figure 6.
Hypercapnia-induced BOLD response. No differences in hypercapnia-induced BOLD activity were observed in the mesocorticolimbic system of saline and cocaine pretreated animals. Abbreviations: mPFC, medial prefrontal cortex; NAC, nucleus accumbens; VTA, ventral tegmental area.
DISCUSSION
The present study supports our previous findings showing cocaine-induced positive BOLD responses across the mesocorticolimbic dopamine system (Febo et al, 2004). Monoamine synaptic terminals, which are the primary targets for the actions of cocaine, are extensively distributed throughout the cerebral cortex and subcortical brain structures that showed increased BOLD responses in this study. A major finding of the present study is that 7 days of pretreatment with cocaine significantly reduces the BOLD response to the drug. Although the lower BOLD response to cocaine appeared to be a generalized and nonspecific effect, several brain areas of acutely and repeatedly treated rats did not show differences in BOLD signal intensity (namely, the dorsal prefrontal cortex, cingulate, and somatosensory cortex). In addition, the lower BOLD response was not associated with differences in cerebrovascular reactivity between the two treatment groups, as measured by brief exposure to hypercapnia. Thus, our data support a reduced neuronal response to cocaine instead of a purely cerebrovascular perturbation upon repeated psychostimulant administration. The reduced cocaine-induced BOLD effect, which was observed across several mesocorticolimbic brain areas, could partly comprise the neuroadaptations that underlie cocaine sensitization.
What accounts for the alterations in BOLD signal intensity observed following cocaine administration? Cocaine can have neuronal actions as well as exert vasoactive effects. Studies employing [14C]iodoantipyrene autoradiography have shown cocaine-induced elevations in regions cerebral blood flow (Stein and Fuller, 1992). Increased flow of oxygenated blood to regions of enhanced neuronal activity and metabolism likely contributes to alterations in BOLD observed in this study and Febo et al (2004). One explanation could be that cocaine-induced BOLD signal changes are largely a result of the energetic requirements for the replenishment of neurotransmitter stores and restabilization of membrane ionic gradients (Attwell and Iadecola, 2002) that were perturbed during drug administration. It is possible that several neurotransmitter mechanisms contribute to the BOLD signal changes, since cocaine binds to dopamine, norepinephrine, and serotonin transporters. Elucidation of the neurotransmitter mechanisms involved in generating the BOLD signal in response to cocaine, and other drugs of abuse, will be important for further interpretation of pharmacological MRI data.
We observed that rats given repeated cocaine showed a blunted BOLD response to subsequent exposure. The lower BOLD response to cocaine was observed mainly within brain areas previously postulated to comprise the neural circuitry involved in the induction and expression of behavioral sensitization in rats (Pierce and Kalivas, 1997). Repeated cocaine administration has major effects on synaptic and metabolic activity within these brain areas that could underlie behavioral sensitization to cocaine (Porrino, 1993; Ungless et al, 2001) and could also account for our present findings. The effects of cocaine on local cerebral metabolic rates for glucose (lCMRglu) have been extensively studied (Porrino et al, 1988) and appear to depend on the duration of the abstinence period. Hammer et al (1993) report a decrease in basal cerebral glucose utilization in limbic, extrapyramidal, and cortical sites, 24 h after the last 3-h self-administration session. This was also observed at 6 and 72 h after a 12-h cocaine ‘binge’ self-administration session (Hammer et al, 1993). Another study found that lCMRglu increases with 8 days of repeated cocaine administration; however, measurements were taken only 45 in after the final injection (Pontieri et al, 1995). Positron emission tomography using [18F]fluorodeoxyglucose (FDG) has shown that metabolic activity is elevated in the frontal cortex of cocaine addicts within 1 week of abstinence, but it decreases with longer periods (Volkow et al, 1991). Thus, with short periods of withdrawal from cocaine administration there is an increase in brain metabolism, whereas reductions may be observed with a longer abstinence. Our present data are consistent with these studies. One explanation for the decreased BOLD response to cocaine found in this study, and the decrease glucose metabolism reported by others (Hammer et al, 1993), could be related to reductions in basal and cocaine-stimulated synaptic monoamine concentrations. This could result from alterations in monoamine uptake transporter function (Kalivas and Duffy, 1993), particularly those involved in regulating dopamine (Imperato et al, 1992) and serotonin concentrations (Parsons et al, 1996). Indeed, reduced dopamine transporter function has been observed following chronic cocaine administration in the rat (Chefer et al, 2003). Alternatively, the reduced BOLD response could in fact reflect differences in basal cerebral blood flow between drug naïve and repeatedly exposed rats. However, our hypercapnia data do not support this effect. More direct measurements of BOLD and CBF, using arterial spin labeling techniques, will be needed to explore this possibility.
