

Abstract
In active Graves' orbitopathy (GO), proinflammatory cytokines predominate. Circulating thyroid stimulating hormone (TSH)-receptor antibodies (TRAb) have been correlated with GO clinical activity and severity. In preliminary studies rituximab (RTX), an anti-CD 20 monoclonal antibody, has induced clinical improvement of active GO without a change in serum anti-thyroid antibodies. We have studied whether RTX in GO acts by affecting proinflammatory cytokines and thyroid and orbital-directed antibodies. Ten patients with GO were treated with RTX, administered twice intravenously (i.v.) (1000 mg) at days 1 and 15, and 20 with methylprednisolone, administered weekly i.v. (500 mg), for 16 weeks. Patients were studied before treatment, at B cell depletion and at 4, 8, 16, 20, 30 and 50 weeks. Peripheral lymphocytes, serum interleukin (sIL)-6, sIL-6r, chemokine (C-X-C motif) ligand 10 (CXCL10), TRAb and stimulating antibodies (TSAb) and autoantibodies against orbital calsequestrin, collagen XIII and flavoprotein subunit of succinate dehydrogenase (FP-SDH) were measured at baseline and after treatment. Serum IL-6 and sIL-6R concentrations did not change after RTX [P = not significant (n.s.)]. Serum CXCL10 increased after RTX at B cell depletion and at 30 weeks (P < 0·003). Serum TSAb did not change in relation to TRAb, nor did antibodies against orbital antigens (P = n.s.). In conclusion, this study shows that RTX in GO does not affect humoral reactions. The observed increase of serum CXCL10 concentrations at B cell depletion may result from cell lysis. We suggest that RTX may exert its effect in GO by inhibiting B cell antigen presentation.
Keywords: CD20, cytokines, Graves'orbitopathy, rituximab, TSH-receptor antibodies
Introduction
Graves' disease (GD) is a multi-organ-specific autoimmune disease involving the thyroid, the orbit and the skin. The hyperthyroidism of GD results from a B cell-driven mechanism leading to sustained production of immunoglobulin G (IgG) stimulating the thyroid stimulating hormone (TSH) receptor (TSAb) on the thyrocytes [1]. Graves' orbitopathy (GO) is the most frequent extrathyroidal manifestation of GD and is thought to be due to immunological cross-reactivity between thyroid and orbital tissue antigens, although its precise pathogenesis has not yet been clarified. The immune reactions in orbital tissues appear to be orchestrated by resident immune cells or cells recruited from the bone marrow through the expression and release of cytokines. It has been shown that in the active phase of GO there is predominant production of proinflammatory and T helper type 1 (Th1)-derived cytokines such as interleukin (IL)-6 and IL-1, and interferon (IFN)-γ-induced chemokines, such as chemokine (C-X-C motif) ligand 10 (CXCL10), while Th2-derived cytokines, including IL-4, IL-5 and IL-10, are associated more probably with the inactive phase of GO [2,3]. In previous work, IL-6 and the soluble receptor (sIL-6R), which acts as an IL-6R agonist, have been found to be elevated in the serum of patients with active GO independently of the thyroid function or treatment [4] when compared to IL-10 and tumour necrosis factor (TNF)-α. More recently, Antonelli et al. [5] reported that serum CXCL10 concentrations are elevated in patients with active GO and decreased in long-standing disease, thus suggesting that this may result from the negative feedback of T helper type 2 (Th2) cytokines on IFN-γ production. One candidate autoantigen involved in thyroid orbit cross-reactivity is the TSH receptor, which has been shown to be expressed on orbital pre-adipocyte fibroblasts in vitro and on other tissues [6]. Circulating TSH-receptor antibodies (TRAb), both TSH receptor binding antibodies and TSAb, have been found to correlate significantly with GO clinical activity [7]. Tsui et al. have observed very recently that orbital fibroblasts of Graves' disease patients co-express TSH receptor and IGF-1 receptor that could bind autoantibodies in TAO [8]. This might result in stimulation of fibroblast growth and prolongation of T cell survival, thereby perpetuating the autoimmune process. More recently, it has been reported that at each time-point of the disease course, patients with severe GO have higher serum TRAb levels than those with mild–moderate GO [9]. However, the lack of variation of serum TRAb titres in relation to disease stabilization [10] may not be consistent with a major role of TRAb in GO pathogenesis. Several studies have focused upon other putative orbital autoantigens, derived mainly from eye muscles [11]. Serum antibodies against orbital antigens such as calsequestrin, XIII collagen and the flavoprotein subunit of succinate dehydrogenase (FP-SDH), have been measured in patients with GO, and it is not clear whether these antibodies have a pathogenic role or appear as a epiphenomenon [12].
