

Abstract
The cellular changes during ageing are incompletely understood yet immune system dysfunction is implicated in the age-related decline in health. The acquired immune system shows a functional decline in ability to respond to new pathogens whereas serum levels of cytokines are elevated with age. Despite these age-associated increases in circulating cytokines, the function of aged macrophages is decreased. Pathogen-associated molecular pattern receptors such as Toll-like receptors (TLRs) are vital in the response of macrophages to pathological stimuli. Here we review the evidence for defective TLR signalling in normal ageing. Gene transcription, protein expression and cell surface expression of members of the TLR family of receptors and co-effector molecules do not show a consistent age-dependent change across model systems. However, there is evidence for impaired downstream signalling events, including inhibition of positive and activation of negative modulators of TLR induced signalling events. In this paper we hypothesize that despite a poor inflammatory response via TLR activation, the ineffective clearance of pathogens by macrophages increases the duration of their activation and contributes to perpetuation of inflammatory responses and ageing.
Keywords: ageing, macrophage, reactive oxygen species, Toll-like receptors
Introduction
Ageing is a biological phenomenon which occurs in all organisms. That dysfunction of the immune system occurs has been well described [1]; a decline in the normal functioning of the acquired (or adaptive) immune system [2] with a shift in the T cell population from naive T cells to memory T cells [3] leads to reduced immunization efficacy and poor acquired immune response to new pathogens. The ageing innate immune response is more complex, with increased neutrophil recruitment but decreased specific activity [1]. Similarly, a poor acute phase protein response has been described in ageing mice in response to inflammatory challenge and is associated with significant tissue damage [4,5]. Paradoxically, circulating levels of the inflammatory cytokines, particularly of the acute phase protein inducer interleukin (IL)-6, are enhanced, although it is unclear whether downstream signalling following ligand–receptor interaction maintains its efficiency with ageing [4,5]. Cross-talk exists between the innate immune system and the acquired immune system through shared receptors; Toll-like receptor (TLR)-mediated priming of T cell and B cell responses enhances development [6–8] of the acquired immune response, therefore age-related dysfunction of TLR function could potentially impact upon the effectiveness of both the innate and acquired immune systems. Collectively, the ageing immune system results in increased risk of infection and reduces ability to remove infectious pathogens, resulting in an increased risk of infection-dependent morbidity and mortality [9]. Age-associated defects in the acquired immune system and the inability to eliminate foreign pathogens or cancerous cells leads to a proinflammatory environment with elevated circulating cytokine levels [10,11]; this associates with an increased risk of a diverse range of disease such as rheumatoid arthritis, cardiovascular disease and cancers, arising in part from a failure to resolve acute inflammatory events [12]. The purpose of this review is to focus upon human ageing in the absence of disease or chronic pathophysiology.
Free radicals and ageing
According to the free radical theory of ageing [13], reactive oxygen species (ROS) are responsible for sustained damage to DNA, proteins and lipids; and the accumulation of damaged biological macromolecules is responsible for biological ageing and age-associated pathologies. ROS are produced as normal by-products of molecular respiration from the electron transport chain [14] and their production by the phagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase constitutes a critical part of the first line of defence against pathogens. Chronic granulomatous disease patients who lack specific components of NADPH oxidase and fail to produce significant ROS during the respiratory burst suffer persistent bacterial infection, demonstrating the importance of the respiratory burst for pathogen removal.
The phagocytic respiratory burst in isolated neutrophils from older adults is reported variably as increased and decreased [1]; the discrepancy here may relate to ligand differences between studies, e.g. CD14- versus CD16-mediated activation, and raises the intriguing possibility that intracellular signalling pathways may be affected differentially by age. Moreover, mitochondrial respiratory chain integrity is diminished and ROS leakage increased during ageing, due principally to cytochrome oxidase dysfunction [15].
Both the mitochondrion and NADPH oxidase isoforms are potential sources of ageing-related excessive ROS production [16,17]. The reduction or abrogation of age-associated pathologies via anti-oxidant supplementation [18] or by overexpression of cellular anti-oxidant defence enzymes in mitochondria [19,20] supports this concept, although this wisdom has been challenged recently in mice [4]; the overexpression of major anti-oxidant enzymes was shown to be insufficient to extend their lifespan. A number of species-specific differences in the innate immunity and macrophage biology have been documented that are relevant to ageing and warrant caution in the extrapolation of observations between species (reviewed previously [21,22]).
TLRs and ligand specificity
First described in 1997 [23], TLRs are a recently discovered family in the transduction of an inflammatory stimulus and mediate many aspects of the immune response, including major histocompatibility complex (MHC) expression, antibody production and the expression of cytokines, chemokines and adhesion molecules (reviewed in [24]). Currently, 13 TLRs (10 human), activating a signalling cascade in response to structurally distinct stimuli, have been described: bacterial lipoproteins are recognized by either TLR-1/2 or TLR-2/6 heterodimer complexes, viral double-stranded RNA is recognized by TLR-3, the endotoxin lipopolysaccharide (LPS) from Gram-negative bacteria is recognized by TLR-4, TLR-5 recognizes bacterial flagella proteins, whereas TLRs 7–9 detect pathogenic nucleic acids summarized in Table 1 (adapted from [24]).
Table 1.
