

Abstract
An autoantigenic role for collagen type I (CI) has been suggested previously in diffuse cutaneous systemic sclerosis (dcSSc). Whether CI is indeed capable of affecting the immune system in dcSSc is not known. Patients with early (3 years or less) or late (>3 years) dcSSc and healthy controls donated blood. Peripheral blood mononuclear cells (PBMC) were cultured with or without CI, and expression of genes known for their involvement in autoimmune and inflammatory processes was assessed using cDNA arrays; results were confirmed by real-time polymerase chain reaction and enzyme-linked immunosorbent assay for selected genes. Patients with early and late dcSSc were similarly different from healthy controls in basal gene expression. When cultured with CI, PBMC from patients with early dcSSc differed from healthy controls in expression of 34 genes, whereas PBMC from patients with late dcSSc differed from healthy controls in expression of only 29 genes. Direct comparisons of matched PBMC samples cultured with and without CI revealed differences in expression of eight genes in healthy controls, of five genes in patients with early dcSSc, and no differences in patients with late dcSSc. Thus, PBMC from patients with dcSSc respond differently than do PBMC from healthy controls when cultured with CI. Exposure to CI in culture of PBMC from patients in the early stage of dcSSc in contrast to PBMC from patients with late-stage dcSSc evokes a greater degree of activation of immune-related genes, suggesting that CI is more dominant as an autoantigen in early versus late dcSSc.
Keywords: autoimmunity, collagen, gene expression, inflammation, scleroderma
Introduction
Immune activation, microvascular injury, excessive platelet aggregation to collagen type I (CI) and excessive matrix deposition with increased numbers of myofibroblasts are the hallmark characteristics of systemic sclerosis (SSc or scleroderma). Activated T cells and soluble mediators of the adaptive immune system are thought to play a central role in disease pathogenesis [1–5]. In the skin, cellular infiltration precedes dermal fibrosis and consists of activated T lymphocytes, plasma cells and macrophages [6–9]. Lymphocytes are also present in the inflammatory infiltrate of affected internal organs, such as the lungs [10–14]. There is evidence to suggest that the activation of T cells in SSc is antigen-driven and not merely a non-specific response to local cytokines. Analysis of the T cell receptor repertoire in skin biopsies of patients with SSc has revealed that T cells have undergone clonal expansion in response to antigenic stimulation [15]. Also, the presence of a dominant T cell clone in skin biopsies obtained from a patient at different time-points and from different skin regions implies that the antigen is persistently present and distributed widely [15]. Several putative antigens have been implicated in disease pathogenesis, including CI, which is the most abundant of all collagens in humans. It is the most abundant collagen, for example, in skin, heart and intestines, all of which are affected in SSc. Early studies from our laboratory demonstrated that peripheral blood mononuclear cells (PBMC) from the majority of patients with SSc produce chemotactic cytokines when cultured with human skin CI, skin CI obtained from lathyritic chicks and chick collagen α1(I) and α2(I) chains [16]. More recently we have demonstrated that PBMC from patients with SSc exhibit significant production of interleukin (IL)-10 and/or interferon (IFN)-γ in response to in vitro culture with collagen α1(I) and α2(I) chains [17]. Immunity to type I collagen has also been described in other fibrotic conditions, such as idiopathic pulmonary fibrosis [18] and bleomycin-induced pulmonary fibrosis [19]. Our recent study [20] demonstrated that CD4+ memory (CD45RO+) T cells from PBMC of one-third of the tested patients with SSc but not from healthy controls or patients with rheumatoid arthritis proliferate and produce cytokines IL-2, IFN-γ, granulocyte–macrophage colony-stimulating factor (GM–CSF) and tumour necrosis factor (TNF)-α in response to in vitro stimulation with CI.
In the present study we sought to profile the changes in the expression of immunity- and inflammation-related genes induced by in vitro stimulation with CI of PBMC from patients with diffuse cutaneous (dc) SSc compared to healthy controls. Genomic profiling of cells from patients with SSc has been performed by several groups in the past, and the results revealed substantial changes in gene expression profiles between patients with SSc and controls [12,13,21,22]. However, previous studies have several limitations, some of which are addressed by this study. First, some of the previous studies focused upon purified cell populations, such as monocytes or lymphocytes [21] or pulmonary CD8+ T cells [12]. Although important for characterization of the cell types in the disease, such studies did not reflect the complexity of interactions between antigen-presenting cells and lymphocytes. Scleroderma lesions involve infiltrates of various cell types [6–10], and unseparated PBMC would represent more realistically the infiltrates occurring in the disease, including the co-operative response of participating cell types to stimulation with a potential (auto) antigen, in this case CI. Therefore, the goal of this study was not to characterize separate cell types statically, but to assess the combined response of cells to CI stimulation. Secondly, even if natural non-separated populations of cells were investigated [13,17], none of the previous studies addressed response to CI stimulation, which is a major limitation considering that scleroderma is simultaneously an autoimmune and a connective tissue disease. Thirdly, this study included only patients with dcSSc, and only patients who did not have debilitating major organ damage that would require treatment with immunosuppressive drugs, corticosteroids or non-steroidal anti-inflammatory drugs (NSAIDs). Therefore, the results of this study are more representative of the disease process and not of the effects of treatments. Fourthly, we have included patients with early (i.e. ≤3 years duration) and late (i.e. >3–10 years duration) dcSSc, covering the spectrum of disease duration.
