

Abstract
Subcutaneous administration of intravenous immunoglobulin G (IgG) preparations provides an additional level of patient convenience and more options for patients with poor venous access or a history of intravenous IgG reactions. An open-label, pharmacokinetic trial (n = 32) determined the non-inferiority of the subcutaneous versus intravenous route of 10% caprylate/chromatography purified human immune globulin intravenous (IGIV-C; Gamunex®) administration by comparing the steady-state area under the concentration-versus-time curve (AUC) of total plasma IgG in patients with primary immunodeficiency disease. Patients on stable IGIV-C received two intravenous infusions (administered 3 or 4 weeks apart). Seven to 10 days after the second intravenous infusion, all patients switched to a weekly infusion of subcutaneous IGIV-C, with the dose equal to 137% of the previous weekly equivalent intravenous dose, for up to 24 weeks. Samples for pharmacokinetic analysis were collected during steady state for intravenous and subcutaneous IGIV-C treatments. The AUC0-τ geometric least-squares mean ratio was 0·89 (90% confidence interval, 0·86–0·92) and met the criteria for non-inferiority. The overall mean steady-state trough concentration of plasma total IgG with subcutaneous IGIV-C was 11·4 mg/ml, 18·8% higher than intravenous IGIV-C (9·6 mg/ml). Subcutaneous IGIV-C was safe and well tolerated. Subcutaneous IGIV-C infusion-site reactions were generally mild/moderate and the incidence decreased over time. No serious bacterial infections were reported. Weekly subcutaneous IGIV-C infusion using 137% of the weekly equivalent intravenous immunoglobulin dose provides an AUC comparable to intravenous administration, thus allowing patients to maintain the same IgG preparation/formulation if switching between intravenous and subcutaneous infusions.
Keywords: Gamunex®, intravenous IgG, pharmacokinetics, primary immunodeficiency disease, subcutaneous IgG
Introduction
Treatment of primary immunodeficiency disease (PIDD) with immunoglobulin G (IgG) replacement therapy has been well recognized, and various routes of administration (e.g. intravenous, subcutaneous or intramuscular) have been evaluated for long-term management of these disorders [1–3]. Although intravenous immunoglobulin (IVIg) therapy has been the standard of care in the United States for more than 25 years, data on the efficacy and safety of subcutaneous administration of newer formulations of IgG [4–8] have provided support for the increasing acceptance of this alternative route of administration. Subcutaneous administration is particularly helpful for patients with poor venous access and for those who experience severe intravenous infusion-related reactions. Furthermore, administration of subcutaneous IgG at home has improved health-related quality of life and patient satisfaction versus administration of IVIg in a hospital or a physician's office [9–13].
Since the introduction of IgG replacement therapy, immunologists have monitored IVIg-treated patients by measuring trough IgG serum concentrations. Although the generally accepted minimum IgG trough concentration (Ctrough) for effective protection against infection in patients with PIDD is ≥5 mg/ml, treatment of those patients with baseline IgG concentrations >2 mg/ml should target an IgG concentration equal to the pretreatment trough concentration plus 3 mg/ml to account for the contribution of the inadequately functional, endogenously produced IgG, to the total measured IgG [2,14,15]. Newer data suggest that target IgG trough concentrations should be individualized based on patient response [16]. It has been postulated, however, that the area under the concentration-versus-time curve (AUC) may be a more robust measure of equivalent drug exposure when comparing two different treatment modalities because the AUC measures the total drug exposure over the entire dosing interval. However, historically, published studies and routine clinical practice have monitored IgG Ctrough concentrations because of the relationship between IgG trough concentrations and protection against infection [14,15,17,18]. Most patients with PIDD receive IVIg every 3–4 weeks, while subcutaneous IgG is typically administered weekly. Because of more frequent administration of lower doses and slower absorption from the extravascular fluid space, subcutaneous administration minimizes peak and trough IgG fluctuations between infusions [3].
The efficacy and safety of immune globulin intravenous (human), 10% caprylate/chromatography purified (IGIV-C; Gamunex®; Talecris Biotherapeutics, Inc., Research Triangle Park, NC, USA) in PIDD has been established in three controlled trials [19–21], and IGIV-C is currently indicated as replacement therapy for patients with PIDD. The current IGIV-C formulation can also be administered subcutaneously, but the appropriate intravenous to subcutaneous dose conversion to maintain comparable AUCs has not been established. The primary objective of this study was to provide guidance to those who treat patients with PIDD by evaluating prospectively the suitability of a selected dose conversion factor for the intravenous to subcutaneous transition by assessing the steady-state AUC of plasma total IgG for subcutaneously administered IGIV-C compared with intravenously administered IGIV-C.
Methods
Patients
Patients aged 13–75 years were recruited from November 2006 to August 2008 from eight centres in the United States and Canada. Eligible patients had a confirmed diagnosis of PIDD for at least 6 months and had previously received or were currently receiving IgG replacement therapy (documented plasma IgG concentration of ≥5 mg/ml within the previous 3 months with current IgG therapy). Patients were excluded from the study if they had an acquired medical condition known to cause secondary immune deficiency (e.g. human immunodeficiency virus infection or lymphoma); were receiving immunosuppressants, immunomodulators or corticosteroids (>1 mg/kg/day prednisolone or equivalent for >30 days); or had a history of bleeding disorders or blistering skin disease. The study was approved by the institutional review boards and ethics committees of all participating centres, and all adult patients or patient legal guardians provided written informed consent. In addition, all patients <18 years of age provided assent.