Treatment with rituximab (RTX), a monoclonal chimeric murine/human antibody that binds the CD20 antigen and causes B cell depletion, has been providing evidence on the role of B cells in systemic and organ-specific autoimmunity. To date, it is not known if the therapeutic effect of RTX in autoimmune diseases is mediated by modifications of the humoral immune response. In 2007 we were the first to report in a study significant improvement of clinically active GO and also of proptosis after RTX when compared to intravenous (i.v.) methylprednisolone [13]. Conversely, we were not able to observe significant changes of serum anti-thyroglobulin antibodies (TgAb), thyroperoxidase antibodies (TPOAb) and TRAb in relation to the consequent peripheral B cell depletion. Circulating TRAb and TgAb were correlated only significantly and negatively with the progressive attainment of euthyroidism during follow-up. Although our findings have been confirmed recently by Chong et al. [14], data on RTX in GO are scarce and derived mainly from single case reports or from case–control studies on small series of patients. In this work we have studied the pattern of serum proinflammatory cytokines, IL-6, sIL-6R and the chemokine CXCL10 of serum TSAb in relation to TRAb and of circulating autoantibodies to previously described orbital antigens in response to treatment with RTX of patients with active GO, based on data obtained from an open-label study.
Patients and methods
Patients
Ten patients with GD, eight female and two male, aged 31–54 years [mean ± standard error (s.e.) 46·6 ± 2·2], of whom eight had active GO and two only lid signs, were studied in our thyroid eye clinic [15]. At the time of RTX therapy, two patients were newly hyperthyroid and untreated; eight patients were euthyroid, four on methimazole (MMI), three in remission after previous anti-thyroid therapy and one patient on L-thyroxine (L-T4) for 12 months, after previous thyroidectomy. The mean ± s.e. clinical activity score (CAS) [16] was 4 ± 0·6. Severity was classified by NOSPECS [17]: four patients had class 20, four 2a and two 2b, while mean ± s.e. proptosis was 21·1 ± 0·72 mm; diplopia was intermittent in two patients, inconstant in one and constant in one, with six patients having no diplopia. For comparison, parameters of humoral immunity were also studied in a group of 20 patients (17 female, three male, mean ± s.e. age 55·4 ± 2·9) submitted to standard immunosuppressive steroid therapy for active GO, as described in previous work [13]. In this group, 19 patients had GD and were euthyroid, 16 were on anti-thyroid therapy, two had disease remission and one was on L-T4 after thyroidectomy. One patient had euthyroid Graves' orbitopathy. Three of these patients had mild, 13 had moderate and four had severe eye disease, respectively.
Clinical assessment and therapeutic protocol
All patients were studied by a complete ophthalmological examination, as reported previously [18]. RTX was administered according to the protocol recommended for the treatment of autoimmune diseases, which consists of two i.v. infusions of 1000 mg of RTX at a 2-week interval. RTX infusion was always preceded by the administration of paracetamol (1 g) and chlorphenamine (10 mg) orally in order to prevent possible allergic reactions. The study protocol was approved by the ethics committee of our institution and the patients gave informed consent for therapy. Patients were studied before treatment, at the time of B cell depletion and 4, 8, 16, 20, 30 and 50 weeks after RTX administration. In the control group, 500 mg of methylprednisolone were administered i.v. once a week for 16 weeks. These patients were studied before and 20 weeks after therapy and followed-up to 12 months after treatment.
Peripheral blood lymphocyte analysis
Peripheral blood lymphocytes were studied at baseline and 2, 4, 8, 16, 20, 30 and 50 weeks after RTX. Cytofluorimetric analysis was performed with the standard immunophenotypic panel (CD3+, CD3+4+, CD3+8+, CD3+DR+, CD20+, CD19+5+, CD56+16+3) on aliquots of approximately 105 lymphocytes, submitted to standard triple-staining procedures in order to carry out immunogating with CD45. Thus, pairs of monoclonal antibodies to subpopulations of T, B and natural killer (NK) cells were analysed and processed in the flow cytometer (BD Facscan, CellQuest software; Becton-Dickinson, San Jose, CA, USA).
Measurement of IL-6, sIL-6R serum cytokines and CXCL10 chemokine
Serum IL-6, sIL-6r and CXCL10 concentrations were measured using a high-sensitivity, commercially available kit (Quantikine R&D Systems, Minneapolis, MN, USA). Detection sensitivity was 1·56 U/l for IL-6, 31·2 U/l for sIL-6r and 3·9 U/l for CXCL10. Tests were performed in duplicate. Quality control pools of low, normal and high concentrations for all parameters were included in each assay.