It is important to note that the present experimental design involves the variation of the environment where cocaine was administered and modifications in the route of administration. Both conditions surrounding psychostimulant administration and the route of injection can affect the behavioral response to these drugs (Browman et al, 1998), and can vary the magnitude of neuronal activity (Porrino, 1993). External stimuli, such as a peripheral injections and the cage environment where cocaine pretreatment was given, are not expected to significantly contribute to evoking neuronal activity in the present study. However, the ICV route of cocaine administration activates the mesocorticolimbic dopaminergic system in a reproducible manner. The results of this study likely represent a direct measure of the brain response to the drug itself and not brain activity in response to drug-associated cues.
This study provides the first evidence of a reduced BOLD response to cocaine following repeated exposure to the drug. The lower BOLD activity involved several cortical, limbic, and thalamic circuits that may play a role in the greater behavioral reactivity to cocaine observed with sensitization. Indeed, our present data are consistent with a previous BOLD fMRI study in human cocaine-dependent subjects, both in terms of the neuroanatomical regions activated by cocaine and the temporal BOLD response to the drug (Breiter et al, 1997). However, a significant advance in our study is that we were able to compare cocaine-induced BOLD responses of repeatedly exposed and drug naïve rats. The combined use of BOLD imaging with animal models of drug addiction will be useful in determining the neuronal pathways associated with this mental disease.
Acknowledgments
This work was partially funded by a National Institute of Health NIDA Grant (R01 DA13517) to Craig F Ferris and an NINDS Grant (Specialized Neuroscience Research Program U54 NS39405) to Annabell C Segarra. An NIDA Minority Supplement Award provided support for Marcelo Febo. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIDA and NINDS.
References
- Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002;25:621–625. doi: 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- Bozarth MA, Gerber GJ, Wise RA. Intracranial self-stimulation as a technique to study the reward properties of drugs of abuse. Pharmacol Biochem Behav. 1980;13(Suppl 1):245–247. doi: 10.1016/s0091-3057(80)80037-2. [DOI] [PubMed] [Google Scholar]
- Breiter HC, Gollub RL, Weisskoff RM, Kennedy DN, Makris N, Berke JD, et al. Acute effects of cocaine on human brain activity and emotion. Neuron. 1997;19:591–611. doi: 10.1016/s0896-6273(00)80374-8. [DOI] [PubMed] [Google Scholar]
- Browman KE, Badiani A, Robinson TE. The influence of environment on the induction of sensitization to the psychomotor activating effects of intravenous cocaine in rats is dosedependent. Psychopharmacology. 1998;137:90–98. doi: 10.1007/s002130050597. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Zakharova I, Shippenberg TS. Enhanced responsiveness to novelty and cocaine is associated with decreased basal dopamine uptake and release in the nucleus accumbens: quantitative microdialysis in rats under transient conditions. J Neurosci. 2003;23:3076–3084. doi: 10.1523/JNEUROSCI.23-07-03076.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ER, Ugurbil K, Kim SG. Effect of basal conditions on the magnitude and dynamics of the blood oxygenation level-dependent fMRI response. J Cereb Blood Flow Metab. 2002;22:1042–1053. doi: 10.1097/00004647-200209000-00002. [DOI] [PubMed] [Google Scholar]
- Febo M, Segarra AC, Tenney JR, Brevard ME, Duong TQ, Ferris CF. Imaging cocaine-induced changes in the mesocorticolimbic dopaminergic system of conscious rats. J Neurosci Methods. 2004;139:167–176. doi: 10.1016/j.jneumeth.2004.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeders NE, Smith JE. Cortical dopaminergic involvement in cocaine reinforcement. Science. 1983;221:773–775. doi: 10.1126/science.6879176. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Moratalla R, Robertson HA. Amphetamine and cocaine induce drug-specific activation of the c-fos gene in striosome-matrix compartments and limbic subdivisions of the striatum. Proc Natl Acad Sci USA. 1990;87:6912–6916. doi: 10.1073/pnas.87.17.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer RP, Jr, Pires WS, Markou A, Koob GF. Withdrawal following cocaine self-administration decreases regional cerebral metabolic rate in critical brain reward regions. Synapse. 1993;14:73–80. doi: 10.1002/syn.890140110. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Ljungdahl A, Fuxe K, Johansson O. Dopamine nerve terminals in the rat limbic cortex: aspects of the dopamine hypothesis of schizophrenia. Science. 1974;184:177–179. doi: 10.1126/science.184.4133.177. [DOI] [PubMed] [Google Scholar]