Materials and methods
Patients and controls
Patients whose PBMC were analysed in this study were enrolled in the multi-centre, randomized, double-blind placebo-controlled trial of oral type I collagen treatment of patients with dcSSc according to a protocol approved by a Data Safety and Monitoring Board, institutional review boards at all participating centres, by the National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIAMS) and Food and Drug Administration (FDA) [23]. Patients with ‘early phase’ (≤3 years disease duration) and ‘late phase’ (>3 and ≤10 years disease duration) were enrolled at 12 different sites in the United States. The analyses of blood samples were approved by all these agencies. Detailed description of the entrance and exclusive criteria demographic and clinical assessments are described in a separate publication [23]. The PBMC samples for this study were selected at random and used if a sufficient amount of good quality RNA was available. The selected patients had clinical characteristics that were similar (P > 0·05) to the overall cohort in the trial [23]. All blood samples were drawn at baseline of the study before placebo or oral collagen were administered. Patients did not receive immunomodulatory therapies, steroids, herbal therapies, megavitamins or NSAIDs. At baseline modified Rodnan skin score (MRSS), health assessment questionnaire disability index (HAQ-DI) Short Form (SF) 36 mental summary component (SF-Men) and physical summary component (SF-Phy), physician's global assessment (Phy GA), patients global assessment (Pat GA), pain visual analogue scale (Pain-VAS), forced vital capacity (FVC), carbon monoxide diffusion capacity (DLCO), systolic blood pressure (SBP), diastolic blood pressure (DBP) and body weight were determined as described previously [23]. As a control, blood was obtained for study from eight healthy volunteers of similar age, sex and race as the patients with dcSSc from participating centres. The control donors were from the different participating centres. The blood samples from patients and controls were processed in an identical fashion as described below. Informed consent was obtained from all patients and controls.
PBMC cultures
All blood samples from all volunteers were processed in a similar fashion at all participating centres. Blood was drawn from volunteers into 10-ml glass vacutainer tubes (#366480; Becton Dickinson, Franklin Lakes, NJ, USA) containing sodium heparin as an anti-coagulant and diluted within 4 h 1 : 4 with RPMI-1640 medium containing 100 U penicillin and 100 µg/ml streptomycin (Gibco, Grand Island, NY, USA) in 50-ml sterile polypropylene conical tubes (Falcon 352070 tubes; Becton Dickinson). Tubes of diluted blood were wrapped in large amounts of paper, placed in a styrofoam box containing a cold pack and shipped by overnight airfreight to the University of Tennessee Health Science Center for further processing for culture. The time between drawing blood samples and arrival to the Memphis site did not exceed 24 h for any of the tested samples. The samples from the Memphis site were diluted similarly; tubes were wrapped in large amounts of paper and placed with a cold pack in a styrofoam box that set out for 24 h and was occasionally turned upside-down before blood was separated on histopaque and set up in culture. Upon arrival, 10 ml histopaque cushion was layered under the diluted blood, which was then subjected to isopyric centrifugation at room temperature with slow acceleration over 5 min to 200 g, at which force the centrifugation was continued for an additional 35 min. The PBMC within the histopaque layer were recovered by careful pipetting, diluted in 35 ml RPMI-1640 and centrifuged for 10 min at 200 g at room temperature. The PBMC pellet was recovered, resuspended in 40 ml RPMI-1640 medium and centrifuged once again for 10 min at 200 g. The PBMC pellet was then resuspended in Dulbecco's high glucose minimum essential medium (Gibco) containing 9% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, non-essential amino acids, 1 mM sodium pyruvate (Gibco) and 2 mM Gluta MAX (Gibco). The PBMC concentration was adjusted to 4 × 106/ml and 450 µl of the suspended PBMC were dispensed into individual wells of 48-well tissue culture plates (Nunc, Roskilde, Denmark). The PBMC were cultured for 24 h at 37°C in a humidified atmosphere containing 5% CO2 with 50 µl phosphate buffered saline (PBS) or 5 µg of bovine CI solution in PBS added. The PBMC were solubilized in TRI reagent (Sigma Chemical Co., St Louis, MO, USA) and stored at −70°C until gene expression analysis.
Native CI was prepared as described previously [24]. Briefly, fetal bovine skin was used to prepare CI. The skin was washed exhaustively and homogenized in cold water. The suspension was centrifuged at 10 000 g for 30 min followed by resuspension in cold water and recentrifugation. Finally, the pellet was resuspended in 0·5 M acetic acid, the pH was adjusted to 2·5 with formic acid and pepsin was added (enzyme/substrate = 1:200). Pepsin digestion was performed for 48 h at 4°C, and the supernatants were harvested by centrifugation. The pepsin digestion was repeated two additional times, and the pooled pepsin-solubilized collagen in the combined extracts was purified by using salt precipitation [CI and CIII precipitate at low concentration (0·7 M) from diluted acid solutions]. CIII precipitates at 1·7 M NaC1, while CI precipitates at 2·5 M NaCl at neutral pH [25]). The purity of CI was monitored by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and amino acid analysis. Lack of lipopolysaccharide (LPS) contamination was confirmed with E-Toxate Limulus amoebocyte lysate tests (Sigma).
Gene expression analyses
Human autoimmune and inflammatory response cDNA-based gene arrays from SuperArray (Frederick, MD, USA) were used. The arrays had 400 spotted probes, 367 of which were single spots for individual gene targets, and the rest represented positive and negative controls. The cDNA synthesis and labelling procedures, hybridization of labelled cDNA to the arrays, washing procedures, image analyses and numerical data readouts were performed according to the manufacturer's recommendations and as described previously [12,13,26]. Briefly, the amount of RNA used for the assays, hybridization conditions and timing and the image development were standardized to allow for easy comparison between the hybridized arrays. Chemiluminescent images of hybridized membranes were scanned using a Storm Imaging System (Molecular Dynamics, Sunnyvale, CA, USA), spot densities and background values digitized using ImageQuant Software (Molecular Dynamics) and background subtracted from the spot densities. The following normalization procedure was applied to each array. First, all non-expressed genes were removed. Then, the interquartile range (25–75%) of all remaining non-zero densities for that array was identified, and the mean value of that range was calculated. Finally, all values from each array were then normalized to the mean value of the interquartile range of non-zero non-saturated spot densities for that array. All arbitrarily low values (<2) resulting from such normalization were then assigned a value of 2. Data from all arrays normalized in this fashion were compiled into the ‘normalized database’. For subsequent statistical analyses, all genes with low expression (normalized density values below 15 across the database) or with saturated density values across the database were eliminated, resulting in 127 selected genes. This database of selected genes was used for statistical analyses that were performed using HDBSat (http://www.ssg.uab.edu/hdbstat). Subsets of relevant data were isolated for each pairwise comparison, and only those genes were considered further for which a mean of at least one group was greater than 15 and the fold-difference of means between the compared groups was greater than 1·5 in either direction. Differences between groups were calculated accounting for false discovery rate (FDR) [27]. Only the differences significant at FDR <0·05 were considered. Hierarchical clustering of genes and samples was performed based on thus normalized, filtered and additionally log-base-2-transformed data, utilizing the CIMminer tool (http://discover.nci.nih.gov/cimminer). The average linkage clustering method was used with correlation distance as a similarity measure.
