

Abstract
Common variable immunodeficiency disorders (CVIDs) are a heterogeneous group of diseases characterized by hypogammaglobulinaemia and consequent susceptibility to infection. CVID patients commonly develop a variety of additional manifestations for which the causative factors are not fully understood. Two such manifestations are granulomatous disease and enteropathy. Because the ability to predict complications would aid clinical management, we continue to search for possible disease modifier genes. NOD2 acts a microbial sensor and is involved in proinflammatory signalling. Particular mutations of the NOD2 gene are associated with Crohn's disease including gly908arg, leu1007finsc and arg702trp polymorphisms. We hypothesized that NOD2 polymorphisms may be a disease modifier gene towards an enteropathic or granulomatous phenotype within CVIDs. Sequence-specific primers returned genotypes for 285 CVID patients from centres across the United Kingdom and Europe. We present the frequencies of the different phenotypes of patients within our international cohort. Arg702trp polymorphisms were significantly less frequent than wild-type (WT) (P = 0·038) among international CVID patients with splenomegaly. Gly908arg polymorphisms were more prevalent than WT in UK patients with autoimmune disorders (P = 0·049) or enteropathy (P = 0·049). NOD2 polymorphisms were not more prevalent than WT in CVID patients with clinical phenotypes of granulomata. UK allele frequencies of 0·014, 0·056 and 0·026 were found for gly908arg, arg702trp and leu1007finsc NOD2 polymorphisms, respectively. These do not differ significantly from UK immunocompetent controls confirming, as expected, that in addition these NOD2 polymorphisms do not confer susceptibility to CVIDs per se.
Keywords: antibody deficiency, common variable immunodeficiency, disease susceptibility/resistance/polymorphisms, immunodeficiency – primary, immunogenetics
Introduction
Common variable immunodeficiency disorders (CVIDs) are the most common forms of clinically significant primary immune deficiencies in humans, with a prevalence estimated at minimum to be one in 30 000 [1–3]. CVIDs are characterized by markedly reduced levels of serum immunoglobulins and subsequent increased susceptibility to infection [4]. CVIDs remain a diagnosis of exclusion and patient cohorts demonstrate highly variable phenotypes [5]. Patients usually present with increased susceptibility to infections, especially encapsulated bacteria, affecting mainly the upper and lower respiratory tract and the gastrointestinal tract [6]. CVID patients are more susceptible to autoimmune diseases, unexplained enteropathy, polyclonal lymphoproliferation and granulomata for which the causative factors (genetic and/or environmental) are not fully understood. The genes involved in the susceptibility of these polygenic diseases remain to be defined [3,7].
Granulomatous disease occurs in 8–22% of CVID patients, depending on the patient cohort, with large variations in the prevalence of this complication between countries [5]. The cause(s) of these non-caseating granulomatous infiltrations is unknown, although recurrent and persistent infections, dysregulated T cell function or macrophage activation have been suggested [8]. The early phase in the development of granulomata is characterized by the accumulation and activation of T helper type 1 (Th1)-type lymphocytes, involving proinflammatory cytokines, namely tumour necrosis factor (TNF) [9], interleukin (IL)-1β[10], IL-2 [11], IL-6, IL-8 [12] and interferon (IFN)-γ[13]. Although the histology of CVID enteropathy is similar to that of coeliac disease, namely villous blunting, intraepithelial T cells in association with epithelial apoptosis, these findings persist despite adherence to a gluten-free diet.
Genes that affect the production of these cytokines could act as potential disease modifier genes for the granulomatous complications of CVIDs [1]. The finding that Nod-like receptors (NLRs) are involved in the granulomata of Crohn's disease and sarcoidosis led to the hypothesis that polymorphisms in these genes might also act as disease modifiers in the varying clinical phenotypes seen in patients with CVIDs.
Nucleotide-binding oligomerization domain (NOD)2, also known as caspase recruitment domain (CARD) 15, is a 163319 base pair gene comprised of 12 exons (GenBank Accession number AJ303140, http://www.ncbi.nlm.nih.gov). It maps to the IBD1 locus in the pericentromeric region of chromosome 16 [14] and encodes a protein composed of two NH2-terminal CARDs, a nucleotide-binding domain (NBD) and 10 COOH-terminal leucine-rich repeats (LRRs). It has been shown to be expressed in macrophages, neutrophils and dendritic cells, as well as Paneth cells of the small intestine [15]and bronchial cells in the lungs [16].