Study design
This open-label, single-sequence, crossover trial comprised a screening phase, a run-in phase (for patients not currently at steady-state levels using IGIV-C), an intravenous treatment phase and a subcutaneous treatment phase. During the screening phase (≤28 days), patients were categorized into three groups depending on their most recent IgG treatment history (Fig. 1). Group 1 included patients who were receiving a stable (≥3 months) dose of IGIV-C (Gamunex), 200–600 mg/kg, every 3 or 4 weeks. Patients in group 1 directly entered the intravenous treatment phase of the study, with the first infusion coinciding with the patients' next regularly scheduled intravenous administration. Group 2 included patients who were receiving 200–600 mg/kg of IVIg, other than IGIV-C, every 3 or 4 weeks. Patients in group 2 entered a 3-month run-in period and received intravenous IGIV-C at a dose and dosing interval equivalent to their previous IVIg therapy. After the run-in period, patients in group 2 were entered into the intravenous treatment phase. Group 3 included patients receiving IgG therapy via non-intravenous routes of administration (e.g. subcutaneous or intramuscular), patients not currently receiving IgG treatment but who had received it in the past, patients receiving any IVIg but not at a stable dose, patients receiving any IVIg but not in the dose range of 200–600 mg/kg, and patients receiving any IVIg but not at a scheduled interval of every 3 or 4 weeks. Patients in group 3 entered a 4-month run-in period and received intravenous IGIV-C at a dose of 200–600 mg/kg every 3 to 4 weeks, as determined by the investigator. After the run-in period, patients in group 3 were entered into the intravenous treatment phase.
Fig. 1.
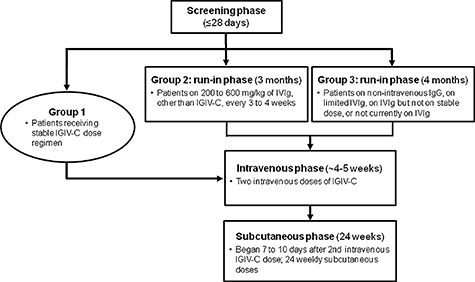
Study design. In this open-label crossover study, patients were categorized into three groups depending on their most recent immunoglobulin G (IgG) treatment history. Patients in group 1 were receiving a stable (≥3 months) dose of immune globulin intravenous (human), 10% caprylate/chromatography purified (IGIV-C), 200–600 mg/kg, every 3 or 4 weeks. Group 1 directly entered the intravenous phase, with the first infusion coinciding with the patients’ next regularly scheduled administration. Patients in group 2 were receiving 200–600 mg/kg of intravenous immunoglobulin (IVIg), other than IGIV-C, every 3 or 4 weeks. Group 2 entered a 3-month run-in period and received intravenous IGIV-C at an equivalent dose and dosing interval as the patients’ previous IVIg therapy. Group 2 was then eligible to enter the intravenous phase. Group 3 included patients receiving IgG therapy via non-intravenous routes of administration (e.g. subcutaneous or intramuscular), patients not currently receiving IgG treatment but who had received it in the past, patients receiving any IVIg but not at a stable dose, patients receiving any IVIg but not in the dose range of 200–600 mg/kg, and patients receiving any IVIg but not at a scheduled interval of every 3 or 4 weeks. Group 3 entered a 4-month run-in period and received intravenous IGIV-C at a dose of 200–600 mg/kg every 3–4 weeks. Group 3 was then eligible to enter the intravenous phase.
In the intravenous treatment phase, patients received two intravenous infusions of IGIV-C. In group 1 (no run-in period), patients received an intravenous IGIV-C dose equivalent to their regular dose. Patients in groups 2 and 3 received the same dose of intravenous IGIV-C administered during the run-in period. At the next scheduled infusion (3 or 4 weeks), each of these patients received a second intravenous infusion (booster) to ensure that they would have adequate plasma total IgG concentrations prior to entry in the subcutaneous phase of the study.
Seven to 10 days after the second intravenous dose (booster), patients entered the subcutaneous treatment phase and received weekly subcutaneous IGIV-C for up to 24 weeks. The subcutaneous dose of IGIV-C was calculated using the following formula:
 |
The conversion factor of 1·37 was determined based on unpublished animal data and published bioavailability data for subcutaneously administered IgG [4,22–24]. Subcutaneous IGIV-C was administered at single or multiple locations using the CADD-Legacy® PLUS Pump (Smiths Medical MD, Inc., St Paul, MN, USA) with a recommended infusion rate of ≤20 ml/h per infusion site. Premedication to manage potential infusion-related reactions prior to intravenous or subcutaneous administration of IGIV-C was not permitted during the study.
Assessments
Blood samples for pharmacokinetic assessments of plasma total IgG concentrations were collected during the intravenous treatment phase, at the first infusion: immediately pre- and post-intravenous infusion; at 1 h post-infusion; and at days 1, 2, 3, 5, 7, 14, 21 and 28 (day 28 for 4-week dosing schedule only) post-intravenous infusion. Blood samples for pharmacokinetic assessments of plasma total IgG concentrations were also collected during the subcutaneous treatment phase at week 17: immediately pre- and post-subcutaneous infusion and at days 1, 3, 4, 5 and 7 post-infusion. Plasma trough IgG concentrations, IgG subclass concentrations and serum titres of antibodies against Haemophilus influenzae and Streptococcus pneumoniae were determined at screening (IgG Ctrough only); during the run-in period; during the intravenous treatment phase before each infusion; and at weeks 5, 9, 13, 17, 21 and 24/final visit during the subcutaneous treatment phase. Safety and tolerability were determined by analysing adverse events, including serious bacterial infections and local infusion-site reactions, clinical laboratory parameters and vital signs. Definitions and criteria to confirm a diagnosis of serious bacterial infections, categorized as an adverse event, were established prospectively as per US Food and Drug Administration guidance [25], and were to include any episodes of bacteraemia or sepsis, bacterial meningitis, osteomyelitis or septic arthritis, pneumonia or visceral abscess, if observed.