Selected data were validated by SYBR green quantitative real-time polymerase chain reaction (PCR) with confirmed primers for type II activin receptor (SABiosciences, Frederick, MD, USA) and by measuring monocyte chemotactic protein-1 (MCP-1) levels in cell culture supernates by Multiplex enzyme-linked immunosorbent assay (ELISA) (Luminex, Austin, TX, USA). The supplier of the PCR primers prevalidated their specificity and guaranteed lack of primer dimer formation. We validated the primers further by (1) ensuring that a single PCR product and no primer dimers were observed electrophoretically after amplification; and (2) by sequencing the PCR product and confirming its sequence identity with target. Reverse transcriptase and real-time PCR reactions were performed using the respective kits provided by the supplier of the primers (SABiosciences). All RNA samples were processed in a similar fashion and tested in triplicate. StepOne Plus Real Time PCR system and software (Applied Biosystems, Foster City, CA, USA) were used according to the manufacturer's recommendations using the 2ΔΔCt method. All data were normalized to the 18S rRNA reference sequence.
Results
Characteristics of patient and control populations
The groups of 24 patients with dcSSc and eight healthy control volunteers were similar in gender distribution (18 females and six males with SSc versus seven females and one male control, P = 0·459, χ2 test), as was race distribution (18 Caucasians, five African Americans and one other race in the SSc group versus five Caucasians, two African Americans and one other race in the healthy control group, P = 0·655, χ2 test). Healthy controls were younger than the patients (mean age 53·8 ± 11·4 in SSc patients and 39·9 ± 12·9 in healthy controls, P = 0·020, unpaired two-tailed t-tests). The patients with dcSSc were divided further into groups with early (3 years and shorter, n = 13) and late (longer than 3 years, n = 11) disease. These groups of patients did not differ in demographic characteristics, biochemical profiles or measures of disease severity (Table 1).
Table 1.
Clinical characteristics of the patients.*
| Characteristic | Early SSc (n = 13) | Late SSc (n = 11) | P† |
|---|---|---|---|
| Mean ± s.d. disease duration, years | 2·0 ± 0·7 | 7·4 ± 2·0 | <0·001 |
| Mean ± s.d. age, years | 56·9 ± 10·2 | 50·3 ± 12·2 | 0·171 |
| Gender, no. female/male | 11/2 | 7/4 | 0·237 |
| Race, no. Caucasian/African American/other | 11/1/1 | 7/4/0 | 0·170 |
| Patient global assessment ± s.d. | 37·5 ± 27·7 | 37·8 ± 20·9 | 0·978 |
| Physician's global assessment ± s.d. | 41·4 ± 22·0 | 49·3 ± 20·1 | 0·368 |
| Health Assessment Questionnaire Disability Index (HAQ-DI) ± s.d. | 1·2 ± 0·6 | 1·3 ± 0·6 | 0·536 |
| Pain visual analogue scale ± s.d. | 31·6 ± 24·4 | 34·5 ± 23·2 | 0·764 |
| Modified Rodnan skin score (MRSS) ± s.d. | 25·1 ± 6·3 | 28·3 ± 8·4 | 0·311 |
| Leucocyte count ± s.d., thousands per microlitre | 7·2 ± 2·2 | 8·3 ± 2·5 | 0·271 |
| Lymphocytes, % ± s.d. | 23·9 ± 6·8 | 26·9 ± 8·4 | 0·379 |
| Monocytes, % ± s.d. | 5·9 ± 3·1 | 5·2 ± 2·4 | 0·508 |
| Neutrophils, % ± s.d. | 66·7 ± 9·6 | 64·5 ± 10·8 | 0·624 |
There was no difference between these groups of patients in weight (P = 0.484); total cholesterol (P = 0.807); haematocrit (P = 0.119); creatinine (P = 0.372); haemoglobin (P = 0.189); forced vital capacity % predicted (P = 0.295); forced expiratory volume in 1 s % predicted (P = 0.928); diffusing capacity for carbon monoxide % predicted (P = 0.266); systolic and diastolic blood pressures (P = 0.775 and P = 0.286, respectively); and Short Form 36 physical, mental and index (P = 0.426, P = 0.469 and P = 0.998, respectively).
The significance of difference was calculated with χ2 and unpaired two-tailed t-tests for categorical and continuous variables, respectively. S.d.: standard deviation.
Differences in basal gene expression
Gene expression profiles were compared among patients with early or late dcSSc or healthy controls with and without stimulation with CI in cell culture. Thus, independent variables were diagnosis of dcSSc versus healthy controls, disease duration (early versus late dcSSc) and presence or absence of stimulation with CI in cell culture. This approach implies a possibility of numerous interactions between these independent variables, many of which may be biomedically meaningless. To ensure focus upon biomedically meaningful comparisons, we chose to perform pairwise analyses instead of multivariate analysis of variance (anova).
Initial analyses compared gene basal gene expression in patients with early dcSSc versus healthy controls and, separately, in patients with late dcSSc versus healthy controls. The patterns of basal gene expression in patients differed dramatically from those in healthy controls (Table 2, Fig. 1). As shown in Table 2, patients with early dcSSc were different in expression of 27 genes (19 with elevated and eight with decreased expression), whereas patients with late dcSSc were different in expression of 25 genes (21 with elevated and four with decreased expression) from healthy controls. Twenty of these genes were the same in the early dcSSc versus healthy controls and in the late dcSSc versus healthy controls comparisons (Table 2). Cluster analyses revealed that changes in expression of numerous genes occurred in a co-ordinated fashion (Fig. 1).
Table 2.