NOD2 acts as a cytosolic pathogen recognition protein to activate the cell or induce apoptosis [17]. NOD2 polymorphisms have been shown to be associated with Crohn's disease, a granulomatous condition of the digestive tract [18–21]. The most prevalent associated NOD2 polymorphisms are leu1007fsinsc (SNP13 in exon 11), gly908arg (SNP12 in exon 8) and arg702trp (SNP8 in exon 4) found in the LRR region of the gene and representing 31%, 18% and 32%, respectively, of the total Crohn's disease-associated polymorphisms [19]. The remaining 19% of polymorphisms come from 27 additional rare variants considered as disease-causing mutations, also located primarily in the LRR region of the gene. NOD2 polymorphisms are strong predictors of ileal disease, but are not associated with perianal or colonic disease [22]. Relative risk is greatest for the leu1007fsinsc variant [22]; however, NOD2 polymorphisms are neither necessary nor sufficient for the development of Crohn's disease.
NOD2 polymorphisms are also associated with other chronic inflammatory disorders, including Blau syndrome (BS) and early-onset sarcoidosis (EOS). EOS and inheritable BS share the characteristic clinical features of juvenile-onset systemic granulomatous syndrome that mainly affects skin, joints and eyes. Patients with CVIDs are sometimes misdiagnosed as having sarcoidosis, although sarcoidosis is not readily confused clinically with CVID provided that serum immunoglobulin measurements are taken and infection history taken. NOD2 polymorphisms related to BS and EOS affect the central nucleotide binding domain of NOD2, rather than the C-terminal portion associated with Crohn's disease [23,24]. Other conditions associated with NOD2 mutations include psoriatic arthritis [25] and graft-versus-host disease [26].
Drewe and Powell reported an expanding phenotype of inflammatory diseases associated with NOD2 mutations and in particular a case with CVID [27]. Evidence is presented that disorders of the innate immune axis are of interest in CVIDs whether through disease-causing or disease-altering mechanisms [28]. We hypothesize that NOD2 polymorphisms, focusing upon those associated with Crohn's disease, might contribute to the granulomatous and/or enteropathic CVID phenotypes. We investigated the frequency of the most common NOD2 polymorphisms associated with Crohn's disease in a cohort of CVID patients to see if there is any association with granulomata or gut involvement, in the hope that this might provide a prognostic marker for these complications in addition to suggesting a disease modification mechanism.
Methods
Patient cohort
A total of 299 unselected CVID patients were recruited from: the John Radcliffe Hospital, Oxford (75), the Royal Free Hospital, London (99), Queens Medical Centre, Nottingham (20), Northern General Hospital, Sheffield (24), Path Links, Scunthorpe (6), St Anne University Hospital, Brno, Czech Republic (33) and Medical School Hannover, Hannover, Germany (42). Ethical approval was obtained from the South West Research Ethics Committee (MREC/04/6/18) for the UK samples and from institutional review boards in the international centres. Samples were processed blind.
A clinical data sheet was completed by the managing clinicians for all patients included in the study and validated independently to ensure that recruited subjects fulfilled European Society for Immunodeficiencies (ESID) diagnostic criteria [4] and correct identification of complications including bronchiectasis (radiologically confirmed), autoimmunity (with details of site), granuloma (location and method of diagnosis i.e. biopsy proven), lymphoid interstitial pneumonitis (LIP) (and method of diagnosis i.e. radiological or histological), splenomegaly (confirmed radiologically >11 cm on ultrasound or palpable) and enteropathy (confirmed by histology).
Amplification of gene region using polymerase chain reaction (PCR)
Primers for the exon regions of interest were purchased from Sigma-Genosys (Haverhill, Suffolk, UK) (Table 1). Primers were made up to final concentrations as detailed in Table 1 with a 796 base pair control amplicon derived from a conserved region of DRB1 so that a positive control was run within each reaction. Three µl of this primer mix was added to appropriate pre-oiled [10 µl mineral oil (Sigma-Aldrich Company Ltd, Dorset, UK)] wells of a 96-well PCR plate (Corning Incorporated, Kennebunk, ME, USA). Biotaq polymerase (Bioline, London, UK) was used at a concentration of 1 µl Taq in 208 µl TDMH (TDMH is our in-house name for the PCR buffer used; it is derived from Tris-buffer, dNTPs, Magnesium and H2O) (kindly donated by the tissue typing laboratory, Churchill Hospital, Oxford). Patient genomic DNA purified from peripheral blood was added to this Taq/TDMH mixture at a ratio of 1:23 (up to 1:8 in the event of PCR failure) and 5 µl of this mixture was then added appropriately to the 96-well PCR plate to make a reaction volume of 18 µl. PCR amplification was performed using the GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, CA, USA). Molecular grade water (Sigma) in place of primers as a negative control was run with each sample.