Summary of Toll-like receptor (TLR) ligand specificity and adaptor signalling molecules, adapted from [24].
| TLR | Location of TLR | PAMPs recognized by TLR | Co-receptor(s) | Signalling adaptor | Transcription factor(s) | Effector cytokines induced |
|---|---|---|---|---|---|---|
| TLR-1/2 | Plasma membrane (cell surface) | Triacyl lipopeptides (bacteria and mycobacteria) | Hetrodimer of TLR-1/2 forms a functional receptor | TIRAP, MyD88 | NF-κB | Inflammatory cytokines (TNF-α, IL-6, etc.) |
| TLR-2 | Plasma membrane (cell surface) | Peptidoglycan (Gram-positive bacteria), LAM (mycobacteria), haemagglutinin (measles virus), phospholipomannan (Candida), glycosylphosphophatidyl inositol mucin (Trypanosoma) | CD36, RP105 | TIRAP, MyD88 | NF-κB | Inflammatory cytokines (TNF-α, IL-6, etc.) |
| TLR-3 | Endosome | ssRNA virus (WNV), dsRNA virus (reovirus), RSV, MCMV | TRIF | NF-κB, IRF3,7 | Inflammatory cytokines (TNF-α, IL-6, etc.), type I IFNs | |
| TLR-4 | Plasma membrane (cell surface) | LPS (Gram-negative bacteria), mannan (Candida), glycoinositolphospholipids (Trypanosoma), envelope proteins (RSV and MMTV) | MD2, CD14, LBP, RP105 | TIRAP, MyD88, TRAM and TRIF | NF-κB, IRF3,7 | Inflammatory cytokines (TNF-α, IL-6, etc.), type I IFNs |
| TLR-5 | Plasma membrane (cell surface) | Flagellin (flagellated bacteria) | MyD88 | NF-κB | Inflammatory cytokines (TNF-α, IL-6, etc.) | |
| TLR-6/2 | Plasma membrane (cell surface) | Diacyl lipopeptides (mycoplasma), LTA (Streptococcus), zymosan (Saccharomyces) | Hetrodimer of TLR-6/2 or dectin-1 forms a functional receptor | TIRAP, MyD88 | NF-κB | Inflammatory cytokines (TNF-α, IL-6 etc.) |
| TLR-7 | Endosome | ssRNA viruses (VSV, influenza virus) | MyD88 | NF-κB, IRF7 | Inflammatory cytokines (TNF-α, IL-6, etc.), type I IFNs | |
| TLR-8 | Endosome | ssRNA from RNA virus | MyD88 | NF-κB, IRF7 | Inflammatory cytokines (TNF-α, IL,-6 etc.), type I IFNs | |
| TLR-9 | Endosome | dsDNA viruses (HSV, MCMV), CpG motifs from bacteria and viruses, haemozoin (plasmodium) | MyD88 | NF-κB, IRF7 | Inflammatory cytokines (TNF-α, IL,-6 etc.), type I IFNs | |
| TLR-11 | Plasma membrane (cell surface) | Uropathogenic bacteria, profillin-like molecule (Toxoplasma gondii) |
CpG: cytosine guanine; IFN: interferon; IL: interleukin; IRF: interferon regulatory factor; LAM: lymphangioleiomyomatosis; LTA: lipoteichoic acid; MCMV: mouse cytomegalovirus; MMTV: mouse mammary tumour virus; NF: nuclear factor; PAMP: pathogen-associated molecular patterns; RSV: respiratory syncytial virus; TNF: tumour necrosis factor; TIRAP: TIR domain containing adaptor protein; TRAF: tumour necrosis factor receptor associated factor; WNV: West Nile virus.
TLRs are found on cells of the innate immune system including monocytes, macrophages, dendritic cells, epithelial and endothelial cells. TLRs are related closely to the IL-1 receptor, both of which contain cytoplasmic Toll/IL-1 receptor (TIR) domains. Although TLRs show a high level of intracellular structural similarity to the IL-1 receptor, the extracellular domains are distinctly different [25], with TLRs having a number of leucine-rich repeats in the place of the immunoglobulin (Ig)G-like domains found on the extracellular portion of the IL-1 receptor.
TLR activation requires the recruitment of a number of adaptor molecules. All TLRs, with the exception of TLR-3, require myeloid differentiation primary response gene 88 (MyD88) to function [24]. MyD88 is important in the expression of LPS-induced IL-1 and is also recruited by the IL-1 receptor via a TIR domain, where MyD88-deficient mice are resistant to LPS-induced endotoxaemia [24]. In addition, MyD88−/− mice are protected from LPS-induced sepsis due to the inability to produce cytokines and proinflammatory proteins in the presence of slow-activated nuclear factor κB (NF-κB) [24]. The slow and fast pathways are considered distinct from one another, even though some downstream elements are common to both pathways, and are termed the MyD88-dependent pathway (fast: MyD88 and Mal) [26], and the MyD88-independent pathway (slow: TRAM and TRIF) [26,27]. The MyD88-dependent pathway is involved in the immediate responses to a bacterial infection and stimulates bactericidal compound production such as reactive nitrogen species (RNS), nitric oxide (NO), via inducible nitric oxide synthase (iNOS) activation, and proinflammatory cytokines such as tumour necrosis factor (TNF)-α. In contrast, the MyD88-independent pathway activates after approximately 30 min from the original LPS stimulation due to TRAM/TRIF signalling from endosomes following receptor internalization (Fig. 1).
Fig. 1.