Significant fold differences (P < 0·05) in gene expression between peripheral blood mononuclear cells (PBMC) from patients with early diffuse cutaneous systemic sclerosis (dcSSc) (E) or late dcSSc (L) and controls (C) after culture in control medium (M) or medium with added type I collagen (CI). Positive values indicate fold higher and negative indicate fold lower levels of gene expression in indicated dcSS samples compared to samples from controls.
| Gene | UniGene | OMIM | Full gene name | E versus C in M | L versus C in M | E versus C in CI | L versus C in CI |
|---|---|---|---|---|---|---|---|
| A20 | Hs.211600 | 191163 | Tumour necrosis factor, alpha-induced protein 3 | –2·8 | –2·0 | ||
| ACVR2 | Hs.470174 | 102581 | Activin A receptor, type II | +4·1 | +4·6 | +5·3 | +6·1 |
| ALK3 | Hs.524477 | 601299 | Bone morphogenetic protein receptor, type IA | +2·9 | +3·8 | +3·4 | +4·0 |
| ALK5 | Hs.494622 | 190181 | Transforming growth factor, beta receptor I | –3·1 | |||
| AMH | Hs.112432 | 600957 | Anti-Mullerian hormone | +2·2 | +2·5 | +3·2 | |
| AMHR2 | Hs.659889 | 600956 | Anti-Mullerian hormone receptor, type II | +2·2 | +2·4 | +1·8 | +1·8 |
| BAD | Hs.370254 | 603167 | BCL2-antagonist of cell death | +2·8 | +2·5 | ||
| CALM2 | Hs.643483 | 114182 | Calmodulin 2 (phosphorylase kinase, delta) | +2·6 | +2·5 | ||
| CBP | Hs.459759 | 600140 | CREB binding protein | +2·7 | +3·3 | +2·2 | +2·5 |
| CCL2 | Hs.303649 | 158105 | Chemokine (C-C motif) ligand 2 | +3·7 | +3·3 | +2·7 | +3·1 |
| CCL5 | Hs.514821 | 187011 | Chemokine (C-C motif) ligand 5 | +2·5 | +2·4 | +2·1 | +1·9 |
| CCR10 | Hs.278446 | 600240 | G protein-coupled receptor 2 | –2·0 | |||
| CCR5 | Hs.450802 | 601373 | Chemokine (C-C motif) receptor 5 | +2·4 | +3·0 | +3·6 | +2·7 |
| CDKN1A | Hs.370771 | 116899 | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | –3·1 | –1·8 | –3·0 | –2·3 |
| CDKN2B | Hs.72901 | 600431 | Cyclin-dependent kinase inhibitor 2B | +3·9 | +5·1 | +3·5 | +3·9 |
| CREB | Hs.584750 | 123810 | CAMP responsive element binding protein 1 | +2·8 | +2·5 | +2·3 | +2·0 |
| CTLA4 | Hs.247824 | 123890 | Cytotoxic T-lymphocyte-associated protein 4 | +3·1 | +3·5 | +3·1 | +2·8 |
| CX3CL1 | Hs.531668 | 601880 | Chemokine (C-X3-C motif) ligand 1 | –1·8 | –2·1 | –1·9 | |
| CXCL2 | Hs.590921 | 139110 | Chemokine (C-X-C motif) ligand 2 | +2·1 | +2·3 | +3·0 | +2·5 |
| CXCL3 | Hs.89690 | 139111 | Chemokine (C-X-C motif) ligand 3 | +1·8 | |||
| EGR1 | Hs.326035 | 128990 | Early growth response 1 | +4·5 | +4·8 | ||
| EGR2 | Hs.1395 | 129010 | Early growth response 2 | +6·2 | +8·2 | ||
| FasL | Hs.2007 | 134638 | Tumour necrosis factor (ligand) superfamily, member 6 | +2·7 | +2·2 | ||
| FCER1A | Hs.897 | 147140 | Fc fragment of IgE, high affinity I, receptor for; alpha polypeptide | +4·0 | |||
| FOSL1 | Hs.283565 | 136515 | FOS-like antigen 1 | +2·4 | +2·3 | +2·1 | |
| GATA4 | Hs.431009 | 603693 | Zinc finger protein, multitype 2 | –1·7 | –2·0 | ||
| GM-CSF | Hs.1349 | 138960 | Colony stimulating factor 2 (granulocyte–macrophage) | –3·0 | –2·7 | ||
| GSK3A | Hs.466828 | 606786 | Glycogen synthase kinase 3 alpha | –1·9 | –1·6 | –1·6 | |
| IGF1 | Hs.160562 | 147440 | Insulin-like growth factor 1 (somatomedin C) | +1·8 | |||
| IL11 | Hs.467304 | 147681 | Interleukin 11 | +2·1 | |||
| IL12Rβ2 | Hs.567294 | 601642 | Interleukin 12 receptor, beta 2 | –2·8 | |||
| NCK2 | Hs.529244 | 604930 | NCK adaptor protein 2 | +2·1 | +2·8 | +2·0 | +2·8 |
| NFKB2 | Hs.73090 | 164012 | Nuclear factor of kappa light polypeptide gene enhancer in B cells 2 | +1·7 | +1·6 | +1·9 | |
| NFRKB | Hs.530539 | 164013 | Nuclear factor related to kappa B binding protein | +2·8 | +2·6 | +3·1 | |
| SELE | Hs.89546 | 131210 | Selectin E | –1·9 | –2·1 | –2·1 | –2·2 |
| SELL | Hs.719894 | 153240 | Selectin L | –4·8 | –4·7 | ||
| STAT1 | Hs.699271 | 600555 | Signal transducer and activator of transcription 1 | +2·4 | +2·0 | +2·4 | +2·8 |
| TLR4 | Hs.174312 | 603030 | Toll-like receptor 4 | –2·8 | |||
| TLR7 | Hs.659215 | 300365 | Toll-like receptor 7 | –2·0 | –1·6 | ||
| TLR8 | Hs.660543 | 300366 | Toll-like receptor 8 | +2·2 | +3·2 | +2·0 | |
| TRAF1 | Hs.531251 | 601711 | TNF receptor-associated factor 1 | +3·1 | +3·9 | +2·0 | +2·2 |
| TRAF5 | Hs.523930 | 602356 | TNF receptor-associated factor 5 | +4·3 | +4·2 |
Fig. 1.