Data analysis
The primary pharmacokinetic assessment was to compare the week 17 steady-state AUC of plasma total IgG over the regular dosing interval of subcutaneous IGIV-C (subcutaneous τ; 7 days) with the AUC of intravenous IGIV-C over the regular dosing interval (intravenous τ; 21 or 28 days). Any patient who received study medication and had sufficient plasma total IgG concentration-versus-time data to calculate AUC for either the intravenous or subcutaneous phase was included in the pharmacokinetics analyses (pharmacokinetic population). Data for patients receiving intravenous IGIV-C every 3 weeks or 4 weeks were pooled. Pharmacokinetic parameters of plasma total IgG were determined by a non-compartmental model using WinNonlin® Professional, version 4·1 (Pharsight Corporation, Mountain View, CA, USA). AUC at steady state over a specific dosing interval was determined by a combination of linear (trapezoid arising from increasing concentrations) and logarithmic (trapezoid arising from decreasing concentrations) trapezoidal methods.
To compare the AUC between subcutaneous and intravenous administration, a linear mixed effect model was used to obtain the geometric least-squares mean (LSM) ratio and its 90% confidence interval (CI). Non-inferiority was concluded if the lower boundary of the 90% CI was larger than the standard bioequivalence range of 0·80. Plasma IgG Ctrough, antibody titres and safety data were assessed using descriptive statistics. The IgG population consisted of any patient who had received any amount of IGIV-C and had plasma total IgG Ctrough data, regardless of whether the plasma total IgG concentration data were adequate for calculation of pharmacokinetic parameters. Any patient who received any amount of IGIV-C was included in the safety population. Data are reported for the pharmacokinetic population and as mean ± standard deviation unless indicated otherwise.
To confirm that the subcutaneous dose conversion factor of 1·37 was adequate and to allow for conversion factor modification if needed, an interim pharmacokinetic analysis was performed for the first six patients who had completed week 17 of the subcutaneous treatment phase of the study. The criteria for a potential dose conversion factor change were established prospectively. If the AUC geometric LSM ratio for subcutaneous versus intravenous IGIV-C had decreased below 10% of the desired ratio of 1·0 and the mean subcutaneous IGIV-C Ctrough had decreased below 5 mg/ml in >3 of the six patients, then a dose conversion factor increase would have been implemented. However, based on the results of the interim analysis, the dose conversion factor of 1·37 was considered adequate and no changes were required.
Results
Of the 35 enrolled patients, three patients were withdrawn from the study during the run-in phase (one because of an adverse event, one was lost to follow-up and one withdrew consent). All 32 patients in the intravenous treatment phase and 26 patients in the subcutaneous treatment phase had sufficient pharmacokinetic data for AUC determination and were included in the pharmacokinetic population. The majority of the patients in the pharmacokinetic population were female (78%) and white (97%), with a median age of 44·0 years (range 13–68 years). The baseline plasma total IgG concentration was 9·1 ± 2·9 mg/ml (mean ± standard deviation). The majority (88%) of patients had been treated with IVIg during the 3 months prior to enrolment [IGIV-C (n = 15) or other (n = 13)], none had been receiving intramuscular immunoglobulin and 66% of patients had a dosing frequency of every 4 weeks at screening. Of the 32 patients who completed the intravenous treatment phase, 25 completed the subcutaneous treatment phase. Of the seven patients who discontinued, two discontinued because of an adverse event (migraine in one patient and arthralgia, hyperhidrosis, fatigue, nausea and myalgia in one patient), with the remaining discontinuing because of non-medical reasons (e.g. non-compliance, consent withdrawn).
Mean plasma total IgG concentrations following intravenous IGIV-C infusion were initially higher than concentrations observed after subcutaneous administration of IGIV-C (Fig. 2). However, at approximately 14 days post-infusion, the mean plasma total IgG concentration with intravenous IGIV-C decreased below the concentration projected for subcutaneous administration of IGIV-C. Overall, subcutaneous administration provided a more consistent steady-state IgG concentration between doses compared with intravenous administration, and mean trough IgG concentrations were higher during the subcutaneous administration phase compared with the intravenous administration phase of the study. The mean adjusted AUC0-τ values for subcutaneous and intravenous IGIV-C administration were 6858 mg h/ml and 7640 mg h/ml, respectively (Table 1). The AUC0-τ geometric LSM ratio (subcutaneous : intravenous: 6706 : 7549) was 0·89 (90% CI, 0·86–0·92). The lower boundary of the 90% CI for the ratio of the AUC was larger than 0·80, thus establishing that subcutaneous IGIV-C was not inferior to intravenous IGIV-C.
Fig. 2.