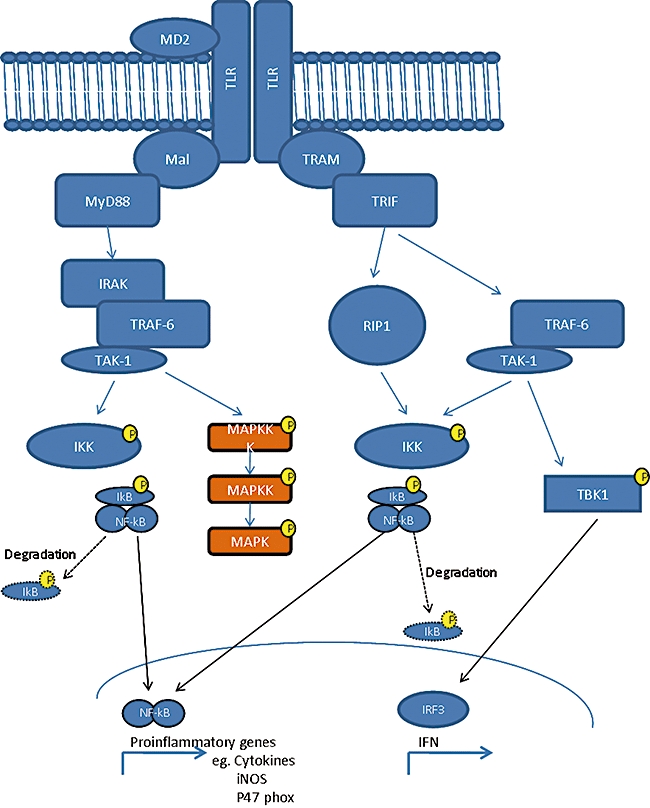
Toll-like receptor (TLR)-mediated signalling pathways and adaptor proteins. Ligation of TLR results in receptor dimerization and subsequent recruitment of Mal and myeloid differentiation factor 88 (MyD88), interleukin 1 receptor associated kinase (IRAK) and tumour necrosis factor receptor associated factor (TRAF)-6 molecules lead to activation of nuclear factor (NF)-kB and mitogen-activated protein kinase (MAPK) pathways. This results in transcriptional activation of proinflammatory genes including cytokines. A delayed response is also activated by TLR dimerization, probably activated after endosomal internalization of the TLR-4 homodimer. This involves recruitment of TRAM and TRIF resulting in a delayed NF-kB activation and transcription via interferon regulatory factor (IRF)3 activation of late genes such as interferon.
Macrophage TLR function in ageing
Age-associated defects in the acquired immune system associate with increased recruitment of phagocytic cells [1]. Macrophages are vital in the immune response and are the major producers of RNS and cytokines in response to microbial infectious stimuli such as LPS, lipoteichoic acid (LTA) and pathogenic nucleic acids, and endogenous danger signals such as oxidized proteins and lipids, TNF-α and heat shock proteins (HSPs) [28]. However, ageing in mice is associated with an increase in the number of bone marrow macrophages that have an impaired ability to generate or release cytokines. The increase in macrophage numbers may reflect a compensation for their reduced function [29]. Studies in mice show that circulating IL-6 is increased significantly with age upon stimulation with LPS, although its downstream signalling capacity appears limited [19,30].
Stimulation of macrophages explanted from ageing mice with LPS increases the anti-inflammatory cytokine IL-10 and reduces endotoxin-induced production of the proinflammatory cytokines TNF-α, IL-1β, IL-6 and IL-12 [31–34], nitric oxide and expression of inducible nitric oxide synthase [34]. Reduced TLR-mediated function in alveolar macrophages from the ageing rat in response to the TLR-4 ligand LPS [35] is shown by significant decreases in ROS and RNS production.
A similar pattern is observed in humans; in vivo, cytokine production is increased with age [36–38]; however, production of IL-6 and TNF-α was decreased with the TLR-1/2 heterodimer ligand N-palmitoyl-S-[2,3-bis(palmitolyoxy)-(2R,S)-propyl]-Cys-[S]-Ser-[S]-Lys(4)trihydrochloride (Pam3CSK4) in macrophages from aged [74·6 (65–89) years] compared to young [25·7 (22–30) years] individuals [39]. This apparent paradox of low cellular production and high plasma cytokine levels may be resolved by considering that with age, macrophages remain activated for longer and by multiple stimuli; have an increased lifespan; or if proinflammatory cytokine production switches from the macrophage to another cell type during the ageing process; cells that are not typically involved in the innate immune response such as T cells, B cells and epithelial cells are able to produce cytokines in response to TLR ligands.
There are a number of TLR modulators that are elevated during ageing and include circulating hormones, free fatty acids and immunoglobulins [27]. These play important regulatory roles in switching off signalling responses to pathogenic stimuli, for example by an increase in prostaglandin E2 (PGE2) production that in turn triggers intracellular cyclic AMP-dependent protein kinase A (PKA) activation and reduces the expression of TLR-4 [27]. Moreover, a number of endogenous TLR ligands that have been identified and are known to increase during ageing include oxidized proteins and lipids, advanced glycation end-products (AGEs) [40] and HSPs [28], may modify the macrophage response to exogenous TLR ligands.
TLR expression in ageing
The molecular mechanisms for the reduction in TLR function may be explained by decreased functional protein expression. Laing et al. [41] investigated the expression of TLRs 1, 2, 4, 5 and 6 in peritoneal macrophages from young (8–10 weeks) and old (>18 months) BALB/cByJL mice. Basal level of TLR-5 transcription alone was altered in the aged group, showing a relative expression of 0·06 of mRNA transcript compared to the young group. Only TLR-5 transcription was altered when macrophages were activated with the Gram-negative bacterium Porphyromonas gingivalis. However, although TLR-5 transcription was reduced in macrophages from the aged group, this was not reflected in reduced receptor expression at the cell surface (Liang et al. [41]). In contrast, the level of TLR-2 surface expression was reduced by a significant, amount, although no changes in mRNA levels of TLR-2 were observed, highlighting the importance of studying functional receptor levels and not just the transcript.
mRNA levels of TLRs 1–9 are reduced in unstimulated splenic macrophages and also in peritoneal macrophages from old (18–24 months) compared to young (2–3 months) C57BL/6 mice [33]. In contrast, Renshaw et al. [33] demonstrated an associated decrease in cell surface expression of TLR-4 which correlated with a functional reduction in TLR activity as reduced IL-6 and TNF-α production in response to LPS, Poly (I:C), zymosan A, Staphylococcus aureus, flagellin, and oligodeoxynucleotide cytosine guanine (ODN CpG). Renshaw et al. [33] showed that after TLR-9 (which was not analysed by Liang et al.), TLR-5 showed the largest alteration in transcript level, suggesting that assay sensitivity could explain the discordant mRNA data.
Another study [31] in mouse models has shown that although LPS-induced TNF-α production is reduced with age, the levels of TLR-4 cell surface expression are not altered; these authors suggest that the mitogen-activated protein kinase (MAPK) signalling pathways downstream of TLR activation are affected by age (discussed below), but that the reduction in TLR functionality is not due to defects at the TLR level.