Measurement of serum anti-TSH-receptor and orbital antibodies and TSH concentrations
Serum TRAb were measured in nine of 10 patients using a third-generation TRAK human lumitest (Brahms AG, Henningsdorf/Berlin, Germany) and each sample was tested in duplicate. The normal range of TRAb values is 0–1·5 U/l. Serum TSAb activity was tested in the Chinese hamster ovary-TSH receptor (CHO-TSHr) JP26 stable cell line [19] by determining cyclic adenosine monophosphate (cAMP) accumulation in a radioimmunological assay, as reported previously [20]. Cells were incubated with 5% serum in hypotonic medium. TSAb values in Graves' patients are expressed as the net stimulating activity of TSAb after subtracting cAMP accumulation of a pool of negative control sera, obtained from a series of 10 patients under TSH-suppressive therapy. The sensitivity of the assay was 6·8 pmol/ml cAMP and the intra-assay CV was 11·6%. The anti-orbit antibodies against eye muscle and orbital tissue were measured by enzyme-linked immunosorbent assay (ELISA) with calsequestrin, collagen XIII and the FP-SDH collagen as antigens. Each sample was tested in duplicate using four wells, two wells containing the antigen of interest and two bovine serum albumin (BSA)-coated wells. Each plate also contained one blank well, one positive and negative control. The final sample, expressed as U/l, was obtained as the subtraction of mean BSA and blank from the mean sera [12]. Serum TSH concentrations were measured using the AutoDELFIA technique (Perkin–Elmer-Life Sciences, Wallac Oy, Turku, Finland).
Statistical analysis
The changes of serum antibody levels and of cytokines and chemokine concentrations in response to RTX therapy were analysed by repeated-measures analysis of variance (anova). Parameters of correlations were studied by non-parametric Spearman's rank test. Statistical significance was defined as P < 0·05. Values are all shown as mean ± s.e.
Results
Effects of RTX on B and T lymphocytes
RTX induced peripheral B cell depletion in all but one patient after the first of the two administered doses (2 weeks). All patients tolerated treatment well, with occurrence of minor hypersensitivity reactions in three of 10 patients at first infusion. One patient, after RTX, had total peripheral CD20+ cell depletion, but persistence of 3–5% CD19+ cells in the circulation [21], indicating incomplete depletion. The mean duration of peripheral B cell depletion was 16·7 ± 2·1 weeks. RTX therapy had no effect on peripheral total CD4, CD8 and CD3 cells at any time-point of therapy or follow-up. Of interest is the observation of a slight, non-significant decrease of peripheral DR+CD3+ cells of about 21% from baseline at about 16 weeks, and subsequent normalization at 50 weeks (not shown).
Effects of RTX on serum IL-6 and sIL-6r
Baseline serum IL-6 and sIL-6R concentrations were 30·6 ± 26·4 and 485·3 ± 40·3 pg/ml, respectively. After RTX, serum IL-6 concentrations did not change significantly (P = n.s.), nor did their values correlate with peripheral B cell depletion, despite the observed slight decrease (Fig. 1a). Similar findings were also observed for serum sIL-6R concentrations (P = n.s.) (Fig. 1b). No significant changes of serum IL-6 and sIL-6R concentrations were found in patients treated with steroid therapy, as shown in Fig. 2b and c.
Fig. 1.
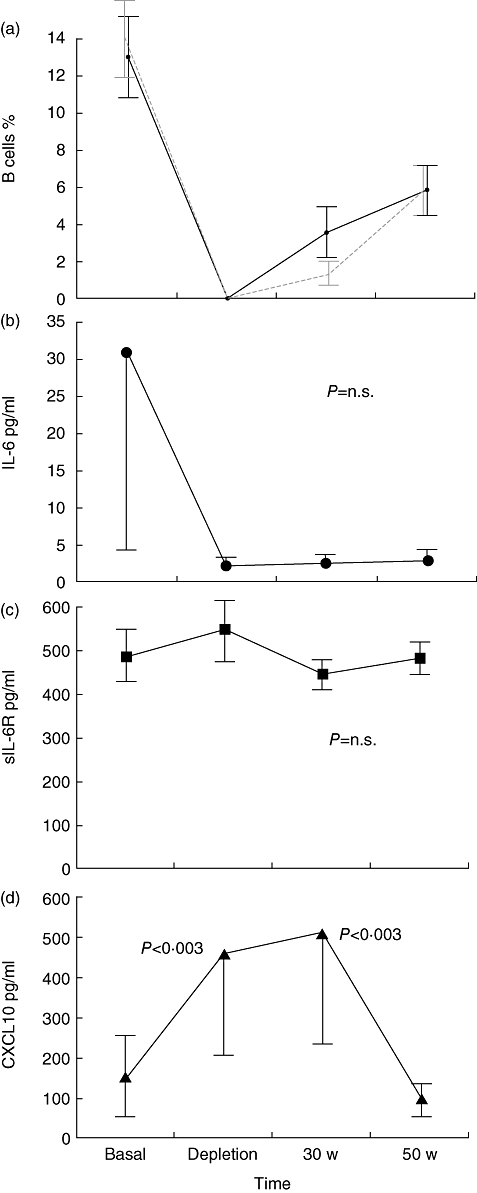
Effects of RTX on peripheral CD 20+ ( ) and CD 19+ (
) and CD 19+ ( ) cells (a), serum IL-6 (b), sIL-6R (c) and chemokine (C-X-C motif) ligand 10 (CXCL10) (d) concentrations in basal condition, at B cell depletion, at 30 and 50 weeks of follow-up. Data are shown as mean ± standard error.