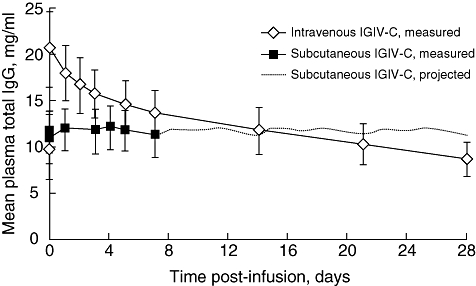
Steady-state plasma total immunoglobulin G (IgG) concentration-versus-time curves during intravenous or subcutaneous administration of immune globulin intravenous (human), 10% caprylate/chromatography purified (IGIV-C). Projected total IgG concentrations for subcutaneous IGIV-C (days 7–28) were based on measured concentrations over 0–7 days obtained during week 17 of the subcutaneous phase. Data are reported for the pharmacokinetic population and as mean ± standard error.
Table 1.
Plasma total IgG pharmacokinetic data for intravenous and subcutaneous IGIV-C in patients with PIDD.*
| Parameter | Intravenous IGIV-C (n = 32) | Subcutaneous IGIV-C (n = 26) |
|---|---|---|
| AUC0-τ, mg h/ml, mean | 7640 | 6858† |
| Cmax, mg/ml, mean ± s.d. | 21·1 ± 3·9 | 12·2 ± 2·4 |
| Ctrough, mg/ml, mean ± s.d. | 9·6 ± 2·1‡ | 11·4 ± 2·3§ |
| Cave, mg/ml, mean ± s.d. | n.d. | 11·6 ± 2·4 |
| tmax, h, mean ± s.d. | 2·9 ± 1·1 | 55·0 ± 48·6 |
| t1/2, h, mean ± s.d. | 735·5 ± 138·5 | n.d.¶ |
Data are reported for the pharmacokinetic population, with the exception of Ctrough data (IgG population).
Adjusted steady-state AUC based on intravenous dosing schedule (every 3 versus 4 weeks).
Mean Ctrough for intravenous IGIV-C was calculated from data for two intravenous infusions.
Mean Ctrough for subcutaneous IGIV-C was calculated from data for weeks 13, 17, 18, 19 and 21 for 28 patients (all patients who received study medication and had Ctrough data).
Not determined because of relatively constant plasma concentrations over 7-day dosing interval. AUC0-τ, area under the concentration-versus-time curve over the dosing interval; Cave, mean steady-state IgG concentration; Cmax, maximum observed IgG concentration; Ctrough, trough (predose) plasma IgG concentration; IgG, immunoglobulin G; IGIV-C, immune globulin intravenous (human), 10% caprylate/chromatography purified; n.d., not determined; PIDD, primary immunodeficiency disease; s.d., standard deviation; t1/2, apparent elimination half-life; tmax, time to Cmax.
Additional parameters of plasma total IgG were evaluated to help establish a pharmacokinetic profile of subcutaneous administration of IGIV-C (Table 1). The overall mean steady-state Ctrough of plasma total IgG determined during weeks 13–21 of the subcutaneous treatment phase was 11·4 mg/ml. This was 18·8% higher than the 9·6 mg/ml mean steady-state Ctrough following intravenous administration. Furthermore, the mean plasma total IgG Ctrough during subcutaneous administration of IGIV-C was relatively constant, starting as early as week 5. Both routes of administration provided a plasma total IgG Ctrough that was much higher than the minimum desired target of 5 mg/ml (Fig. 3). Furthermore, the Ctrough for the IgG subclasses remained relatively constant between intravenous and subcutaneous treatment phases, with slightly higher values observed during the subcutaneous treatment phase (Table 2).
Fig. 3.
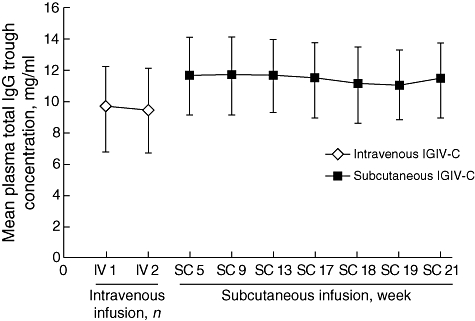
Plasma total immunoglobulin G (IgG) trough concentrations during intravenous or subcutaneous administration of immune globulin intravenous (human), 10% caprylate/chromatography purified (IGIV-C), by visit. Data are reported for the IgG population and as mean ± standard error.
Table 2.
Mean serum IgG subclass trough concentrations during intravenous and subcutaneous administration of IGIV-C in patients with PIDD.*
| Ctrough, mg/ml, mean ± s.d. |
|||
|---|---|---|---|
| IgG subclass | Intravenous treatment phase | Subcutaneous treatment phase† | Mean change from intravenous to subcutaneous, % |
| IgG1 | 5·2 ± 1·2 | 6·3 ± 1·5 | 20·2 |
| IgG2 | 3·3 ± 0·8 | 3·8 ± 0·9 | 15·2 |
| IgG3 | 0·7 ± 1·2 | 0·7 ± 1·2 | 12·3 |
| IgG4 | 0·4 ± 0·2 | 0·4 ± 0·2 | 11·1 |
Data are reported for the IgG population.
Mean Ctrough for subcutaneous administration of IGIV-C was calculated from data for weeks 13, 17 and 21, if available. Ctrough, trough (predose) plasma IgG concentration; IgG, immunoglobulin G; IGIV-C, immune globulin intravenous (human), 10% caprylate/chromatography purified; PIDD, primary immunodeficiency disease; s.d., standard deviation.