- Imperato A, Mele A, Scrocco MG, Puglisi-Allegra S. Chronic cocaine alters limbic extracellular dopamine. Neurochemical basis for addiction. Eur J Pharmacol. 1992;212:299–300. doi: 10.1016/0014-2999(92)90349-9. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P. Time course of extracellular dopamine and behavioral sensitization to cocaine. I. Dopamine axon terminals. J Neurosci. 1993;13:266–275. doi: 10.1523/JNEUROSCI.13-01-00266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Pierce RC, Cornish J, Sorg BA. A role for sensitization in craving and relapse in cocaine addiction. J Psychopharmacol. 1998;12:49–53. doi: 10.1177/026988119801200107. [DOI] [PubMed] [Google Scholar]
- Ludwig R, Bogdanov G, King JA, Allard A, Ferris CF. A dual RF resonator system for high-field functional magnetic resonance imaging of small animals. J Neurosci Methods. 2004;132:125–135. doi: 10.1016/j.jneumeth.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lee TM, Kay AR, Tank DW. Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc Natl Acad Sci USA. 1990;87:9868–9872. doi: 10.1073/pnas.87.24.9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain In Stereotaxic Coordinates. 3. Academic Press; Boston: 1997. [Google Scholar]
- Parsons LH, Koob GF, Weiss F. Extracellular serotonin is decreased in the nucleus accumbens during withdrawal from cocaine self-administration. Behav Brain Res. 1996;73:225–228. doi: 10.1016/0166-4328(96)00101-5. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Kalivas PW. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res Brain Res Rev. 1997;25:192–216. doi: 10.1016/s0165-0173(97)00021-0. [DOI] [PubMed] [Google Scholar]
- Pontieri FE, Mainero C, La Riccia M, Passarelli F, Orzi F. Functional correlates of repeated administration of cocaine and apomorphine in the rat. Eur J Pharmacol. 1995;284:205–209. doi: 10.1016/0014-2999(95)00441-m. [DOI] [PubMed] [Google Scholar]
- Porrino LJ. Functional consequences of acute cocaine treatment depend on route of administration. Psychopharmacology (Berl) 1993;112:343–351. doi: 10.1007/BF02244931. [DOI] [PubMed] [Google Scholar]
- Porrino LJ, Domer FR, Crane AM, Sokoloff L. Selective alterations in cerebral metabolism within the mesocorticolimbic dopaminergic system produced by acute cocaine administration in rats. Neuropsychopharmacology. 1988;1:109–118. doi: 10.1016/0893-133x(88)90002-4. [DOI] [PubMed] [Google Scholar]
- Post RM, Rose H. Increasing effects of repetitive cocaine administration in the rat. Nature. 1976;260:731–732. doi: 10.1038/260731a0. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11:1598–1604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- Sicard K, Shen Q, Brevard ME, Sullivan R, Ferris CF, King JA, et al. Regional cerebral blood flow and BOLD responses in conscious and anesthetized rats under basal and hypercapnic conditions: implications for functional MRI studies. J Cereb Blood Flow Metab. 2003;23:472–481. doi: 10.1097/01.WCB.0000054755.93668.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EA, Fuller SA. Selective effects of cocaine on regional cerebral blood flow in the rat. J Pharmacol Exp Ther. 1992;262:327–334. [PubMed] [Google Scholar]
- Strupp JP. Stimulate: A GUI based fMRI analysis software package. Neuroimage. 1996;3:S607. [Google Scholar]
- Thierry AM, Blanc G, Sobel A, Stinus L, Golwinski J. Dopaminergic terminals in the rat cortex. Science. 1973;182:499–501. doi: 10.1126/science.182.4111.499. [DOI] [PubMed] [Google Scholar]
- Thompson JK, Peterson MR, Freeman RD. Single-neuron activity and tissue oxygenation in the cerebral cortex. Science. 2003;299:1070–1072. doi: 10.1126/science.1079220. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Uslaner JM, Crombag HS, Ferguson SM, Robinson TE. Cocaine-induced psychomotor activity is associated with its ability to induce c-fos mRNA expression in the subthalamic nucleus: effects of dose and repeated treatment. Eur J Neurosci. 2003;17:2180–2186. doi: 10.1046/j.1460-9568.2003.02638.x. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wolf AP, Hitzemann R, Dewey S, Bendriem B, et al. Changes in brain glucose metabolism in cocaine dependence and withdrawal. Am J Psychiatry. 1991;148:621–626. doi: 10.1176/ajp.148.5.621. [DOI] [PubMed] [Google Scholar]