) cells (a), serum IL-6 (b), sIL-6R (c) and chemokine (C-X-C motif) ligand 10 (CXCL10) (d) concentrations in basal condition, at B cell depletion, at 30 and 50 weeks of follow-up. Data are shown as mean ± standard error.
Fig. 2.
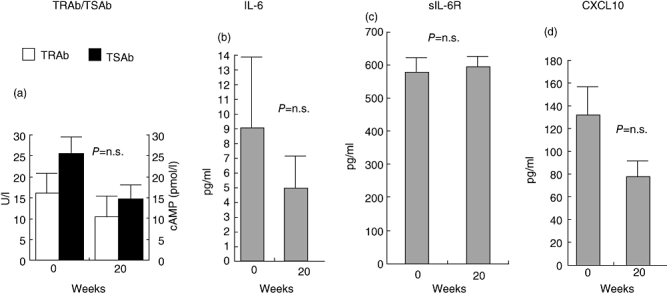
Effects of intravenous glucocorticoids on thyroid stimulating hormone (TSH)-receptor antibodies, TBII and TSAb (a), interleukin (IL)-6 (b), serum interleukin (sIL)-6-R (c) and chemokine (C-X-C motif) ligand 10 (CXCL10) (d) at baseline and at 20 weeks after treatment. Data are shown as mean ± standard error.
Effects of RTX on serum CXCL10
Basal serum CXCL10 concentrations were 151·6 ± 93·5 pg/ml. A significant increase of CXCL10 was observed in patients treated with RTX at the time at CD20+ cell depletion (2 weeks) and at 30 weeks (P < 0·003). At 50 weeks serum CXCL10 concentrations returned to baseline levels (Fig. 1c). No significant changes of serum CXCL10 concentrations were observed in patients treated with steroids (Fig. 2d).
Effects of RTX on serum TRAb and orbital antibodies
As shown in previous studies [13], circulating TPOAb did not change after RTX, whereas mean serum levels of TRAb did not change significantly (anova; P = n.s., Fig. 3b), and correlated only slightly negatively with time at about 75 weeks of follow-up (Spearman's r = –0·33, P < 0·01; not shown), in relation to the attainment of euthyroidism, but not to RTX-induced B cell depletion.
Fig. 3.
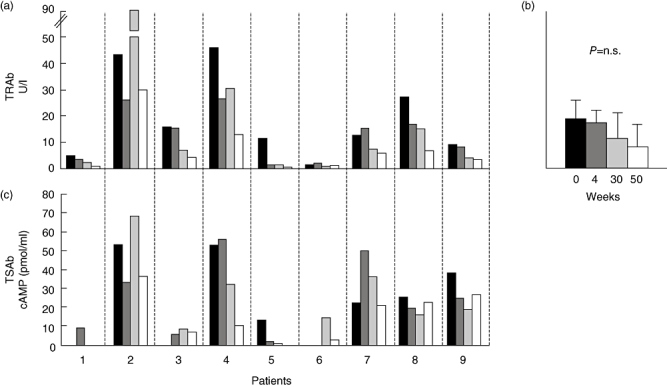
Effect of rituximab (RTX) on thyroid stimulating hormone (TSH)-receptor antibodies, TRAb (a) and TSAb (c) at baseline ( ), at CD20 B cell depletion (
), at CD20 B cell depletion ( ), at 30 (
), at 30 ( ) and 50 (□) weeks of follow-up in each treated patient. In (b) mean serum TRAb were analyzed by analysis of variance (P = not significant) at the same time-points of the study.
) and 50 (□) weeks of follow-up in each treated patient. In (b) mean serum TRAb were analyzed by analysis of variance (P = not significant) at the same time-points of the study.
In order to study whether RTX might have affected distinctively the subpopulation of TSAb within TRAb, as postulated recently [22], we measured and correlated serum TSAb after RTX to serum TRAb in nine of 10 patients in the study (Fig. 3). Of the 10 patients in the study, six were on MMI. Of these, only two were hyperthyroid and became euthyroid throughout the duration of the study, whereas the other four were already euthyroid at the time they were given RTX. Serum TRAb did not decrease significantly in the six patients before 30 weeks from treatment. Although, in euthyroid patients, baseline TSAb were not measurable, we found overall no distinct effect of RTX on TSAb in any of the patients in the study and no difference in relation to changes in the corresponding serum TRAb (Fig. 3a and c). In order to exclude that stimulation of CHO-TSHr cells might have been caused by elevated serum TSH, we also measured serum TSH concentrations in the same sera (Fig. 3c). Only one patient (no. 4) had significant elevation of serum TSH concentrations (26 mU/l) upon LT-4 discontinuation for scintigraphic evaluation of the thyroid residue after total thyroidectomy, and only a slight effect on the TSAb assay. Of note, both serum TRAb and TSAb levels did not change significantly (anova; P = n.s.) in control patients treated with standard steroid therapy (Fig. 2a), similar to what we have reported in patients treated with RTX. The mean baseline serum antibody levels against the three orbital antigens, calsequestrin, XIII collagen and FP-SDH, were 0·24 ± 0·06, 0·09 ± 0·03 and 0·13 ± 0·02 U/l, respectively, and no significant change was observed after RTX (P = n.s.) (Fig. 4).