Cell surface expression of TLR1 is reduced by 36%, but the cell surface expression of TLR-2 is not altered significantly with age [39]. This TLR defect appeared to be isolated to TLR-1, as TLR-2 ligands elicited no age-dependent alteration in function. Further investigation showed no significant change in whole cell expression levels of TLR-1 protein. The reduced cell surface TLR-1 expression coupled with the lack of change to total protein levels of TLR-1 suggests that some process which is essential for cell membrane targeting may be subject to age-mediated dysfunction.
Trafficking is affected by the degree of post-translational modification (PTM). All TLRs undergo N-glycosylation [42,43] and defects of N-glycoslyation activity have been implicated in ageing [44], but as yet are not associated with TLR dysfunction in ageing. Other essential PTMs required for TLR-1 function are the formation of a number of disulphide bridges [43,45,46]. Formation of disulphide bridges is controlled within the endoplasmic reticulum (ER) by the action of protein disulphide isomerase (PDI), and recent reports suggest that enhanced ROS and RNS associated with ageing may induce a nitrosyl post-translational modification to PDI impairing its function; an effect that has been linked to the misfolding of the NMDA receptor [47]. It is clear that an age-related decline and activity of key molecular chaperones and folding enzymes in the ER compromises proper protein folding. These changes, together with an impaired adaptive unfolded protein response, have been implicated in receptor dysfunction in the nervous system and implications for TLRs have not yet been explored [48].
The importance of TLR expression in age-related TLR dysfunction remains unclear. More extensive human studies should be conducted with conformation-sensitive antibodies in order to establish the importance of TLR surface expression in aged individuals. Also the role of PTMs on TLRs, and their contribution to functional activity and expression, should be investigated in the context of ageing.
TLR adaptor molecules and age
TLR-induced macrophage activation requires recruitment of a number of adaptor molecules. In the case of TLR-4, extracellular co-stimulatory molecules include soluble (s)CD14 and membrane-bound (m)CD14, MD-2 and LPS binding protein (LBP). Serum levels of LBP are unaltered in aged mice or humans. However, expression of CD14 on macrophages is reduced in mice, as are mRNA levels of MD-2, a key regulator for LPS-induced TLR-4 activation.
A number of intracellular molecules have been identified as essential for TLR-mediated signalling, including MyD88, which is essential in the signal transduction from all TLRs with the exception of the endosomally located TLR-3 (Fig. 1). MyD88 adaptor-like protein [Mal; also called TIR domain containing adaptor protein (TIRAP)] is also required for MyD88-dependent TLR signalling and is used by all TLRs except TLR-3. MyD88/Mal are associated with IL-1 receptor-associated kinase (IRAK)-1 and -4; upon MyD88/Mal activation IRAK-1/-4 are phosphorylated and dissociate from the MyD88/Mal complex eliciting a cascade of phosphorylation events. IRAK-1 and IRAK-4 themselves have serine/threonine kinase activity and upon dissociation from MyD88/Mal activate TNF receptor-associated factor (TRAF)-6 by phosphorylation. TRAF6 phosphorylates transforming growth factor (TGF)-β-activated kinase (TAK)-1 which, in turn, activates the mitogen-activated protein kinase (MAPK) and NF-kB pathways (discussed below).
Ineffective signalling from TLR via adaptor molecules in the age-associated decline in adaptive immunity presents an attractive mechanism for failure to elicit cytokine production in response to ligand activation in light of the conflicting data regarding age-dependent changes in TLR function but not expression. Microarray analysis of mRNA from young (4 months) murine macrophages compared to aged macrophages (20–22 months) demonstrated a decrease in both basal and LPS-stimulated levels of MyD88 [34]; however, the same study showed an increase in mRNA levels of Mal confirmed by both microarray analysis and polymerase chain reaction (PCR) that was not confirmed at the protein level. Levels of IRAK-1 were also up-regulated in response to LPS stimulation but not basally in aged mice compared to young mice. In contrast, the level of the IRAK-1 substrate, TRAF6, was decreased significantly. Another potential target for age-related decline in signal transduction from the receptor involves MD-2, which has an absolute requirement for two cysteine residues, probably to form a disulphide bridge with TLR-4 after ligand binding [49]. Given the altered intracellular redox environment associated with ageing that favours irreversible thiol oxidation due to glutathione insufficiency, oxidative PTMs to MD-2 are also probable candidates to explain an age-associated decline in response to extracellular ligands [50].
TLR mRNA analysis suggests altered expression with age, but protein expression and therefore the functional impact of these alterations in transcriptional regulation of TLR adaptor molecules has yet to be confirmed.
Although few data are available on age-related MyD88-independent dysfunction, LPS-stimulated IFN-γ production in human monocytes is decreased with age [51,52]. Also members of the family of IFN regulatory factors have been shown to be increased in response to TLR-9 activation [53].
Suppressors of TLR signalling in age
Excessive activation or aberrant stimulation of TLR signalling pathways can contribute to endotoxic shock and chronic inflammatory diseases, such as periodontitis [31]. Therefore, a number of physiological negative regulatory mechanisms exist to ‘switch off’ TLR signals.
One of the major ways in which TLR signalling is regulated is by the controlled disassembly and degradation of adaptor molecules associated with the activated TLR complex. One such adaptor molecule which is removed after TLR activation is mal; mal contains a TIR domain and is therefore recruited upon ligand-induced TLR dimerization; mal acts as a molecular bridge to aid in the recruitment of MyD88. Following activation of TLR and recruitment of mal, mal undergoes Bruton's tyrosine kinase (Btk)-induced phosphorylation, causing it to become a substrate for suppressor of cytokine signalling (SOCS)-1 dependent ubiquitination. Polyubiquitinated mal is then directed for degradation via the 26S proteasome. The contribution of the SOCS family of proteins in age-mediated TLR dysfunction is at present unclear, but increased levels of SOCS are reported in rodent brain tissue [54], human muscle tissue [55] and human neutrophils [56] supporting an age-associated accelerated degradation of the TLR signalling complex.