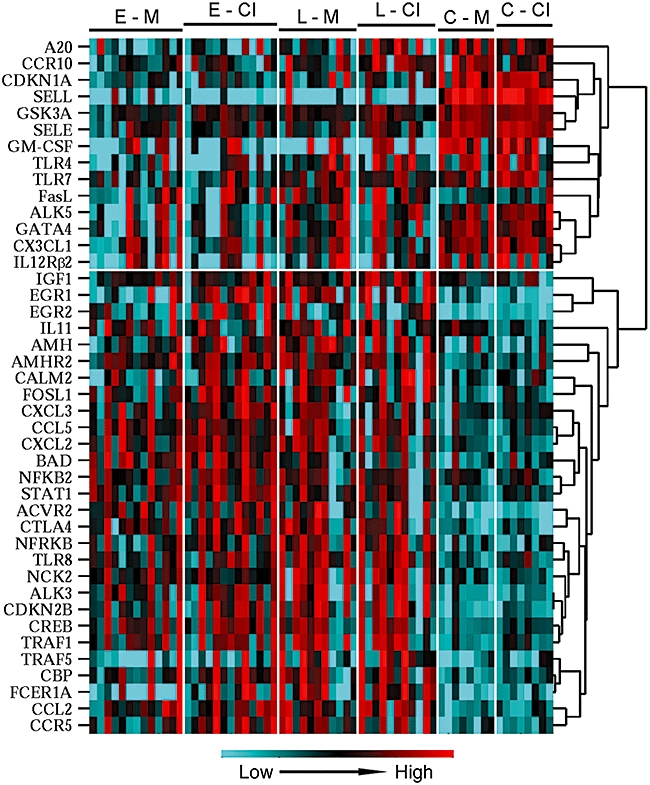
Cluster analysis using average linkage clustering by correlation of gene expression normalized to the mean expression levels of each gene in all samples. Volunteer groups are indicated on top: E, early diffuse cutaneous systemic sclerosis (dcSSc); L, late dcSSc; and C, healthy controls, whose cells were cultured in M, control cell culture medium or collagen type I (CI), same medium with type I collagen added. Colour-coding of relative gene expression levels from lower to higher is shown at the bottom.
To confirm further that gene expression in PBMC from patients with early and late dcSSc did not differ significantly at baseline, direct comparisons between these groups were performed. Comparison of PBMCs from patients with late dcSSc and patients with early dcSSc revealed differences in basal expression of only two genes: expression of suppressor of cytokine signalling 7 (SOCS7) was higher is patients with late dcSSc, whereas expression of Bcl-2-binding protein, also known as Bcl-2 antagonist of cell death (BAD), was higher in patients with early dcSSc. Considering that early disease is often more active, such limited differences were somewhat unexpected, and suggested that early disease may not entail a more active immune response without additional antigenic stimulation.
Effect of in vitro stimulation with CI on gene expression in cultured PBMC
The effect of stimulation with CI on gene expression in PBMC from patients with dcSSc was analysed using three approaches. First, gene expression was compared in CI-stimulated PBMC from patients with early dcSSc against CI-stimulated PBMC from healthy controls and, separately, in CI-stimulated PBMC from patients with late dcSSc against CI-stimulated PBMC from healthy controls. Secondly, direct comparison of matching (cultured with and without CI) samples was performed in each of the volunteer groups (healthy controls, early dcSSc, late dcSSc). Thirdly, cluster analyses were performed to determine whether genes differed between the groups in a co-ordinated fashion. The results of these three approaches to data analyses are described in detail below.
Gene expression in PBMC from patients with early dcSSc cultured with CI differed from gene expression in PBMC from healthy controls cultured with CI with regard to 34 genes (Table 2, compared to the differences in expression of only 27 genes in non-stimulated PBMC between these volunteer groups). Gene expression in PBMC from patients with dcSSc with late disease cultured with CI differed from gene expression in PBMC from healthy controls cultured with CI with regard to 29 genes (Table 2, compared to the differences in expression of 25 genes in non-stimulated PBMC between these volunteer groups). Of note, numerous genes that were different between patients with dcSSc and healthy controls at baseline remained different after stimulation with CI: 23 of 42 genes listed in Table 2 showed differences in at least three comparisons shown. Nevertheless, these analyses suggested that culturing with CI induced substantial changes in gene expression in PBMC from patients with early dc SSc, but rather limited changes in PBMC from patients with late dcSSc.
Comparison of matched PBMC samples cultured with CI versus cultured without CI revealed that culture of PBMC with CI caused changes in expression of eight genes in healthy controls (n = 8) (Table 3). Culture with CI of PBMC from patients with early dcSSc (n = 13) resulted in changes in expression of five genes, of which only two (RUNX2 and EMAP2) overlapped with the previous comparison (Table 3). There was no difference in gene expression when PBMC from patients with late dcSSc were cultured with or without CI (n = 11). These observations suggest that PBMC from patients with early dcSSc respond, whereas PBMC from patients with late dcSSc do not respond to stimulation with CI; and that either respond to CI stimulation differently from PBMC from healthy controls.
Table 3.
Effect of in vitro stimulation with collagen I (CI) on gene expression in cultured peripheral blood mononuclear cells (PBMC) based on comparison of matched samples (PBMC from the same volunteers, cultured with CI versus cultured without CI) in healthy controls (Ctrls), patients with early dcSSc (Early) and patients with late dcSSc (Late). Significant fold differences (P < 0·05) are shown. Positive values indicate fold higher and negative indicate fold lower levels of gene expression in indicated CI-stimulated cultures compared with cells cultured in control medium (M).
| Gene | UniGene | OMIM | Full gene name | CI versus M Ctrls | CI versus M Early | CI versus M Late |
|---|---|---|---|---|---|---|
| IFN-β | Hs.93177 | 147640 | Interferon β1 | +1·6 | ||
| IL11 | Hs.467304 | 147681 | Interleukin 11 | −1·7 | ||
| SAA1 | Hs.632144 | 104750 | Serum amyloid A1 | +2·6 | ||
| TLR7 | Hs.659215 | 300365 | Toll-like receptor 7 | +1·5 | ||
| TLR8 | Hs.660543 | 300366 | Toll-like receptor 8 | +1·6 | ||
| TRAF1 | Hs.531251 | 601711 | TNF receptor-associated factor 1 | +1·6 | ||
| RUNX2 | Hs.535845 | 600211 | Runt-related transcription factor 2 | +2·9 | +2·1 | |
| EMAP-2 | Hs.591680 | 603605 | Endothelial monocyte-activating polypeptide 2 | +3·1 | +5·8 | |
| EGR2 | Hs.1395 | 129010 | Early growth response 2 | +1·8 | ||
| FOSL1 | Hs.283565 | 136515 | FOS-like antigen 1 | +1·6 | ||
| JAK2 | Hs.656213 | 147796 | Janus kinase 2 | +2·2 |
Cluster analyses revealed that changes in expression of numerous genes occurred in a co-ordinated fashion (Fig. 1). The results of cluster analyses also show visually that the differences between patients and controls are more profound than the differences induced by culturing with CI.