Fig. 4.
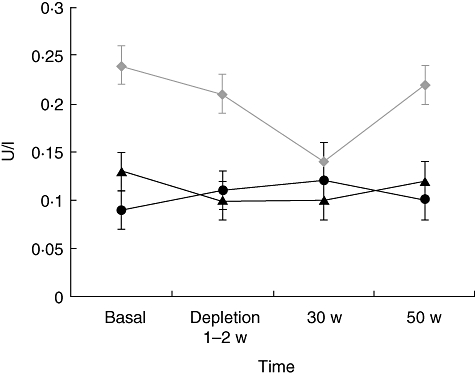
Effects of rituximab (RTX) on antibodies against orbital antigens: collagen XIII ( ), calsequestrin (
), calsequestrin ( ) and flavoprotein subunit of succinate dehydrogenase (FP-DSH) (
) and flavoprotein subunit of succinate dehydrogenase (FP-DSH) ( ) at baseline, at CD20 B cell depletion, at 30 and 50 weeks of follow-up. No significant changes were observed. Data are shown as mean ± standard error.
) at baseline, at CD20 B cell depletion, at 30 and 50 weeks of follow-up. No significant changes were observed. Data are shown as mean ± standard error.
Discussion
Our findings, with the limitation of being observed in an uncontrolled study, show that the therapeutic effect of RTX in patients with GO is not mediated through changes in humoral immunity. We have reported previously unchanged serum TRAb levels in Graves' hyperthyroid patients after RTX, not due to either MMI or the duration of treatment, and we postulated that this drug might exert its immunosuppressive function by modifying the cytokine milieu in the orbit [13] and that the therapeutic effect might result from a distinct response on intraorbital and thyroid lymphocytic infiltrates [23,24]. The rapid improvement of inflammatory signs after RTX might be due to a putative effect on the orbital proinflammatory cytokine pattern, perhaps involving the IL-6 secretion system in which sIL-6R acts as an IL-6 agonist, as has been shown previously [4]. In the present study, although we have confirmed the increase of IL-6 and sIL-6R in patients with active GO, we did not find RTX therapy to affect these cytokines and any relationship to peripheral B cell depletion. This finding suggests that RTX affects orbital inflammation in GO without interfering with the proinflammatory cytokine milieu. Serum CXCL10 concentrations have also been shown to be elevated during the active inflammatory phase of GO and to decrease when the disease becomes inactive. In this study we have observed a significant increase of serum CXCL10 concentrations at the time of B cell depletion and 30 weeks after RTX, when the disease was inactive. Despite the known high variability of serum CXCL10 measurement, an increase of this chemokine at the time of B cell depletion might be a consequence of the massive B cell lysis induced by RTX, mediated possibly by complement, as observed in other diseases [25], but we cannot explain the increased concentrations observed at 30 weeks.