Table 1.
NOD2 primer mixes for sequence specific primer polymerase chain reaction (PCR).
| Polymorphism | NCBI reference | SNP position | Sense primer 5′–3′ | Sense conc. µg/ml | Anti-sense primer 5′–3′ | AS conc. µg/ml | Amplicon size |
|---|---|---|---|---|---|---|---|
| Arg702trp | rs2066844 | 2104C | TGAGAAGGCCCTGCTCC | 20 | AGAGTTGTAGTCCAGCTGCAG | 10 | 278 |
| 2104T | CTGAGAAGGCCCTGCTCT | 10 | 10 | ||||
| Gly908trg | rs2066845 | 2722G | GGCCTTTTCAGATTCTGGG | 2.5 | GACATTTCCAAGTCACCCAG | 5 | 242 |
| 2722C | GGCCTTTTCAGATTCTGGC | 20 | 20 | ||||
| Leu1007finsc | rs2066847 | 3020 | CCCTCCTGCAGGCCCT | 10 | AACCGCAGAAGGTCTGATC | 10 | 377 |
| 3030Ci | CCCTCCTGCAGGCCCC | 10 | 10 |
NCBI: National Centre for Biotechnology Information; SNP: single nucleotide polymorphism.
Agarose gel electrophoresis
Synthesis of appropriately sized products was confirmed by running on agarose gel containing 3·3 × 10−4g/l ethidium bromide (Sigma) alongside a DNA ladder (Sigma). Gels were viewed under ultraviolet light and photographed (Transluminator UVP, Upland, CA, USA).
Genetic sequencing
A random representation of each genotype was sequenced using an Applied Biosystems 3730 DNA Analyser with Sequencing Analyser version 5·2 software (Applied Biosystems) for genotype confirmation.
Calculation of allele frequency and statistical analysis
Allele frequency was calculated using the formula below:
 |
Fisher's exact test (GraphPad Prism® version 4) was used to compare allele frequencies. P-values < 0·05 were considered as significant.
Results
Complications among international cohort of CVID patients
Frequencies of complications within our CVID cohort are given in Fig. 1 and Table 2. Details of complications requested from managing clinicians (see Materials and methods) were validated from the information given and if necessary confirmed by independent review. The clinical data were finally 99·3% complete. Subsequent to validation, eight patients were unselected as they did not meet CVID diagnostic criteria.
Fig. 1.
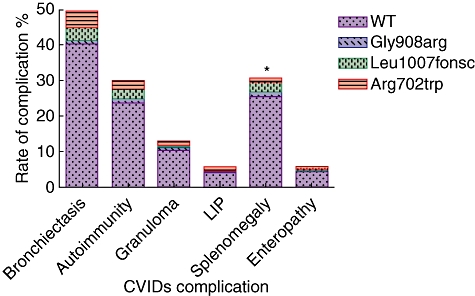
Frequency of disease complications and NOD2 polymorphisms among 285 international common variable immunodeficiency (CVID) patients. *Arg702trp polymorphisms are significantly (P = 0·038) reduced within CVIDs with splenomegaly. Gly908arg polymorphisms appear more frequent among CVIDs with enteropathy; however, this has not reached statistical significance here (P = 0·072).
Table 2.
Prevalence of complications and NOD2 polymorphisms among 285 international common variable immunodeficiency (CVID) patients.
| Complication |
||||||
|---|---|---|---|---|---|---|
| NOD2 genotype | Bronchiectasis | Autoimmunity | Granuloma | LIP | Splenomegaly | Enteropathy |
| Gly908Arg | 4 | 4 | 2 | 1 | 4 | 2 |
| Leu1007finsC | 10 | 7 | 1 | 0 | 8 | 1 |
| Arg702Trp | 14 | 7 | 3 | 2 | 3 | 0 |
| Total with at least one mutation | 26* | 17* | 6 | 3 | 14* | 3 |
| WT | 112 | 64 | 30 | 13 | 71 | 13 |
| Total | 138 | 81 | 36 | 16 | 85 | 16 |
Patients within these groups had multiple NOD2 polymorphisms. LIP: lymphoid interstitial pneumonitis; WT: wild-type.