Increased levels of IRAK-M mRNA have been observed with increased age in mice [57]. During TLR signalling, IRAK-1 associates with TRAF6 resulting in activation via phosphorylation. In contrast, IRAK-M, although sharing much homology to IRAK-1, lacks catalytic kinase activity and during TLR signalling IRAK-M inhibits the association of IRAK-1 with TRAF6, thereby inhibiting downstream TLR-induced MAPK and activation. IRAK-M, unlike other members of the IRAK family, is restricted in its expression to monocytes and macrophages, whereas other IRAKs are expressed ubiquitously throughout the body. It remains to be determined whether IRAK-M protein levels map the changes reported previously at the mRNA level, i.e. are increased during ageing, as this would probably translate into reduced downstream signalling from the receptor.
Other mechanisms of TLR negative regulation include modulation by the reactive intermediate 4-hydroxynonenal (4HNE) [58]. 4HNE is a product of lipid peroxidation and is found to be increased during ageing as well as in a number of pathologies, including Alzheimer's disease and cancer. LPS-induced TNF-α, IL-6 and IL-1β responses are reduced when monocytes are treated with 4HNE; this is accompanied by decreased NF-κB activation and reduced phagocytic activity. To effect this change, 4HNE forms adducts with TLR4 at reactive cysteine residues, consequently inhibiting TLR4 dimerization and therefore subsequent signalling [58].
These examples of inhibitors of TLR activation and signalling are part of a large and diverse group of mechanisms established to maintain balance in the immune system; dysfunction of TLR signalling could arise from either decreased activation of components of TLR signalling or increased activity of negative regulators of TLR function, and evidence in favour of increased modulation of negative regulation of TLR signalling is emerging.
TLR signalling
Subsequent to activation of the TLR and recruitment of adaptors to the complex, TRAF6 phosphorylates TAK-1 which in turn activates the MAPK cascade, a pleiotropic signalling pathway that is activated by many inflammatory stimuli.
MAPK
The MAPK family is comprised of three groups: extracellular regulated kinase 1/2 (ERK 1/2), c-jun N-terminal kinase (JNK) and p38 MAPK. MAPKs are activated via phosphorylation of a specific threonine-X-tyrosine motif of which the amino acid separating the two phosphorylation sites differs between the different families of MAPK and is glutamate, proline or glycine in ERK, JNK or p38 MAPK families, respectively. Members of the MAPK family are phosphorylated and activated by proline-directed dual specific MAPK kinases (MAPKK, MEK), which are themselves activated via phosphorylation reactions that are mediated by MAPKK kinase (MAPKKK, MEKK) enzymes, TAK-1 is a TLR-activated MEKK. In the activation of p38 MAPK, TAK-1 activates MEK3/6 which, in turn, activates p38 MAPK. TAK-1-mediated JNK activation is mediated via MEK7 (reviewed in [59]). Figure 1 indicates the integration of TLR activation to the MAPK cascade.
The role of MAPK pathways in TLR-induced cytokine production is vital; genetic manipulation or pharmacological inhibition of MAPK proteins decreases the LPS response. MAPK phosphorylation is reduced during ageing [31], and when normalized to a housekeeping gene such as β-actin confirms that total protein levels of JNK and p38 MAPK are reduced in aged individuals compared to young adults. The reduced MAPK function in age appears to be twofold; first there is a reduction in phosphorylated JNK and p38 when normalized to total protein levels of JNK and p38, and also the total protein levels are reduced. This has been confirmed by examining the phosphorylation of MAPK-activated protein kinase (MAPKAPK) 2, a substrate for p38 MAPK, which was shown to be reduced in LPS-stimulated macrophages from young mice compared to aged mice [31].
MAPKs are regulated negatively by dual specific phosphatases (DUSPs). To date, a single report has described a decrease in DUSP-10 mRNA levels in a murine model of ageing and if translated to protein, this would suggest an enhanced signalling response via the MAPK cascade [60]. However, this is inconsistent with the observed reports of no change to or a reduction in MAPK signalling with age. No specific investigation has been conducted into the function of negative regulators of MAPKs focusing specifically on the macrophage, nor is it known whether the generic changes observed at the mRNA level are translated into changes in protein expression or function.
NF-κB
NF-κB is one of the most important transcriptional regulators of the innate immune response and also has important roles in controlling cell death (Phillips et al. [61]). It is activated by a number of different stimuli ranging from exogenous bacterial and viral pathogens to endogenous ligands such as TNF-α via the TNF-α receptor and AGEs via the receptor for AGE (RAGE).
Activation of NF-κB is initiated via the activation of inhibitor of κB (IκB) kinase (IKK) complex. The IKK complex is formed of a number of proteins: IKKα, IKKβ and a scaffold protein called NF-κB essential modulator (NEMO). The IKK complex is activated by phosphorylation of Ser177 and Ser181 and in turn activates IκB [62].
The association of NF-κB with IκB sequesters the dimeric NF-κB complex in the cytoplasm of the cell. NF-κB activation requires phosphorylation of the IκBα molecule on ser32 and ser36 [63]. This dual phosphorylation causes dissociation from the NF-κB molecule and targets IκB for ubiquitin conjugation and subsequent proteasomal degradation. The dissociation of IκB from NF-κB unmasks a nuclear localization signal resulting in the translocation of NF-κB to the nucleus. Upon translocation to the nucleus NF-κB binds to NF-κB binding elements, where it induces transcription of many genes associated with inflammation such as TNF-α, IL-1β, iNOS and cyclo-oxygenase-2 (COX-2) [61].