It has been proposed that RTX may block the production of a new pool of plasma cells as a consequence of B cell depletion located in stromal inflamed sites, thus inhibiting the production of pathogenic antibodies, without affecting plasma cells resident in the bone marrow [26]. Recently, El Fassi and coworkers [22] have reported a significant reduction of serum TPOAb and TSAb, but not of serum TRAb, in patients with Graves' hyperthyroidism mainly without GO and treated with RTX. These authors have postulated that RTX may have a different effect on pathogenic and non-pathogenic autoantibodies, even when directed against the same antigen. In the present work, we have studied both binding and stimulating serum TSH-receptor antibodies and we have not been able to observe any specific effect of RTX on TSAb, which appeared to be unchanged and to fluctuate with an identical pattern compared with TRAb in either hyperthyroid or euthyroid patients, whether or not treated with MMI. These data are consistent with other reports in which decreased serum levels of autoantibodies do not correlate with a positive clinical response to RTX in various autoimmune diseases [27–32]. Autoantibodies against previously recognized orbital antigens have been measured in patients with active disease [33,34] as markers of orbital inflammation, and perhaps not pathogenic factors, in the initiation of GO. In this study, serum antibodies against orbital antigens also did not change in relation to RTX-induced peripheral B cell depletion. All these findings suggest that RTX does not have an effect on either thyroid or orbital autoantibodies, while acting in a distinct way on the compartments of lymphocytic infiltrates of the thyroid and the orbit, as we [23] and others [14,24] have reported recently. It is plausible that even at the orbital level RTX does not affect antibody-producing B cells, but presumably some earlier-occurring immune reaction. Indeed, the generally rapid therapeutic effect of RTX observed in autoimmune disease argues against an antibody-mediated action. Because RTX binds the CD20 antigen expressed on B cells but not on immunoglobulin (Ig)-secreting plasma cells, a putative effect on antibody production would be expected to occur after 6–12 weeks [35]. Among the possible explanations for the fairly rapid action of RTX in GO we may consider the ‘decoy immunocomplex hypothesis’[28], or perhaps the activation of complement in inducing B cell depletion [25]. The first mechanism implies that RTX-opsonized B cells may act as decoys by engaging the Fc receptor on effector cells to divert monocytes or macrophages from pathogenetic interactions with tissue-associated immunocomplexes, thereby reducing disease activity, without affecting autoantibody levels [36]. This mechanism has been found to be important in other autoimmune diseases, such as immune thrombocytopenic purpura [37,38]. Complement-mediated B cell depletion has indeed been observed to be the main mechanism for the effect of RTX on malignant B cells [39–41], and together with opsonization of B cell represents the faster mechanisms of RTX-induced B cell depletion. Although these findings, as well as the previously reported data on the significant improvement of active GO manifestations after RTX, bear the limitation of being observed in an uncontrolled study, they indicate that the effect of RTX in GO does not impact the humoral reactions linked to thyroid autoimmunity. This would support the concept that autoantibodies, including TRAb and TSAb, do not have a primary role in the pathogenesis of the orbital inflammation occurring in the active phase of GO. The observed reduction of peripheral DR+CD3+ 20 weeks after RTX, without changes of T cell subpopulations, may be consistent with a reduced DR expression on T cells and result in a persistent effect of RTX in inhibiting antigen presentation, as outlined above [28]. In conclusion, the effect of RTX in GO may occur through the activation of complement and by the inhibition of many B cell functions, including the pathway of B cell antigen presentation, with consequent decreased activation of pathogenic T cell clones. Currently ongoing randomized controlled trials will help to clarify further the relationship between RTX therapy in GO and its impact on pathophysiological immune mechanisms.
Acknowledgments
This work was supported in part by MURST, Roma and by Fondazione Cà Granda, IRCCS, Milano, Italy.
Disclosure
The authors have nothing to disclose.
References
- 1.Zakarija M, McKenzie JM. The spectrum and significance of autoantibodies reacting with the thyrotropin receptor. Endocrinol Metab Clin North Am. 1988;16:343–63. [PubMed] [Google Scholar]
- 2.Wakelkamp IM, Bakker O, Baldeschi L, Wiersinga WM, Prummel MF. TSH-R expression and cytokine profile in orbital tissue of active vs. inactive Graves' ophthalmopathy patients. Clin Endocrinol (Oxf) 2003;58:280–7. doi: 10.1046/j.1365-2265.2003.01708.x. [DOI] [PubMed] [Google Scholar]