Allele frequency of NOD2 polymorphisms in CVID
Optimized protocols were used to genotype our cohort of 291 confirmed CVID patients for gly908Arg, arg702trp and leu1007finsc polymorphisms of the NOD2 gene. Thirty-six heterozygous (four gly908arg, 21 arg702trp and 11 leu1007fins) polymorphisms, one homozygous gly908arg and one homozygous arg702trp polymorphism were found among our UK cohort giving gly908arg, arg702trp and leu1007finsc allele frequencies of 0·014, 0·056 and 0·026, respectively. Six samples were unusable, providing 285 results for statistical analysis. No significant difference was found when comparing the UK CVIDs from our cohort to published data on NOD2 allele frequencies in 1876 UK control subjects (0·011, 0·047 and 0·027, respectively) [29].
NOD2 polymorphisms and disease complications in CVID
Fisher's exact tests were conducted for NOD2 WT versus the three Crohn's disease associated polymorphisms for each CVID disease complication group defined (Fig. 1). A trend towards a gly908arg polymorphism conferring possible susceptibility towards autoimmunity, granuloma, LIP, splenomegaly and enteropathy is seen; however, these differences were not significant as defined. The most significant finding observed within the international cohort was a possible protective role of an arg702trp polymorphism from splenomegaly (P = 0·038, relative risk RR = 0·374). Interestingly, among the UK CVIDs cohort with a gly908arg polymorphism (n = 5), a significantly increased frequency of autoimmune disease (P = 0·049, RR = 2·400) and enteropathy (P = 0·049, RR = 5·771) are seen in comparison to the WT CVIDs. Autoimmune disease and enteropathy were prevalent in the UK cohort at 34·1 and 7·8%, respectively. No other significant associations were found within the UK subset of the international cohort or the international cohort as a whole between NOD2 genotype and disease complication phenotype.
Discussion
We investigated the presence of those polymorphisms found to be associated with Crohn's disease, namely gly908arg, arg702trp and leu1007finsc in the DNA of 285 CVID patients. These NOD2 polymorphisms were examined as potential disease modifiers within CVIDs. Autoimmune disease and enteropathy were found to be more prevalent in those UK patients with a gly908arg polymorphism. Interestingly, autoimmunity and enteropathy were not found to be associated significantly with each other in a previous European study of CVIDs [5]. Furthermore, the enteropathy of CVID is resistant to gluten withdrawal and so distinct from coeliac disease.
Granuloma and autoimmune disease are associated independently with splenomegaly [5]. As splenomegaly was shown to be less prevalent in the international cohort in CVID patients with arg702trp polymorphisms, this may protect from granuloma development. Enteropathy is associated with splenomegaly, although only weakly [5], suggesting that these may be independent gene associations with each phenotype.
No significant association was seen between NOD2 polymorphisms and other disease complications investigated (bronchiectasis, granulomatous disease, lymphoid interstitial pneumonitis). Because invasive investigations are required for histological diagnosis, variable thresholds of individual clinicians for requesting these investigations may have affected ascertainment, especially as this can be missed if not affecting a crucial anatomical site along with the variability in prevalence of complications between countries [5] and individual physicians' awareness for taking biopsies. As expected, no significant differences in allele frequency were found between CVID patients and geographically matched controls.
Crohn's disease is characterized by a dysregulated mucosal immune response [28]. The precise mechanisms by which NOD2 polymorphisms contribute to this dysregulated mucosal immunity are still under speculation [30]. Proposals include defective regulation of responses to commensal and/or pathogenic bacteria [31–33] or via irregular secretion of α-defensins by Paneth cells in response to NOD2 stimulation [34]. Impaired IL-17 secretion following NOD2 stimulation in Crohn's disease patients may contribute to impaired bacterial clearance [35].
A key proposed role of NOD2 is as a negative regulator of IL-12 stimulated through Toll-like receptor (TLR)-2 [36]. Presence of a Crohn's disease-like polymorphism increases TLR-2-mediated activation of nuclear factor (NF)-kB, resulting in increased production of IL-12 [37]. This leads to a Th1 skew, which is proposed as important in the pathogenesis of Crohn's disease. We hypothesized that this might also be important in the pathogenesis of the T cell infiltration of the epithelium and the interstium in unexplained enteropathy or of T cell activation in granulomata in CVIDs. The expanding pool of interest in the role of the innate immune system in CVIDs has also been supported recently by findings of abnormalities in the TLR-7/-9 pathways [38]. Further functional assays are required to investigate these hypotheses and the interplay of intracellular NOD receptors with TLRs in the recognition of bacteria which are the major pathogens in antibody deficiencies [39].