The 26S proteasome is less active in aged individuals, and this may impact upon the NF-kB pathway; Huber et al. ([64]) demonstrated in a hepatic ischaemia–reperfusion model that inactivation of proteosomal activity led to an increase in tissue damage but a decrease in NF-kB activity with age due to the prevention of the recruitment of ubiquitinated IkBα to the proteasome, thereby preventing release from the complex, nuclear localization and transcriptional activity of NF-kB. Other studies have shown that aged rodents have increased levels of NF-κB binding to DNA in both muscle [65] and brain tissue [66] compared to young animals. Although a number of differences between rodent and human immunology have been well documented, evidence of human tissue-specific increases in NF-κB binding have been documented [67], although to date the effect of age on NF-κB binding in human macrophages has not been reported.
The inflammaging, TLR and infection paradox
The collected evidence for the effect of age on TLR signalling does not suggest an amplification of response to ligand as a mechanism for the increased levels of circulating cytokines with age. Moreover, older adults are more prone to infection and have impaired immune responses. Together, these data suggest that an ineffective clearance of infectious agents and prolonged phagocytic cell activation is the primary driver for increasing the inflammatory response and may arise from ineffective signalling from TLRs. Consequently, the released cytokines and chemokines would serve as signals to further phagocytic recruitment, and further activation of inflammatory responses while ineffective removal of pathogen sustains the stimulus (Fig. 2). We have proposed recently that the disturbance to the cellular redox state will impair cell death pathways and may lead to longer-lived, activated cells [61].
Fig. 2.
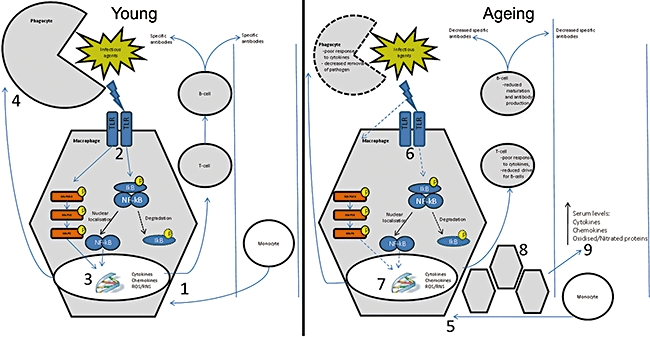
The role of macrophages in the response and removal of bacterial pathogens. Monocytes are first recruited to the site of infection and undergo differentiation to macrophages (1). Toll-like receptor (TLR)-mediated signalling events are initiated (2) leading to increased production of cytokines and reactive oxygen species (ROS) products (3). Phagocytes initiate phagocytosis (4) resulting in the killing and removal of the pathogen. In an aged immune system monocytes are recruited and differentiate into macrophages (5). Subsequent TLR-mediated signalling events are deficient (6) and this results in decreased bactericidal and phagocytic activity (7). More macrophages are recruited (8) and although each cell is less functional, their increased numbers lead to increased serum cytokines levels (9). The ineffective signalling cascades do not result in the killing and removal of the invading pathogen, thereby leading to chronic inflammation and susceptibility to infection.
The inability of older macrophages to mount an effective innate response to challenge poses a significant health risk, notably the persistence of infection and chronicity of inflammation. Restoration of effective bacterial clearance and the phenotypic switching of macrophages to anti-inflammatory cells or their apoptosis are likely to be the key features of an effective strategy to reduce age-associated inflammatory damage.
Acknowledgments
Christopher Dunston is funded by the FP7 MARKAGE project.
Disclosure
None.
References
- 1.Lord JM, Butcher S, Killampali V, Lascelles D, Salmon M. Neutrophil ageing and immunesenescence. Mech Ageing Dev. 2001;122:1521–35. doi: 10.1016/s0047-6374(01)00285-8. [DOI] [PubMed] [Google Scholar]
- 2.Siegrist C-A, Aspinall R. B-cell responses to vaccination at the extremes of age. Nat Rev Immunol. 2009;9:185–94. doi: 10.1038/nri2508. [DOI] [PubMed] [Google Scholar]
- 3.Fagnoni FF, Vescovini R, Passeri G, et al. Shortage of circulating naive CD8+ T cells provides new insights on immunodeficiency in aging. Blood. 2000;95:2860–8. [PubMed] [Google Scholar]
- 4.Gomez CR, Acuna-Castillo C, Perez C, et al. Diminished acute phase response and increased hepatic inflammation of aged rats in response to intraperitoneal injection of lipopolysaccharide. J Gerontol A Biol Sci Med Sci. 2008;63:1299–306. doi: 10.1093/gerona/63.12.1299. [DOI] [PubMed] [Google Scholar]
- 5.Gomez CR, Hirano S, Cutro B, et al. Advanced age exacerbates the pulmonary inflammatory response after lipopolysaccharide exposure. Crit Care Med. 2007;35:246–51. doi: 10.1097/01.CCM.0000251639.05135.E0. [DOI] [PubMed] [Google Scholar]
- 6.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–50. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi EA, Akira S, Nobrega A. Role of TLR in B cell development: signaling through TLR4 promotes B cell maturation and is inhibited by TLR2. J Immunol. 2005;174:6639–47. doi: 10.4049/jimmunol.174.11.6639. [DOI] [PubMed] [Google Scholar]
- 8.Rudd BD, Brien JD, Davenport MP, Nikolich-Zugich J. Cutting edge: TLR ligands increase TCR triggering by slowing peptide-MHC Class I decay rates. J Immunol. 2008;181:5199–203. doi: 10.4049/jimmunol.181.8.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang SY, Mackowiak PA. Infections in the elderly. Clin Geriatr Med. 2007;23:441–56. doi: 10.1016/j.cger.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 10.Bruunsgaard H, Pedersen M, Pedersen BK. Aging and proinflammatory cytokines. Curr Opin Hematol. 2001;8:131–6. doi: 10.1097/00062752-200105000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Forsey RJ, Thompson JM, Ernerudh J, et al. Plasma cytokine profiles in elderly humans. Mech Ageing Dev. 2003;124:487–93. doi: 10.1016/s0047-6374(03)00025-3. [DOI] [PubMed] [Google Scholar]