- 3.Salvi M, Pedrazzoni M, Girasole G, et al. Serum concentrations of proinflammatory cytokines in Graves' disease: effect of treatment, thyroid function, ophthalmopathy and cigarette smoking. Eur J Endocrinol. 2000;143:197–202. doi: 10.1530/eje.0.1430197. [DOI] [PubMed] [Google Scholar]
- 4.Salvi M, Girasole G, Pedrazzoni M, et al. Increased serum concentrations of interleukin-6 (IL-6) and soluble IL-6 receptor in patients with Graves' disease. J Clin Endocrinol Metab. 1996;81:2976–9. doi: 10.1210/jcem.81.8.8768861. [DOI] [PubMed] [Google Scholar]
- 5.Antonelli A, Rotondi M, Ferrari SM, et al. Interferon-gamma-inducible alpha-chemokine CXCL10 involvement in Graves' ophthalmopathy: modulation by peroxisome proliferator-activated receptor-gamma agonists. J Clin Endocrinol Metab. 2006;91:614–20. doi: 10.1210/jc.2005-1689. [DOI] [PubMed] [Google Scholar]
- 6.Bahn RS. Clinical review 157: pathophysiology of Graves' ophthalmopathy: the cycle of disease. J Clin Endocrinol Metab. 2003;88:1939–46. doi: 10.1210/jc.2002-030010. [DOI] [PubMed] [Google Scholar]
- 7.Gerding MN, van der Meer JW, Broenink M, Bakker O, Wiersinga WM, Prummel MF. Association of thyrotrophin receptor antibodies with the clinical features of Graves' ophthalmopathy. Clin Endocrinol (Oxf) 2000;52:267–71. doi: 10.1046/j.1365-2265.2000.00959.x. [DOI] [PubMed] [Google Scholar]
- 8.Tsui S, Naik V, Hoa N, et al. Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: a tale of two antigens implicated in Graves' disease. J Immunol. 2008;181:4397–405. doi: 10.4049/jimmunol.181.6.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckstein AK, Plicht M, Lax H, et al. Thyrotropin receptor autoantibodies are independent risk factors for Graves' ophthalmopathy and help to predict severity and outcome of the disease. J Clin Endocrinol Metab. 2006;91:3464–70. doi: 10.1210/jc.2005-2813. [DOI] [PubMed] [Google Scholar]
- 10.De Bellis A, Sansone D, Coronella C, et al. Serum antibodies to collagen XIII: a further good marker of active Graves' ophthalmopathy. Clin Endocrinol (Oxf) 2005;62:24–9. doi: 10.1111/j.1365-2265.2004.02167.x. [DOI] [PubMed] [Google Scholar]
- 11.Mikozami T, Salvi M, Wall JR. Eye muscle antibodies in Graves' ophthalmopathy: pathogenic or secondary epiphenomenon? J Endocrinol Invest. 2004;27:221. doi: 10.1007/BF03345270. [DOI] [PubMed] [Google Scholar]
- 12.Gopinath B, Musselman R, Adams CL, Tani J, Beard N, Wall JR. Study of serum antibodies against three eye muscle antigens and the connective tissue antigen collagen XIII in patients with Graves' disease with and without ophthalmopathy: correlation with clinical features. Thyroid. 2006;16:967–74. doi: 10.1089/thy.2006.16.967. [DOI] [PubMed] [Google Scholar]
- 13.Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves' disease and associated ophthalmopathy with the anti-CD 20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33–40. doi: 10.1530/eje.1.02325. [DOI] [PubMed] [Google Scholar]
- 14.Khanna D, Chong KK, Afifiyan NF, et al. Rituximab treatment of patients with severe, corticosteroid-resistant thyroid-associated ophthalmopathy. Ophthalmology. 2010;117:133–9. doi: 10.1016/j.ophtha.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.European Group on Graves' Orbitopathy (EUGOGO) Wiersinga WM, Perros P, et al. Clinical assessment of patients with Graves' orbitopathy: the European Group on Graves' Orbitopathy recommendations to generalists, specialists and clinical researchers. Eur J Endocrinol. 2006;155:387–9. doi: 10.1530/eje.1.02230. [DOI] [PubMed] [Google Scholar]
- 16.Mourits MP, Prummel MF, Wiersinga WM, Koorneef L. Clinical activity score as a guide in the management of patients with Graves' ophthalmopathy. Clin Endocrinol (Oxf) 1997;47:9–14. doi: 10.1046/j.1365-2265.1997.2331047.x. [DOI] [PubMed] [Google Scholar]
- 17.Werner SC. Classification of the eye changes of Graves' disease. J Clin Endocrinol Metab. 1969;29:982–4. doi: 10.1210/jcem-29-7-982. [DOI] [PubMed] [Google Scholar]
- 18.Bartalena L, Baldeschi L, Dickinson AJ, et al. Consensus statement of the European group on Graves' orbitopathy (EUGOGO) on management of Graves' orbitopathy. Thyroid. 2008;18:333–46. doi: 10.1089/thy.2007.0315. [DOI] [PubMed] [Google Scholar]
- 19.Persani L, Tonacchera M, Beck-Peccoz P, et al. Measurement of cAMP accumulation in Chinese hamster ovary cells transfected with the recombinant human TSH receptor (CHO-R): a new bioassay for human thyrotropin. J Endocrinol Invest. 1993;16:511–19. doi: 10.1007/BF03348894. [DOI] [PubMed] [Google Scholar]
- 20.Radetti G, Persani L, Moroder W, Cortelazzi D, Gentili L, Beck-Peccoz P. Transplacental passage of anti-thyroid auto-antibodies in a pregnant woman with auto-immune thyroid disease. Prenat Diagn. 1999;19:468–71. [PubMed] [Google Scholar]