The polymorphisms investigated in this study account for 81% of the NOD2 polymorphisms found to be associated with Crohn's disease, but this leaves 19% not yet investigated here [19]. It should also be noted that genetic susceptibility to Crohn's disease is not limited to chromosome 16 and NOD2. Numerous genes have been found to be associated with inflammatory bowel diseases, and genomewide scans have identified additional susceptibility loci [40–44]. Further investigation into the other domains is essential alongside stimulation studies before a role of NOD2 as a disease modifier towards a granulomatous phenotype in CVIDs can be excluded. The importance of environmental factors on the development of complications and the pathogenesis of disease must also continue to be considered, and there are clearly multiple susceptibility factors and disease modifiers for Crohn's and other granulomatous disorders.
Conclusions
From this study it is seen that these major NOD2 polymorphisms are not associated with a granulomata in patients with CVIDs and are unlikely to act as disease-modifying polymorphisms, although there may be a protective role of arg908trp from splenomegaly within international CVID patients and a role of gly908arg polymorphisms towards enteropathic complications in UK CVIDs. Association of gly908arg with autoimmune disease within our UK cohort was also demonstrated and provides a hypothesis for further studies of patients with these complications.
Acknowledgments
This work was supported by funding from the European Union 6th Framework grant [EUROPOLICY SP23-CT-2005-006411], Baxter Healthcare and the Oxford NIHR Biomedical Research Centre Programme. We are grateful to Professor Derek Jewell from the Nuffield department of Clinical Medicine, the John Radcliffe Hospital for his kind donation of positive control DNA and to Professor M. Lee, Professor of Medical Statistics, UCLA, for confirming statistical calculations.
Disclosure
The authors have no conflicts of interest.
References
- 1.Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145:709–27. doi: 10.1111/j.1365-2141.2009.07669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spickett GP. Current perspectives on common variable immunodeficiency (CVID) Clin Exp Allergy. 2001;31:536–42. doi: 10.1046/j.1365-2222.2001.01117.x. [DOI] [PubMed] [Google Scholar]
- 3.Di Renzo M, Pasqui AL, Auteri A. Common variable immunodeficiency: a review. Clin Exp Med. 2004;3:211–17. doi: 10.1007/s10238-004-0027-2. [DOI] [PubMed] [Google Scholar]
- 4.Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) Clin Immunol. 1999;93:190–7. doi: 10.1006/clim.1999.4799. [DOI] [PubMed] [Google Scholar]
- 5.Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–86. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 7.Castigli E, Wilson SA, Garibyan L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–34. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 8.Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997;127:613–17. doi: 10.7326/0003-4819-127-8_part_1-199710150-00005. [DOI] [PubMed] [Google Scholar]
- 9.Bachwich PR, Lynch JP, III, Larrick J, Spengler M, Kunkel SL. Tumor necrosis factor production by human sarcoid alveolar macrophages. Am J Pathol. 1986;125:421–5. [PMC free article] [PubMed] [Google Scholar]
- 10.Hunninghake GW. Release of interleukin-1 by alveolar macrophages of patients with active pulmonary sarcoidosis. Am Rev Respir Dis. 1984;129:569–72. [PubMed] [Google Scholar]
- 11.Müller-Quernheim J, Saltini C, Sondermeyer P, Crystal RG. Compartmentalized activation of the interleukin 2 gene by lung T lymphocytes in active pulmonary sarcoidosis. J Immunol. 1986;137:3475–83. [PubMed] [Google Scholar]
- 12.Takizawa H, Satoh M, Okazaki H, et al. Increased IL-6 and IL-8 in bronchoalveolar lavage fluids (BALF) from patients with sarcoidosis: correlation with the clinical parameters. Clin Exp Immunol. 1997;107:175–81. doi: 10.1046/j.1365-2249.1997.d01-905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson BW, McLemore TL, Crystal RG. Gamma interferon is spontaneously released by alveolar macrophages and lung T lymphocytes in patients with pulmonary sarcoidosis. J Clin Invest. 1985;75:1488–95. doi: 10.1172/JCI111852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hugot JP, Laurent-Puig P, Gower-Rousseau C, et al. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature. 1996;379:821–3. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 15.Ogura Y, Lala S, Xin W, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut. 2003;52:1591–7. doi: 10.1136/gut.52.11.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Opitz B, Püschel A, Schmeck B, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem. 2004;279:36426–32. doi: 10.1074/jbc.M403861200. [DOI] [PubMed] [Google Scholar]