Mean trough antibody titres against H. influenzae and six serotypes of S. pneumoniae were measured during intravenous administration of IGIV-C and subcutaneous administration of IGIV-C (Fig. 4). Mean trough antibody titres increased for antibodies against all serotypes tested and were higher when patients received subcutaneous administration of IGIV-C compared with when they received intravenous administration of IGIV-C. This increase was observed at the first assessment at week 5 in the subcutaneous treatment phase and was maintained to week 21.
Fig. 4.
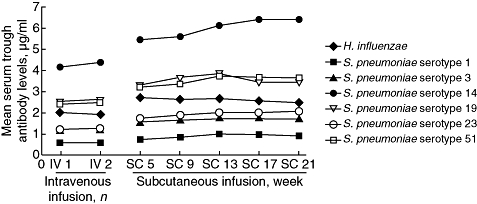
Mean serum trough concentrations of antibodies against Haemophilus influenzae and six Streptococcus pneumoniae serotypes during intravenous or subcutaneous administration of immune globulin intravenous (human), 10% caprylate/chromatography purified. Data are reported for the immunoglobulin G population.
A total of 64 intravenous and 725 subcutaneous infusions were conducted during the respective treatment phases. The majority (86%) of subcutaneous infusions were conducted using a total of four infusion sites (range 2–8 sites), with a mean infusion duration of 2·3 ± 1·1 h (range 0·8–8·3 h). The overall incidence of adverse events was 0·25 events per infusion for the intravenous IGIV-C. The overall incidence of adverse events was higher with subcutaneous IGIV-C (0·82 events per infusion) than with intravenous IGIV-C; however, the incidence of non-infusion-site adverse events (0·24 events per infusion) was 3·5-fold lower than the total incidence of adverse events associated with subcutaneous treatment and was similar to the total incidence of 0·25 events per infusion observed with intravenous administration of IGIV-C.
The incidence of infusion-site reactions with IGIV-C decreased over time during the subcutaneous treatment phase, with a 40% reduction in the percentage of patients with a local infusion-site reaction by week 24 versus week 1 (32% versus 53%, respectively) and a 55% reduction in the number of local infusion-site reactions per infusion (0·44 versus 0·97, respectively). In addition, the rate of non-infusion-site adverse events during the subcutaneous treatment phase, assessed at 4-week intervals, decreased over time from a rate of 0·44 adverse events per infusion during weeks 1–4 to 0·12 adverse events per infusion during weeks 21–24. The majority of adverse events with IGIV-C were mild to moderate in severity in both the intravenous treatment phase (88% of events) and subcutaneous treatment phase (96% of events). Four patients experienced nine severe, but no serious, infusion-site reactions during the subcutaneous treatment phase. The most common adverse events with subcutaneous administration of IGIV-C were infusion-site erythema and infusion-site pain (Table 3). No clinically meaningful changes in safety laboratory parameters were observed during the study. No serious adverse events were reported during the intravenous or subcutaneous treatment phases, with the exception of an unrelated report of alprazolam dependence during the subcutaneous treatment phase.
Table 3.
Adverse events reported in ≥5% of patients in a treatment group, irrespective of causality.*
| Intravenous IGIV-C |
Subcutaneous IGIV-C |
|||
|---|---|---|---|---|
| Adverse event | Patients, n (%) (n = 32) | Events per infusion (n = 64)† | Patients, n (%) (n = 32) | Events per infusion (n = 725)† |
| Any adverse event | 11 (34) | 0·25 | 30 (94) | 0·82 |
| Infusion-site reactions | ||||
| Infusion-site erythema | 0 | 0 | 13 (41) | 0·17 |
| Infusion-site pain | 0 | 0 | 13 (41) | 0·08 |
| Infusion-site swelling | 0 | 0 | 11 (34) | 0·07 |
| Infusion-site extravasation | 0 | 0 | 8 (25) | 0·02 |
| Infusion-site pruritus | 0 | 0 | 8 (25) | 0·07 |
| Infusion-site bruising | 0 | 0 | 7 (22) | 0·02 |
| Infusion-site haemorrhage | 0 | 0 | 5 (16) | 0·02 |
| Infusion-site oedema | 0 | 0 | 4 (12) | 0·07 |
| Non-infusion-site adverse events | ||||
| Sinusitis | 0 | 0 | 8 (25) | 0·02 |
| Upper respiratory tract infection | 2 (6) | 0·03 | 7 (22) | 0·01 |
| Headache | 1 (3) | 0·02 | 6 (19) | 0·05 |
| Diarrhoea | 2 (6) | 0·05 | 5 (16) | 0·01 |
| Fatigue | 0 | 0 | 5 (16) | 0·01 |
| Nausea | 1 (3) | 0·02 | 4 (12) | 0·01 |
| Dizziness | 2 (6) | 0·03 | 0 | 0 |
| Toothache | 2 (6) | 0·03 | 0 | 0 |
Data are reported for the safety population.
Total number of infusions. IGIV-C, immune globulin intravenous (human), 10% caprylate/chromatography purified.
No serious bacterial infections were reported during the intravenous or subcutaneous phases. Twenty-four of 32 patients (75%) experienced an infection or possible signs or symptoms of infection during the 6-month subcutaneous phase, with sinusitis (25%), upper respiratory tract infection (22%), diarrhoea (16%), pharyngolaryngeal pain (9%) and bronchitis (9%) reported most commonly in this patient population. A substantially larger total exposure was observed during the subcutaneous treatment phase versus the intravenous treatment phase (14·5 versus 3·1 patient-years, respectively). Using these values, the infection rate was 4·1 per patient-year for subcutaneous administration of IGIV-C and 2·0 per patient-year for intravenous administration of IGIV-C.