- 12.Flavell SJ, Hou TZ, Lax S, Filer AD, Salmon M, Buckley CD. Fibroblasts as novel therapeutic targets in chronic inflammation. Br J Pharmacol. 2008;153:S241–6. doi: 10.1038/sj.bjp.0707487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harman D. Free radical theory of aging. Mutation. 1992;275:257–66. doi: 10.1016/0921-8734(92)90030-s. [DOI] [PubMed] [Google Scholar]
- 14.Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. 2008;036:976–80. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 15.Cooper JM, Mann VM, Schapira AHV. Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: effect of ageing. J Neurol Sci. 1992;113:91–8. doi: 10.1016/0022-510x(92)90270-u. [DOI] [PubMed] [Google Scholar]
- 16.Johannsen DL, Ravussin E. The role of mitochondria in health and disease. Curr Opin Pharmacol. 2009;9:780–6. doi: 10.1016/j.coph.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birch-Machin MA. The role of mitochondria in ageing and carcinogenesis. Clin Exp Dermatol. 2006;31:548–52. doi: 10.1111/j.1365-2230.2006.02161.x. [DOI] [PubMed] [Google Scholar]
- 18.Ferrari CKB, Torres EAFS. Biochemical pharmacology of functional foods and prevention of chronic diseases of aging. Biomed Pharmacother. 2003;57:251–60. doi: 10.1016/s0753-3322(03)00032-5. [DOI] [PubMed] [Google Scholar]
- 19.Treuting PM, Linford NJ, Knoblaugh SE, et al. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J Gerontol A Biol Sci Med Sci. 2008;63:813–22. doi: 10.1093/gerona/63.8.813. [DOI] [PubMed] [Google Scholar]
- 20.Dai D-F, Santana LF, Vermulst M, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–97. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mestas J, Hughes CCW. Of mice and not men. Differences. 2004;172:2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 22.Schneemann M, Schoeden G. Macrophage biology and immunology: man is not a mouse. J Leukoc Biol. 2007;81:579. doi: 10.1189/jlb.1106702. [DOI] [PubMed] [Google Scholar]
- 23.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 24.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–5. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 25.Xu Y, Tao X, Shen B, et al. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408:111–15. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 26.O'Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 27.Han K-J, Su X, Xu L-G, Bin L-H, Zhang J, Shu H-B. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-ΰB activation and apoptosis pathways. J Biol Chem. 2004;279:15652–61. doi: 10.1074/jbc.M311629200. [DOI] [PubMed] [Google Scholar]
- 28.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/Interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–12. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Udupa K, Xiao H, Lipschitz D. Effect of age on marrow macrophage number and function. Ageing. 1995;7:379–84. doi: 10.1007/BF03324349. [DOI] [PubMed] [Google Scholar]
- 30.Wei J, Xu H, Davies JL, Hemmings GP. Increase of plasma IL-6 concentration with age in healthy subjects. Life Sci. 1992;51:1953–6. doi: 10.1016/0024-3205(92)90112-3. [DOI] [PubMed] [Google Scholar]
- 31.Boehmer ED, Meehan MJ, Cutro BT, Kovacs EJ. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech Ageing Dev. 2005;126:1305–13. doi: 10.1016/j.mad.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 32.Chelvarajan L, Popa D, Liu Y, Getchell TV, Stromberg AJ, Bondada S. Molecular mechanisms underlying anti-inflammatory phenotype of neonatal splenic macrophages. J Leukoc Biol. 2007;82:403–16. doi: 10.1189/jlb.0107071. [DOI] [PubMed] [Google Scholar]
- 33.Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired toll-like receptor expression and function in aging. J Immunol. 2002;169:4697–701. doi: 10.4049/jimmunol.169.9.4697. [DOI] [PubMed] [Google Scholar]
- 34.Chelvarajan RL, Liu Y, Popa D, et al. Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J Leukoc Biol. 2006;79:1314–27. doi: 10.1189/jlb.0106024. [DOI] [PubMed] [Google Scholar]
- 35.Deborah Ruth T, Regina M, Silvia OC, Beatriz M. Age-dependent change in reactive oxygen species and nitric oxide generation by rat alveolar macrophages. Aging Cell. 2003;2:159–64. doi: 10.1046/j.1474-9728.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 36.Brüünsgaard H, Pedersen BK. Age-related inflammatory cytokines and disease. Immunol Allergy Clin North Am. 2003;23:15–39. doi: 10.1016/s0889-8561(02)00056-5. [DOI] [PubMed] [Google Scholar]
- 37.Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004;39:687–99. doi: 10.1016/j.exger.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 38.Rink L, Cakman I, Kirchner H. Altered cytokine production in the elderly. Mech Ageing Dev. 1998;102:199–209. doi: 10.1016/s0047-6374(97)00153-x. [DOI] [PubMed] [Google Scholar]
- 39.van Duin D, Mohanty S, Thomas V, et al. Age-associated defect in human TLR-1/2 function. J Immunol. 2007;178:970–5. doi: 10.4049/jimmunol.178.2.970. [DOI] [PubMed] [Google Scholar]
- 40.Hodgkinson CP, Laxton RC, Patel K, Ye S. Advanced glycation end-product of low density lipoprotein activates the Toll-Like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:2275–81. doi: 10.1161/ATVBAHA.108.175992. [DOI] [PubMed] [Google Scholar]
- 41.Liang S, Domon H, Hosur KB, Wang M, Hajishengallis G. Age-related alterations in innate immune receptor expression and ability of macrophages to respond to pathogen challenge in vitro. Mech Ageing Dev. 2009;130:538–46. doi: 10.1016/j.mad.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.da Silva Correia J, Ulevitch RJ. MD-2 and TLR4 N-linked glycosylations are important for a functional lipopolysaccharide receptor. J Biol Chem. 2002;277:1845–54. doi: 10.1074/jbc.M109910200. [DOI] [PubMed] [Google Scholar]