- 17.Sabbah A, Chang TH, Harnack R, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10:1073–80. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 19.Lesage S, Zouali H, Cézard JP, et al. CARD15/NOD2 mutational analysis and genotype–phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–57. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 21.van Heel DA, McGovern DP, Cardon LR, et al. Fine mapping of the IBD1 locus did not identify Crohn disease-associated. Am J Med Genet. 2002;111:253–9. doi: 10.1002/ajmg.10588. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad T, Armuzzi A, Bunce M, et al. The molecular classification of the clinical manifestations of Crohn's disease. Gastroenterology. 2002;122:854–66. doi: 10.1053/gast.2002.32413. [DOI] [PubMed] [Google Scholar]
- 23.Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 24.Kanazawa N, Okafuji I, Kambe N, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105:1195–7. doi: 10.1182/blood-2004-07-2972. [DOI] [PubMed] [Google Scholar]
- 25.Rahman P, Bartlett S, Siannis F, et al. CARD15: a pleiotropic autoimmune gene that confers susceptibility to psoriatic arthritis. Am J Hum Genet. 2003;73:677–81. doi: 10.1086/378076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holler E, Rogler G, Herfarth H, et al. Both donor and recipient NOD2/CARD15 mutations associate with transplant-related mortality and GvHD following allogeneic stem cell transplntation. Blood. 2004;104:889–94. doi: 10.1182/blood-2003-10-3543. [DOI] [PubMed] [Google Scholar]
- 27.Drewe E, Powell RJ. NOD2 and inflammatory disease. CPD Bull Immunol Allergy. 2004;4:20–2. [Google Scholar]
- 28.Schreiber S. Inflammatory bowel disease: immunologic concepts. Hepatogastroenterology. 2000;47:15–28. [PubMed] [Google Scholar]
- 29.Hugot JP, Zaccaria I, Cavanaugh J, et al. Prevalence of CARD15/NOD2 mutations in caucasian healthy people. Am J Gastroenterol. 2007;102:1259–67. doi: 10.1111/j.1572-0241.2007.01149.x. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto S, Ma X. Role of Nod2 in the development of Crohn's disease. Microbes Infect. 2009;11:912–18. doi: 10.1016/j.micinf.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rath HC, Schultz M, Freitag R, et al. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect Immun. 2001;69:2277–85. doi: 10.1128/IAI.69.4.2277-2285.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53:1–4. doi: 10.1136/gut.53.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feller M, Huwiler K, Schoepfer A, Shang A, Furrer H, Egger M. Long term antibiotic treatment for Crohn's disease: systematic review and meta-analysis of placebo controlled trials. Clin Infect Dis. 2010;50:473–80. doi: 10.1086/649923. [DOI] [PubMed] [Google Scholar]
- 34.Wehkamp J, Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–64. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27:660–9. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Eckmann L, Karin M. NOD2 and Crohn's disease: loss or gain of function? Immunity. 2005;22:661–7. doi: 10.1016/j.immuni.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–8. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 38.Yu JE, Knight AK, Radigan L, et al. Toll-like receptor 7 and 9 defects in common variable immunodeficiency. J Allergy Clin Immunol. 2009;124:349–56. doi: 10.1016/j.jaci.2009.05.019. 356 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Núñez G. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity. 2008;28:246–57. doi: 10.1016/j.immuni.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 40.Cho JH, Nicolae DL, Gold LH, et al. Identification of novel susceptibility loci for inflammatory bowel disease on chromosomes 1p, 3q, and 4q: evidence for epistasis between 1p and IBD1. Proc Natl Acad Sci USA. 1998;95:7502–7. doi: 10.1073/pnas.95.13.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satsangi J, Parkes M, Louis E, et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet. 1996;14:199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- 42.Rioux JD, Silverberg MS, Daly MJ, et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet. 2000;66:1863–70. doi: 10.1086/302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hampe J, Shaw SH, Saiz R, et al. Linkage of inflammatory bowel disease to human chromosome 6p. Am J Hum Genet. 1999;65:1647–55. doi: 10.1086/302677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duerr RH, Barmada MM, Zhang L, Pfützer R, Weeks DE. High-density genome scan in Crohn disease shows confirmed linkage to chromosome 14q11-12. Am J Hum Genet. 2000;66:1857–62. doi: 10.1086/302947. [DOI] [PMC free article] [PubMed] [Google Scholar]