Discussion
Intravenous IGIV-C has been well established as an efficacious, safe and well-tolerated IgG replacement or immunomodulatory therapy [20,21,26–28]. Subcutaneous administration of IgG replacement therapy provides, for some patients, a better treatment experience by eliminating the need for intravenous access and enhancing their sense of independence and control. Healthcare providers, however, need data to guide dosing decisions when switching patients from intravenous to subcutaneous IGIV-C administration. Therefore, a key goal of this study was to verify a dose conversion factor to help guide healthcare providers when calculating the weekly subcutaneous dose for patients switching from intravenous IGIV-C. The results of this pharmacokinetic study confirmed that a conversion factor of 1·37 provides an exposure measured by AUC of IGIV-C administered subcutaneously that was comparable (i.e. non-inferior) to that provided by intravenous IGIV-C administration. While serum trough concentrations are used traditionally as a guide in dosing decisions, the US Food and Drug Administration has provided the pharmaceutical industry with guidance that AUCs should be compared when establishing non-inferiority between subcutaneous and intravenous IgG administration [29]. Therefore, this study compared AUCs of plasma total IgG for subcutaneously and intravenously administered IGIV-C.
This conversion factor was chosen based on previously published results, including data for immune globulin subcutaneous (Vivaglobin®; CSL Behring, GmbH, Marburg, Germany), in which a subcutaneous monthly dose adjustment of 137% of the previous monthly equivalent IVIg dose compensated for the lower bioavailability of subcutaneous IgG [4,22,23]. However, discussions have arisen in the literature regarding the necessity of a conversion factor for switching from an intravenous IgG dose to a subcutaneous IgG dose [4,30]. Data from published studies suggest that AUC equivalence is not necessary for therapeutic equivalence and that a higher Ctrough of subcutaneous IgG, using a conversion factor of 1·37 (137% of intravenous dose), would not confer a substantial increase in protection against infection benefits versus administration of a dose equivalent (100%) to an intravenous IgG dose [4,30].
For example, a similar frequency of infection during subcutaneous dosing has been observed when either a 137% or an equivalent intravenous IgG dose has been assessed. Rates of infection reported in a study by Gardulf et al. [5] (N = 52), in which the monthly subcutaneous IgG dose was equivalent to the monthly intravenous IgG dose, were similar to those reported in a study by Ochs et al. [4] (N = 51), in which the monthly subcutaneous IgG dose was 137% of the monthly intravenous dose. Furthermore, a study published in abstract form (N = 12) evaluated the efficacy of intravenous administration of IGIV-C versus subcutaneous administration of IGIV-C, administered as 100% of the intravenous dose, and concluded that there were no differences in efficacy [31]. In the current pharmacokinetic study, no serious bacterial infections were reported during the intravenous or subcutaneous treatment phases, and mean Ctrough concentrations of IgG subclasses and mean titres of antibodies against H. influenzae and S. pneumoniae with subcutaneous administration of IGIV-C were not lower than values observed for intravenous administration of IGIV-C. Therefore, subcutaneous administration of IGIV-C maintained trough antibody titres against bacterial pathogens and trough concentrations of IgG subclasses at least as well as intravenous administration of IGIV-C.
The overall infection rate of 4·1 per patient-year with subcutaneous administration of IGIV-C was higher than the 2·0 per patient-year observed for intravenous administration of IGIV-C in the current study. However, several factors probably impacted the results. The subcutaneous administration phase was substantially longer in duration than the intravenous administration phase, and resulted in a longer time-frame of exposure in which to observe an event with subcutaneous administration. Furthermore, the seasonality of the study design may have impacted the results. A higher percentage (60%) of subcutaneous infusions occurred during the autumn and winter months, while a higher percentage of intravenous infusions (66%) occurred during the spring and summer months. Thus, caution should be exercised when drawing any conclusions from overall infection rates between administration routes in this pharmacokinetic study.
Subcutaneous administration of IGIV-C exhibited a good pharmacokinetic profile, with generally constant mean plasma total IgG concentrations observed throughout the course of treatment. Concentrations of plasma total IgG during intravenous administration of IGIV-C were initially higher than those during the subcutaneous administration of IGIV-C but declined within 2 weeks to concentrations lower than those observed during the subcutaneous treatment phase. In fact, the mean steady-state plasma IgG Ctrough during subcutaneous administration of IGIV-C was ∼19% higher over the course of treatment compared with intravenous administration of IGIV-C. This percentage difference in Ctrough between subcutaneous and intravenous administration of IGIV-C was lower than the 32% difference reported by Ochs et al. [4], but was higher than both the intravenous IgG dose-equivalent (100%) study with IGIV-C (8% difference; Gamunex) [31] and the dose-equivalent study for immune globulin subcutaneous (11% difference; Vivaglobin) [5,30]. The higher IgG Ctrough observed with subcutaneous administration is probably related to the higher concentration administered and the increased frequency of administration [3,30].
Subcutaneous administration of IGIV-C was well tolerated, with no apparent safety concerns observed during this study. As expected, infusion-site reactions were the most common adverse events observed with subcutaneous administration of IGIV-C. However, infusion-site reactions were generally mild to moderate in severity, did not result in discontinuation from the study and decreased in incidence over time. The safety and tolerability results, including the reduced incidence of infusion-site reactions over time, are consistent with other studies of subcutaneous IgG [4–6,32]. Furthermore, the overall infection rate of 4·1 per patient-year for subcutaneous administration of IGIV-C was low and, although it is difficult to compare studies, was consistent with data for Vivaglobin (4·4 infections per patient-year) [4].