- 21.Salvi M, Vannucchi G, Campi I, et al. Rituximab treatment in a patient with severe thyroid-associated ophthalmopathy: effects on orbital lymphocytic infiltrates. Clin Immunol. 2009;131:360–5. doi: 10.1016/j.clim.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 22.El Fassi D, Banga JP, Gilbert JA, Padoa C, Hegedüs L, Nielsen CH. Treatment of Graves' disease with rituximab specifically reduces the production of thyroid stimulating autoantibodies. Clin Immunol. 2009;130:252–8. doi: 10.1016/j.clim.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Salvi M, Vannucchi G, Campi I, et al. Efficacy of rituximab treatment for thyroid-associated ophthalmopathy as a result of intraorbital B-cell depletion in one patient unresponsive to steroid immunosuppression. Eur J Endocrinol. 2006;154:511–17. doi: 10.1530/eje.1.02119. [DOI] [PubMed] [Google Scholar]
- 24.El Fassi D, Clemmensen O, Nielsen CH, Silkiss RZ, Hegedüs L. Evidence of intrathyroidal B-lymphocyte depletion after rituximab therapy in a patient with Graves' disease. J Clin Endocrinol Metab. 2007;92:3762–3. doi: 10.1210/jc.2007-1238. [DOI] [PubMed] [Google Scholar]
- 25.Golay J, Cittera E, Di Gaetano N, et al. The role of complement in the therapeutic activity of rituximab in a murine B lymphoma model homing in lymph nodes. Haematologica. 2006;91:176–83. [PubMed] [Google Scholar]
- 26.Ferraro AJ, Drayson MT, Savage CO, MacLennan IC. Levels of autoantibodies, unlike antibodies to all extrinsic antigen groups, fall following B cell depletion with Rituximab. Eur J Immunol. 2008;38:292–8. doi: 10.1002/eji.200737557. [DOI] [PubMed] [Google Scholar]
- 27.Sabahi R, Anolik JH. B-cell-targeted therapy for systemic lupus erythematosus. Drugs. 2006;66:1933–48. doi: 10.2165/00003495-200666150-00004. [DOI] [PubMed] [Google Scholar]
- 28.Taylor RP, Lindorfer MA. Drug insight: the mechanism of action of rituximab in autoimmune disease the immune complex decoy hypothesis. Nat Clin Pract Rheumatol. 2007;3:86–95. doi: 10.1038/ncprheum0424. [DOI] [PubMed] [Google Scholar]
- 29.Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–81. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 30.Looney RJ, Anolik J, Sanz I. B cells as therapeutic targets for rheumatic diseases. Curr Opin Rheumatol. 2004;16:180–5. doi: 10.1097/00002281-200405000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Eisenberg R, Looney RJ. The therapeutic potential of anti-CD20 ‘what do B-cells do?’. Clin Immunol. 2005;117:207–13. doi: 10.1016/j.clim.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Sfikakis PP, Boletis JN, Tsokos GC. Rituximab anti-B-cell therapy in systemic lupus erythematosus: pointing to the future. Curr Opin Rheumatol. 2005;17:550–7. doi: 10.1097/01.bor.0000172798.26249.fc. [DOI] [PubMed] [Google Scholar]
- 33.Salvi M, Bernard N, Miller A, Zhang ZG, Gardini E, Wall JR. Prevalence of antibodies reactive with a 64 kDa eye muscle membrane antigen in thyroid-associated ophthalmopathy. Thyroid. 1991;1:207–13. doi: 10.1089/thy.1991.1.207. [DOI] [PubMed] [Google Scholar]
- 34.Gunji K, De Bellis A, Kubota S, et al. Serum antibodies against the flavoprotein subunit of succinate dehydrogenase are sensitive markers of eye muscle autoimmunity in patients with Graves' hyperthyroidism. J Clin Endocrinol Metab. 1999;84:1255–62. doi: 10.1210/jcem.84.4.5640. [DOI] [PubMed] [Google Scholar]
- 35.Male D, Cooke A, Owen M, Trowsdale J, Champion B. Antigen receptor molecules. In: Male D, Cooke A, Owen M, Trowsdale J, Champion B, editors. Advanced immunology. London: Mosby; 1996. pp. 2.1–2.2. [Google Scholar]
- 36.Taylor RP, Lindorfer MA. Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr Opin Immunol. 2008;20:444–9. doi: 10.1016/j.coi.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stasi R, Pagano A, Stipa E, Amadori S. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood. 2001;98:952–7. doi: 10.1182/blood.v98.4.952. [DOI] [PubMed] [Google Scholar]
- 38.Introna M, Golay J, Barbui T. Rituximab: a new therapeutic tool for primary immune thrombocytopenic purpura? Haematologica. 2003;88:482–4. [PubMed] [Google Scholar]
- 39.Harjunpää A, Junnikkala S, Meri S. Rituximab (anti-CD20) therapy of B-cell lymphomas: direct complement killing is superior to cellular effector mechanisms. Scand J Immunol. 2000;51:634–41. doi: 10.1046/j.1365-3083.2000.00745.x. [DOI] [PubMed] [Google Scholar]
- 40.Treon SP, Agus TB, Link B, et al. CD20-directed antibody-mediated immunotherapy induces responses and facilitates hematologic recovery in patients with Waldenstrom's macroglobulinemia. J Immunother. 2001;24:272–9. [PubMed] [Google Scholar]
- 41.Ziller F, Macor P, Bulla R, Sblattero D, Marzari R, Tedesco F. Controlling complement resistance in cancer by using human monoclonal antibodies that neutralize complement-regulatory proteins CD55 and CD59. Eur J Immunol. 2005;35:2175–83. doi: 10.1002/eji.200425920. [DOI] [PubMed] [Google Scholar]