- 43.Jin MS, Kim SE, Heo JY, et al. Crystal structure of the TLR1–TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–82. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 44.Vanhooren V, Desmyter L, Liu XE, et al. N-glycomic changes in serum proteins during human aging. Rejuvenation Res. 2007;10:521–31. doi: 10.1089/rej.2007.0556. [DOI] [PubMed] [Google Scholar]
- 45.Kim HM, Park BS, Kim J-I, et al. Crystal structure of the TLR4–MD-2 complex with bound endotoxin antagonist eritoran. Cell. 2007;130:906–17. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 46.Park BS, Song DH, Kim HM, Choi B-S, Lee H, Lee J-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature. 2009;458:1191–5. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura T, Lipton SA. Emerging roles of s-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid Redox Signal. 2008;10:87–102. doi: 10.1089/ars.2007.1858. [DOI] [PubMed] [Google Scholar]
- 48.Naidoo N. ER and aging – protein folding and the ER stress response. Ageing Res Rev. 2009;8:150–9. doi: 10.1016/j.arr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 49.Ren L, Leung WK, Darveau RP, Jin L. The expression profile of lipopolysaccharide-binding protein, membrane-bound CD14, and Toll-Like receptors 2 and 4 in chronic periodontitis. J Periodontol. 2005;76:1950–9. doi: 10.1902/jop.2005.76.11.1950. [DOI] [PubMed] [Google Scholar]
- 50.Mallis R, Hamann M, AZhang T, Hendrich S, Thomas J. Irreversible thiol oxidation in carbonic anhydrase III: protection by S-glutathiolation and detection in ageing rats. Biol Chem. 2002;383:649–62. doi: 10.1515/BC.2002.067. [DOI] [PubMed] [Google Scholar]
- 51.Goetzl EJ, Huang M-C, Kon J, et al. Gender specificity of altered human immune cytokine profiles in aging. FASEB J. 2010 doi: 10.1096/fj.10-160911. fj.10-160911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Speziali E, Aranha CHM, Teixeira-Carvalho A, et al. Ageing down-modulates liver inflammatory immune responses to schistosome infection in mice. Scand J Immunol. 2010;71:240–8. doi: 10.1111/j.1365-3083.2010.02370.x. [DOI] [PubMed] [Google Scholar]
- 53.Stout-Delgado HW, Yang X, Walker WE, Tesar BM, Goldstein DR. Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J Immunol. 2008;181:6747–56. doi: 10.4049/jimmunol.181.10.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peralta S, Carrascosa JM, Gallardo N, Ros M, Arribas C. Ageing increases SOCS-3 expression in rat hypothalamus: effects of food restriction. Biochem Biophys Res Commun. 2002;296:425–8. doi: 10.1016/s0006-291x(02)00906-3. [DOI] [PubMed] [Google Scholar]
- 55.Léger B, Derave W, De Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 2008;11:163–75. doi: 10.1089/rej.2007.0588. [DOI] [PubMed] [Google Scholar]
- 56.Tortorella C, Simone O, Piazzolla G, Stella I, Cappiello V, Antonaci S. Role of phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways in granulocyte macrophage-colony-stimulating factor failure to delay fas-induced neutrophil apoptosis in elderly humans. J Gerontol A Biol Sci Med Sci. 2006;61:1111–18. doi: 10.1093/gerona/61.11.1111. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Howell EA, Lagoo AS, et al. Differential gene expression of interleukin-1 receptor associated kinase-1 and interleukin-1 receptor associated kinase-M in peripheral blood mononuclear cells of young and aged rats following preconditioning with endotoxin. Shock. 2009;31:55–63. doi: 10.1097/SHK.0b013e3181778ab2. 10.1097/SHK.1090b1013e3181778ab3181772. [DOI] [PubMed] [Google Scholar]
- 58.Kim YS, Park ZY, Kim SY, Jeong E, Lee JY. Alteration of Toll-like receptor 4 activation by 4-hydroxy-2-nonenal mediated by the suppression of receptor homodimerization. Chem Biol Interact. 2009;182:59–66. doi: 10.1016/j.cbi.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 59.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 60.Kim HS, Song M-C, Kwak IH, Park TJ, Lim IK. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J Biol Chem. 2003;278:37497–510. doi: 10.1074/jbc.M211739200. [DOI] [PubMed] [Google Scholar]
- 61.Phillips D, Dias H, Kitas G, Griffiths H. Aberrant reactive oxygen and nitrogen species generation in rheumatoid arthritis (RA): causes and consequences for immune function, cell survival, and therapeutic intervention. Antioxid Redox Signal. 2010;12:743–85. doi: 10.1089/ars.2009.2607. [DOI] [PubMed] [Google Scholar]
- 62.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IB kinase activity through IKK subunit phosphorylation. Science. 1999;284:309–13. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 63.Peters RT, Liao S-M, Maniatis T. IKK[var epsilon] is part of a novel PMA-inducible I[kappa]B kinase complex. Mol Cell. 2000;5:513–22. doi: 10.1016/s1097-2765(00)80445-1. [DOI] [PubMed] [Google Scholar]
- 64.Huber N, Sakai N, Eismann T, et al. Age-related decrease in proteasome expression contributes to defective nuclear factor-kappaB activation during hepatic ischemia/reperfusion. Hepatology. 2009;49:1718–28. doi: 10.1002/hep.22840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helenius M, Hänninen M, Lehtinen SK, Salminen A. Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor-kappa B. Biochem J. 1996;318:603–8. doi: 10.1042/bj3180603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korhonen P, Helenius M, Salminen A. Age-related changes in the regulation of transcription factor NF-[kappa]B in rat brain. Neurosci Lett. 1997;225:61–4. doi: 10.1016/s0304-3940(97)00190-0. [DOI] [PubMed] [Google Scholar]
- 67.Adler AS, Sinha S, Kawahara TLA, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007;21:3244–57. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]