Validation of cDNA array data
Earlier mRNA and protein data from large independent SSc populations in our previous studies and published works of others validate our results, particularly for CCL2/MCP-1 [13], CCL5/regulated upon activation normal T cell expressed and secreted (RANTES) [14] and cytotoxic T lymphocyte antigen 4 (CTLA-4) [28]. To validate the data from these particular cDNA arrays and these particular samples, two genes were selected that were elevated in patients with dcSSc compared with healthy controls, type II activin receptor (ACVR2) and MCP-1 (CCL2). ACVR2 mRNA was quantified by real-time PCR in some of the remaining left-over cDNA samples after array analyses were complete. The real-time PCR data correlated well with the findings in cDNA array experiments for this gene (Fig. 2a). The levels of MCP-1 were tested by ELISA of fresh PBMC supernates after 24 h of cells culture. Only some of the PBMC samples in this assay matched the samples utilized for the array experiments. Therefore, correlative analyses between mRNA (array data) and protein levels (ELISA data) could not be performed. However, mean concentrations of MCP-1 in PBMC supernates from early and late dcSSc groups were each higher than those in PBMC supernates from healthy controls (Fig. 2b).
Fig. 2.
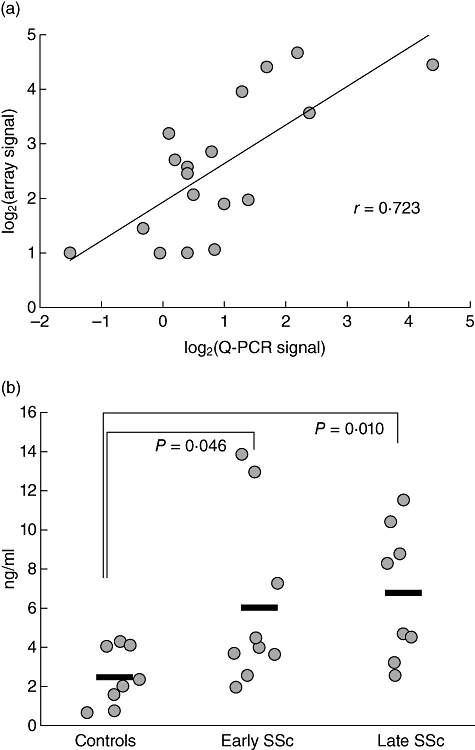
Validation of the array data by real-time polymerase chain reaction of matching cDNA samples for type II activin receptor (AVCR2) mRNA (a) and by enzyme-linked immunosorbent assay for monocyte chemotactic protein-1 (MCP-1) of cell culture supernates from partially matching healthy controls (n = 8), early diffuse cutaneous systemic sclerosis (dcSSc) (n = 9) and late dcSSc (n = 8) patients (p). (b) Individual measures are shown by circles and mean values for each group are shown by horizontal bars.
Discussion
This study assessed two aspects of gene expression in PBMC of patients with dcSSc: (i) differences between early dcSSc, late dcSSc and healthy controls; and (ii) effect of in vitro stimulation with CI on gene expression. Although this study is, by definition, a descriptive survey of gene expression, it provided important mechanistic clues that will be addressed in future targeted investigations of individual genes suggested here for their possible involvement in the pathogenesis of dcSSc. By virtue of the DNA arrays utilized, this study focused upon the genes whose protein products are known to be involved in autoimmune and inflammatory processes. Because of such specific focus, numerous genes were found whose expression was changed in patients or upon culture with CI. Basal expression levels of numerous genes were different in patients with dcSSc compared to healthy controls (see Table 2 and Fig. 1). There were significant differences in the expression levels of chemokines, cytokines and their receptors; intracellular signalling molecules; factors controlling apoptotic pathways and proliferation pathways; and molecules of the innate immune system. These changes are indicative of a substantial imbalance of immune regulation in patients with dcSSc.
The experiments revealed differences in responsiveness to culturing with or without CI between PBMC from patients with late dcSSc, patients with early dcSSc and from healthy controls (see Tables 2 and 3 and Fig. 1). The results suggests that in patients with late disease, the immune system is already activated to a significant extent at the basal level by CI acting as a major autoantigen, so that additional exposure to CI has minimal, if any, further activating effect. In contrast to late dcSSc, PBMC from patients with early dcSSc are responsive to culturing with CI, but that their responsiveness is different from that of PBMC from controls.
The data suggest an important link between the adaptive and innate branches of immunity in scleroderma. Although stimulation with a specific antigen (CI) was used, expression levels of numerous genes involved in the innate immunity were changed (see Tables 2 and 3), notably of Toll-like receptors (TLR)-4, -7 and -8. These observations are in agreement with previous reports of others on the potential roles of TLR-4 [29,30] and other Toll-like receptors [31] in scleroderma.