In conclusion, using a weekly subcutaneous IGIV-C dose of 137% of the weekly equivalent intravenous IGIV-C dose, the resulting AUC will be non-inferior to the AUC of the intravenous dose. In addition, mean trough serum concentrations of IgG, IgG subclasses and H. influenzae and S. pneumoniae antibody titres may be higher than concentrations and titres during intravenous administration. Subcutaneous administration of IGIV-C provides high, stable IgG concentrations and is safe and well tolerated.
Acknowledgments
This study was supported by the Talecris Biotherapeutics Center for Science and Education. Editorial assistance was provided under the direction of the authors by MedThink Communications and PAREXEL with support from the Talecris Biotherapeutics Center for Science and Education.
Disclosure
J. Chen and S. Sorrells are employees of Talecris Biotherapeutics Inc. Dr Irani and Dr Gupta have served on advisory boards and received research grants from Talecris. Dr Stark, Dr Wasserman, Dr Irani, Dr Gupta, Dr Tracy, Dr Tsoukas, Dr Levy and Dr Roberts have served as Investigators on Talecris-sponsored studies.
References
- 1.Kivity S, Katz U, Daniel N, Nussinovitch U, Papageorgiou N, Shoenfeld Y. Evidence for the use of intravenous immunoglobulins – a review of the literature. Clin Rev Allergy Immunol. 2010;38:201–69. doi: 10.1007/s12016-009-8155-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orange JS, Hossny EM, Weiler CR, et al. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2006;117(Suppl.):S525–53. doi: 10.1016/j.jaci.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Bonilla FA. Pharmacokinetics of immunoglobulin administered via intravenous or subcutaneous routes. Immunol Allergy Clin North Am. 2008;28:803–19. ix. doi: 10.1016/j.iac.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Ochs HD, Gupta S, Kiessling P, Nicolay U, Berger M, the Subcutaneous IgG Study Group Safety and efficacy of self-administered subcutaneous immunoglobulin in patients with primary immunodeficiency diseases. J Clin Immunol. 2006;26:265–73. doi: 10.1007/s10875-006-9021-7. [DOI] [PubMed] [Google Scholar]
- 5.Gardulf A, Nicolay U, Asensio O, et al. Rapid subcutaneous IgG replacement therapy is effective and safe in children and adults with primary immunodeficiencies – a prospective, multi-national study. J Clin Immunol. 2006;26:177–85. doi: 10.1007/s10875-006-9002-x. [DOI] [PubMed] [Google Scholar]
- 6.Chapel HM, Spickett GP, Ericson D, Engl W, Eibl MM, Bjorkander J. The comparison of the efficacy and safety of intravenous versus subcutaneous immunoglobulin replacement therapy. J Clin Immunol. 2000;20:94–100. doi: 10.1023/a:1006678312925. [DOI] [PubMed] [Google Scholar]
- 7.Fasth A, Nyström J. Safety and efficacy of subcutaneous human immunoglobulin in children with primary immunodeficiency. Acta Paediatr. 2007;96:1474–8. doi: 10.1111/j.1651-2227.2007.00485.x. [DOI] [PubMed] [Google Scholar]
- 8.Chouksey A, Duff K, Wasserbauer N, Berger M. Subcutaneous immunoglobulin-G replacement therapy with preparations currently available in the United States for intravenous or intramuscular use: reasons and regimens. Allergy Asthma Clin Immunol. 2005;1:120–30. doi: 10.1186/1710-1492-1-3-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolay U, Kiessling P, Berger M, et al. Health-related quality of life and treatment satisfaction in North American patients with primary immunedeficiency diseases receiving subcutaneous IgG self-infusions at home. J Clin Immunol. 2006;26:65–72. doi: 10.1007/s10875-006-8905-x. [DOI] [PubMed] [Google Scholar]
- 10.Kittner JM, Grimbacher B, Wulff W, Jäger B, Schmidt RE. Patients' attitude to subcutaneous immunoglobulin substitution as home therapy. J Clin Immunol. 2006;26:400–5. doi: 10.1007/s10875-006-9031-5. [DOI] [PubMed] [Google Scholar]
- 11.Fasth A, Nyström J. Quality of life and health-care resource utilization among children with primary immunodeficiency receiving home treatment with subcutaneous human immunoglobulin. J Clin Immunol. 2008;28:370–8. doi: 10.1007/s10875-008-9180-9. [DOI] [PubMed] [Google Scholar]
- 12.Gardulf A, Borte M, Ochs HD, Nicolay U the Vivaglobin Clinical Study Group. Prognostic factors for health-related quality of life in adults and children with primary antibody deficiencies receiving SCIG home therapy. Clin Immunol. 2008;126:81–8. doi: 10.1016/j.clim.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Gardulf A, Nicolay U, Math D, et al. Children and adults with primary antibody deficiencies gain quality of life by subcutaneous IgG self-infusions at home. J Allergy Clin Immunol. 2004;114:936–42. doi: 10.1016/j.jaci.2004.06.053. [DOI] [PubMed] [Google Scholar]
- 14.Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenous immunoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet. 1987;1:1075–7. doi: 10.1016/s0140-6736(87)90494-6. [DOI] [PubMed] [Google Scholar]