The results of our recent multi-centre, randomized, double-blind, placebo-controlled trial of oral type I collagen suggested that patients with late-phase dcSSc experienced a significant improvement in their disease after 12 months of oral CI [23]. The dose of oral CI was 500 µg/day; immune cells in the gut-associated lymphoid tissue may have encountered CI concentration at the 10 µg/ml level. In tissues such as skin, lungs, other internal organs and blood vessel walls, the amount of collagen available to interact with antigen-presenting cells would probably also reach this level. This is comparable with the stimulatory concentration of 5 µg CI in culture in the current study. The difference between the current in vitro study and the mentioned trial of oral collagen is that the current study investigated rapid changes in steady-state mRNA levels in isolated PBMC cell culture as an immediate response to CI, whereas the clinical trial addressed the overall disease response after a year of CI therapy. The early in vitro responses are of interest in themselves and responses after prolonged exposure may be different. We are currently analysing our data on gene expression of PBMC from patients treated for 12 months with oral CI versus placebo, in an attempt to bridge our molecular findings with clinical changes in these patients.
This study has certain strengths and weaknesses. The strengths are that we studied only patients with dcSSc, ensuring a homogeneous population; and that the patients did not have major organ damage that required serious treatment, meaning that the confounding effect of immunosuppressive therapies was not a concern in this study. A novel aspect of our study was that we investigated the spectrum of disease duration by comparing patients with early and late dcSSc. The central novelty of this project was that the primary PBMC from the patients and healthy controls were stimulated in vitro with CI in order to investigate the responsiveness to CI as an autoantigen. Although such design allowed for important new data, the cells had to be cultured for 24 h; this approach prevented easy comparisons of our data with the results of others who studied basal gene expression immediately upon obtaining the cells from volunteers. We chose to study unseparated PBMC, as opposed to purified cell subtypes, because such an approach allows for antigen (CI) processing and presentation to lymphocytes in cell culture. It is also possible that the differences in expression profiles were due in part to differences in PBMC composition between patients and controls. However, patients with early dcSSc and late dcSSc did not differ in differential cell counts (see Table 1), nor was there a difference in PBMC composition between the samples cultured with and without CI (matching samples were compared in Table 3). Although our approach did not allow for characterization of purified cell types, it was more representative of the realistic disease process, as the inflammatory infiltrates in patients with SSc are composed of antigen-presenting cells and lymphocytes [6–10], and interactions between these two types of cells are central to autoimmune activation. It is also possible that activation of non-immune collagen-binding receptors, such as integrins, discoidin domain receptors, glycoprotein VI, leucocyte-associated immunoglobulin-like receptor-1 (LAIR) and mannose receptor family, contributed to the observed changes in gene expression [32,33].
In conclusion, expression of genes known for their involvement in autoimmunity and inflammation is dramatically different in PBMC from patients with early dcSSc, late dcSSc and healthy controls. Culturing with collagen type I has a more prominent effect on gene expression in PBMC from patients with early SSc and a rather limited effect on gene expression in PBMC from patients with late dcSSc. Therefore, collagen may be a more dominant autoantigen in early dcSSc, resulting in the changes in the expression of cytokines, chemokines, their receptors, signalling molecules and other factors when PBMC are exposed to collagen. The reasons for this phenomenon are not apparent from our study. It may result, in part, from a decrease in immunity to collagen in patients with late state dcSSc. Indeed, we found that patients with early stage dcSSc, in contrast to late stage dcSSc, have more IFN-γ production by their PBMC when cultured with collagen or collagen fragments [34].
Disclosure
Dr Arnold E. Postlethwaite owns stock or stock options in arGentis Pharmaceuticals. Dr Arnold E. Postlethwaite is named on a patent owned by University of Tennessee Research Corporation for use of Type I collagen oral tolerance to treat scleroderma and fibrosing diseases. The other authors have nothing to disclose.
Acknowledgments
This work was supported by National Institutes of Health (grant numbers N01-AR922422, P50-AR44890); and by the Department of Veterans Affairs (Merit Review awards to S.P.A. and A.E.P.).
References
- 1.Postlethwaite AE. Role of T cells and cytokines in effecting fibrosis. Int Rev Immunol. 1995;12:247–58. doi: 10.3109/08830189509056716. [DOI] [PubMed] [Google Scholar]
- 2.Postlethwaite AE. Bidirectional interactions between T cells and the extracellular matrix. Transpl Immunol. 1997;5:289–91. doi: 10.1016/s0966-3274(97)80010-x. [DOI] [PubMed] [Google Scholar]
- 3.Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol. 2008;83:237–44. doi: 10.1189/jlb.0707504. [DOI] [PubMed] [Google Scholar]
- 4.Atamas SP, White B. The role of chemokines in the pathogenesis of scleroderma. Curr Opin Rheumatol. 2003;15:772–7. doi: 10.1097/00002281-200311000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Atamas SP, White B. Cytokine regulation of pulmonary fibrosis in scleroderma. Cytokine Growth Factor Rev. 2003;14:537–50. doi: 10.1016/s1359-6101(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 6.Manetti M, Neumann E, Muller A, et al. Endothelial/lymphocyte activation leads to prominent CD4+ T cell infiltration in the gastric mucosa of patients with systemic sclerosis. Arthritis Rheum. 2008;58:2866–73. doi: 10.1002/art.23806. [DOI] [PubMed] [Google Scholar]
- 7.Xie Y, Zhang X, Wakasugi S, Makino T, Inoue Y, Ihn H. Immunohistochemical characterization of the cellular infiltrate in localized scleroderma. Int J Dermatol. 2008;47:438–42. doi: 10.1111/j.1365-4632.2008.03615.x. [DOI] [PubMed] [Google Scholar]