- 15.Quartier P, Debré M, De Blic J, et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: a retrospective survey of 31 patients. J Pediatr. 1999;134:589–96. doi: 10.1016/s0022-3476(99)70246-5. [DOI] [PubMed] [Google Scholar]
- 16.Bonagura VR, Marchlewski R, Cox A, Rosenthal DW. Biologic IgG level in primary immunodeficiency disease: the IgG level that protects against recurrent infection. J Allergy Clin Immunol. 2008;122:210–12. doi: 10.1016/j.jaci.2008.04.044. [DOI] [PubMed] [Google Scholar]
- 17.Bonilla FA, Bernstein IL, Khan DA, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94(5 Suppl.)(1):S1–63. doi: 10.1016/s1081-1206(10)61142-8. [DOI] [PubMed] [Google Scholar]
- 18.Eijkhout HW, van Der Meer JWM, Kallenberg CGM, et al. for the Inter-University Working Party for the Study of Immune Deficiencies. The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia: a randomized, double-blind, multicenter crossover trial. Ann Intern Med. 2001;135:165–74. doi: 10.7326/0003-4819-135-3-200108070-00008. [DOI] [PubMed] [Google Scholar]
- 19.Gelfand EW, Hanna K the IGIV-C Increased Maximum Infusion Rate Study Group. Safety and tolerability of increased rate of infusion of intravenous immunoglobulin G, 10% in antibody-deficient patients. J Clin Immunol. 2006;26:284–90. doi: 10.1007/s10875-006-9014-6. [DOI] [PubMed] [Google Scholar]
- 20.Roifman CM, Schroeder H, Berger M, et al. the IGIV-C in PID Study Group. Comparison of the efficacy of IGIV-C, 10% (caprylate/chromatography) and IGIV-SD, 10% as replacement therapy in primary immune deficiency: a randomized double-blind trial. Int Immunopharmacol. 2003;3:1325–33. doi: 10.1016/s1567-5769(03)00134-6. [DOI] [PubMed] [Google Scholar]
- 21.Ballow M, Berger M, Bonilla FA, et al. Pharmacokinetics and tolerability of a new intravenous immunoglobulin preparation, IGIV-C, 10% (Gamunex, 10%) Vox Sang. 2003;84:202–10. doi: 10.1046/j.1423-0410.2003.00286.x. [DOI] [PubMed] [Google Scholar]
- 22.Waniewski J, Gardulf A, Hammarström L. Bioavailability of γ-globulin after subcutaneous infusions in patients with common variable immunodeficiency. J Clin Immunol. 1994;14:90–7. doi: 10.1007/BF01541341. [DOI] [PubMed] [Google Scholar]
- 23.Smith GN, Griffiths B, Mollison D, Mollison PL. Uptake of IgG after intramuscular and subcutaneous injection. Lancet. 1972;1:1208–12. doi: 10.1016/s0140-6736(72)90926-9. [DOI] [PubMed] [Google Scholar]
- 24.Vivaglobin [package insert] Marburg, Germany: CSL Behring GmbH; 2007. [Google Scholar]
- 25.US Department of Health and Human Services. Safety, efficacy, and pharmacokinetic studies to support marketing of immune globulin intravenous (human) as replacement therapy for primary humoral immunodeficiency. Washington, DC: US Department of Health and Human Services; 2008. [Google Scholar]
- 26.Hughes RAC, Donofrio P, Bril V, et al. on behalf of the ICE Study Group. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7:136–44. doi: 10.1016/S1474-4422(07)70329-0. [DOI] [PubMed] [Google Scholar]
- 27.Bril V, Katzberg H, Donofrio P, et al. Electrophysiology in chronic inflammatory demyelinating polyneuropathy with IGIV. Muscle Nerve. 2009;39:448–55. doi: 10.1002/mus.21236. [DOI] [PubMed] [Google Scholar]
- 28.Merkies ISJ, Bril V, Dalakas MC, et al. on behalf of the ICE Study Group. Health-related quality-of-life improvements in CIDP with immune globulin IV 10%: the ICE Study. Neurology. 2009;72:1337–44. doi: 10.1212/WNL.0b013e3181a0fd80. [DOI] [PubMed] [Google Scholar]
- 29.Aebersold P, US Department of Health and Human Services . Intravenous immune globulins in the 21st century: progress and challenges in efficacy, safety, and paths to licensure. Bethesda, MD: US Department of Health and Human Services; 2005. Regulatory requirements for subcutaneous Ig for PID. Available at: http://www.fda.gov/downloads/BiologicsBloodVaccines/NewsEvents/WorkshopsMeetingsConferences/TranscriptsMinutes/UCM054437.pdf (accessed 16 October 2009. [Google Scholar]
- 30.Berger M. Subcutaneous administration of IgG. Immunol Allergy Clin North Am. 2008;28:779–802. doi: 10.1016/j.iac.2008.07.002. vii. [DOI] [PubMed] [Google Scholar]
- 31.Desai S, Poll J, Chouksey A, Berger M. Randomized, crossover study of the efficacy and tolerability of the same dose of Gamunex® given subcutaneously or intravenously. J Allergy Clin Immunol. 2008;121:85. [Google Scholar]
- 32.Gustafson R, Gardulf A, Hansen S, et al. Rapid subcutaneous immunoglobulin administration every second week results in high and stable serum immunoglobulin G levels in patients with primary antibody deficiencies. Clin Exp Immunol. 2008;152:274–9. doi: 10.1111/j.1365-2249.2008.03620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]