- 8.Hebbar M, Gillot JM, Hachulla E, et al. Early expression of E-selectin, tumor necrosis factor alpha, and mast cell infiltration in the salivary glands of patients with systemic sclerosis. Arthritis Rheum. 1996;39:1161–5. doi: 10.1002/art.1780390713. [DOI] [PubMed] [Google Scholar]
- 9.Kraling BM, Maul GG, Jimenez SA. Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology. 1995;63:48–56. doi: 10.1159/000163933. [DOI] [PubMed] [Google Scholar]
- 10.Wells AU, Lorimer S, Majumdar S, et al. Fibrosing alveolitis in systemic sclerosis: increase in memory T-cells in lung interstitium. Eur Respir J. 1995;8:266–71. doi: 10.1183/09031936.95.08020266. [DOI] [PubMed] [Google Scholar]
- 11.Luzina IG, Todd NW, Nacu N, et al. Regulation of pulmonary inflammation and fibrosis through expression of integrins alphaVbeta3 and alphaVbeta5 on pulmonary T lymphocytes. Arthritis Rheum. 2009;60:1530–9. doi: 10.1002/art.24435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luzina IG, Atamas SP, Wise R, et al. Occurrence of an activated, profibrotic pattern of gene expression in lung CD8+ T cells from scleroderma patients. Arthritis Rheum. 2003;48:2262–74. doi: 10.1002/art.11080. [DOI] [PubMed] [Google Scholar]
- 13.Luzina IG, Atamas SP, Wise R, Wigley FM, Xiao HQ, White B. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol. 2002;26:549–57. doi: 10.1165/ajrcmb.26.5.4683. [DOI] [PubMed] [Google Scholar]
- 14.Atamas SP, Yurovsky VV, Wise R, et al. Production of type 2 cytokines by CD8+ lung cells is associated with greater decline in pulmonary function in patients with systemic sclerosis. Arthritis Rheum. 1999;42:1168–78. doi: 10.1002/1529-0131(199906)42:6<1168::AID-ANR13>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 15.Sakkas LI, Xu B, Artlett CM, Lu S, Jimenez SA, Platsoucas CD. Oligoclonal T cell expansion in the skin of patients with systemic sclerosis. J Immunol. 2002;168:3649–59. doi: 10.4049/jimmunol.168.7.3649. [DOI] [PubMed] [Google Scholar]
- 16.Stuart JM, Postlethwaite AE, Kang AH. Evidence for cell-mediated immunity to collagen in progressive systemic sclerosis. J Lab Clin Med. 1976;88:601–7. [PubMed] [Google Scholar]
- 17.McKown KM, Carbone LD, Bustillo J, Seyer JM, Kang AH, Postlethwaite AE. Induction of immune tolerance to human type I collagen in patients with systemic sclerosis by oral administration of bovine type I collagen. Arthritis Rheum. 2000;43:1054–61. doi: 10.1002/1529-0131(200005)43:5<1054::AID-ANR14>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 18.Kravis TC, Ahmed A, Brown TE, Fulmer JD, Crystal RG. Pathogenic mechanisms in pulmonary fibrosis: collagen-induced migration inhibition factor production and cytotoxicity mediated by lymphocytes. J Clin Invest. 1976;58:1223–32. doi: 10.1172/JCI108576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schrier DJ, Phan SH, Ward PA. Cellular sensitivity to collagen in bleomycin-treated rats. J Immunol. 1982;129:2156–9. [PubMed] [Google Scholar]
- 20.Warrington KJ, Nair U, Carbone LD, Kang AH, Postlethwaite AE. Characterisation of the immune response to type I collagen in scleroderma. Arthritis Res Ther. 2006;8:R136. doi: 10.1186/ar2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan H, Fleming J, Pritchard DK, et al. Combined analysis of monocyte and lymphocyte messenger RNA expression with serum protein profiles in patients with scleroderma. Arthritis Rheum. 2008;58:1465–74. doi: 10.1002/art.23451. [DOI] [PubMed] [Google Scholar]
- 22.Tan FK, Zhou X, Mayes MD, et al. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology (Oxf) 2006;45:694–702. doi: 10.1093/rheumatology/kei244. [DOI] [PubMed] [Google Scholar]
- 23.Postlethwaite AE, Wong WK, Clements P, et al. A multicenter, randomized, double-blind, placebo-controlled trial of oral type I collagen treatment in patients with diffuse cutaneous systemic sclerosis. I. Oral type I collagen does not improve skin in all patients but may improve skin in late-phase disease. Arthritis Rheum. 2008;58:1810–22. doi: 10.1002/art.23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang AH, Piez KA, Gross J. Characterization of the alpha-chains of chick skin collagen and the nature of the NH2-terminal cross-link region. Biochemistry. 1969;8:3648–55. doi: 10.1021/bi00837a023. [DOI] [PubMed] [Google Scholar]
- 25.Kang AH, Seyer JM. Compilation of collagen data. In: Fasma GD, editor. Handbook of biochemistry. Cleveland, OH: CRC Press; 1996. pp. 474–89. [Google Scholar]
- 26.Rus V, Atamas SP, Shustova V, et al. Expression of cytokine- and chemokine-related genes in peripheral blood mononuclear cells from lupus patients by cDNA array. Clin Immunol. 2002;102:283–90. doi: 10.1006/clim.2001.5182. [DOI] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 28.Sato S, Fujimoto M, Hasegawa M, et al. Serum soluble CTLA-4 levels are increased in diffuse cutaneous systemic sclerosis. Rheumatology (Oxf) 2004;43:1261–6. doi: 10.1093/rheumatology/keh303. [DOI] [PubMed] [Google Scholar]
- 29.van Lieshout AW, Vonk MC, Bredie SJ, et al. Enhanced interleukin-10 production by dendritic cells upon stimulation with Toll-like receptor 4 agonists in systemic sclerosis that is possibly implicated in CCL18 secretion. Scand J Rheumatol. 2009;38:282–90. doi: 10.1080/03009740802572467. [DOI] [PubMed] [Google Scholar]
- 30.Fineschi S, Goffin L, Rezzonico R, et al. Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of Toll-like receptor 4. Arthritis Rheum. 2008;58:3913–23. doi: 10.1002/art.24049. [DOI] [PubMed] [Google Scholar]
- 31.Lafyatis R, York M. Innate immunity and inflammation in systemic sclerosis. Curr Opin Rheumatol. 2009;21:617–22. doi: 10.1097/BOR.0b013e32832fd69e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogel WF. Collagen-receptor signaling in health and disease. Eur J Dermatol. 2001;11:506–14. [PubMed] [Google Scholar]
- 33.Leitinger B, Hohenester E. Mammalian collagen receptors. Matrix Biol. 2007;26:146–55. doi: 10.1016/j.matbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Postlethwaite AE, Wong WK, Ingels J, Kang AH, Clements P, Furst D. Maximal T cell reactivity to type I collagen (CI) is present during the first 3 years of diffuse systemic sclerosis (SSc) Arthritis Rheum. 2005;52:S462. Abstract. [Google Scholar]