

Abstract
B7-H1 [programmed death-ligand-1 (PD-L1)] is a B7-family member that binds to programmed death-1 (PD-1). Recently, deficiency of PD-L1 has been demonstrated to result in accelerated gastric epithelial cell damage in gastritis, and PD-L1 is suggested to play a critical role in regulating T cell homeostasis. Here, we aimed to gain more insight into gastric PD-L1 expression, regulation and function during Helicobacter pylori infection. PD-L1 expression in human gastric epithelial cells was analysed using Western blotting, quantitative polymerase chain reaction and fluorescence activated cell sorter analysis. Furthermore, co-culture experiments of human gastric epithelial cells with primary human T cells or Jurkat T cells were conducted. PD-L1 expression in primary human gastric epithelial cells was strongly enhanced by H. pylori infection and activated T cells, and augmented markedly by further stimulation with interferon-γ or tumour necrosis factor-α. Moreover, PD-L1 expression in gastric epithelial cells significantly induced apoptosis of T cells. Our results indicate that a novel bidirectional interaction between human gastric epithelial cells and lymphocytes modulates PD-L1 expression in human gastric epithelial cells, contributing to the unique immunological properties of the stomach.
Keywords: apoptosis, B7-H1, gastric epithelial cell, Helicobacter pylori, PD-1, PD-L1
Introduction
Approximately 50% of the world's population is infected with the pathogen Helicobacter pylori, being associated with duodenal and peptic ulcer diseases. The clinical consequences range from asymptomatic gastritis to peptic ulceration and gastric malignancy [1,2]. The outcome of the infection is determined by interactions among H. pylori virulence factors, host gastric mucosal factors and the environment. However, the mechanisms by which host factors cause disease remain unclear. H. pylori colonization induces systemic and mucosal immune responses. There is increased T cell infiltration at the site of infection with H. pylori[3]. Recent studies have suggested that the number of T helper type 1 (Th1) cells is increased selectively during infection [3–6]. Th1 cytokines, such as interferon (IFN)-γ and tumour necrosis factor (TNF)-α, can increase the release of proinflammatory cytokines, augmenting apoptosis induced by H. pylori[7].
B7-like molecules and their cognate receptors constitute important co-stimulatory pathways that control and fine-tune immune responses. In recent years, an array of new members of the B7 family have been identified, including B7-H1 [programmed death-ligand-1 (PD-L1)] and B7-DC (PD-L2) [8]. Both are ligands for programmed death-1 (PD-1), which is expressed on activated T and B cells [9–11]. PD-L1 has been described to be expressed inducibly in a variety of organs [8,11–17]. A number of studies support the role of PD-L1 as a negative regulator of T cell responses and suggest that PD-L1 promotes peripheral tolerance [18].
Here, we analysed whether H. pylori induces overexpression of PD-L1 in human gastric epithelial cells. Our data suggest that a novel bidirectional interaction between human gastric epithelial cells and lymphocytes modulates PD-L1 expression in gastric epithelial cells during H. pylori infection.
Materials and methods
Subjects and culture of primary human gastric epithelial cells
Biopsy specimens of the gastric antrum were obtained from 23 patients undergoing gastric endoscopy for dyspepsia at China Medical University Hospital. The study was approved by the Medical Ethics Committee of China Medical University Medical Center (Taichung, Taiwan) and informed consent was obtained from all patients. The specimens were taken from areas of grossly normal gastric mucosa comprising surface and deep glandular epithelium. Among the 23 patients, 12 were H. pylori-positive and 11 were H. pylori-negative. Human gastric epithelial cells were cultured using a previously described method [19,20]. In brief, gastric biopsy specimens were collected in Leibowitz's L-15 medium (Life Technologies, Grand Island, NY, USA). Gastric cells were isolated enzymatically using collagenase/dispase after the tissue had been mechanically minced into ≤ 1-mm pieces. The tissue was then pelleted by centrifugation at 200 g for 5 min at 4°C and the collagenase/dispase was discarded. The tissue was then washed once in 10 ml of phosphate-buffered saline (PBS) and pelleted by centrifugation. Cells were resuspended in the cell culture medium. Gastric epithelial cells obtained as described above were suspended in 2 ml of Ham's F-12 cell culture medium (Life Technologies) with 10% fetal bovine serum and placed into a six-well tissue culture plate. Primary gastric cells 48 h after being placed in culture were stained with antibodies to cytokeratin 18 (Sigma Chemical Co., St Louis, MO, USA) to test for purity [21]. The cytoplasm of virtually every cell in the colony stained positively for cytokeratin 18, indicating that these cells were of epithelial origin. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] reduction assay (Chemicon International Inc., Temecula, CA, USA) at 48 h revealed that the viability of the epithelial cells was > 95%.
Cell isolation and cell culture
T cells were separated from peripheral blood mononuclear cells and cultured as described previously [22]. Heparinized peripheral venous blood was obtained from healthy donors. T cells were separated by the Rosette separation method (Stem Cell Technologies, Vancouver, British Columbia, Canada). Briefly, non-T cells were selected by a tetrameric antibody mixture against CD14, CD16, CD19, CD56 and glyA bound to erythrocytes. These complexes were separated from the T cells by a Ficoll-Paque gradient. The purity of the isolated human T cells was > 97% as tested by flow cytometry with anti-CD3 monoclonal antibodies (mAb).
The human gastric adenocarcinoma cell line (AGS) was obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS). For some experiments, cells were stimulated with recombinant TNF-α (R&D Systems, Minneapolis, MN, USA; 40 ng/ml) or IFN-γ (Roche, Indianapolis, IN, USA; 100 U/ml). For activation, Jurkat T cells or primary T cells were incubated with anti-CD28 (clone: CD28·2; eBioscience, San Diego, CA, USA; 1 µg/ml/1 × 106) and recombinant interleukin (rIL)-2 (Sigma; 100 IU/ml/1 × 106) in PBS-washed anti-CD3-coated 96-well round-bottomed plates (clone: OKT3; eBioscience; 100 µg/plate). These cells were stimulated for 72 h in 96-well flat-bottomed microtitre plates before adding them to the epithelial cell cultures. In co-cultures of epithelial cells and T cells, the co-cultured suspension T cells were removed prior to reverse transcription (RT); thereafter, only epithelial cell RNA was collected for later use. The ratio of epithelial cells to T cells was 1:1.
Bacterial cultures
H. pylori strain ATCC 26695 bacteria were grown on blood-agar plates (BD Biosciences) at 37°C under microaerophilic conditions. After 48 h in blood agar plates, H. pylori were grown in Brucella broth for 24 h. The bacteria were washed with normal saline, and their concentration was measured by optical density (OD) at 530 nm absorbance using a DU-65 spectrophotometer (Beckman Instruments, Inc., Fullerton, CA, USA). The bacteria were then adjusted to a concentration taking 1 OD equivalent to 2 × 108 bacteria/ml.
Infection of gastric epithelial cells with H. pylori
Before infecting the gastric epithelial cells with H. pylori for 48 h, gastric epithelial cells were washed and medium was replaced with antibiotic-free medium. The bacteria were resuspended in RPMI-1640 medium and used with a cell : bacteria ratio of 1:100.
Analysis of mRNA expression
The PD-L1 expression was analysed by real-time quantitative reverse transcription–polymerase chain reaction (RT–PCR). Total RNA was purified using the Trizol reagent (Invitrogen Life Technologies, Grand Island, NY, USA) following the instructions in the manufacturer's manual. Total RNA (5 µg) was reverse-transcribed with Superscript II RNase H reverse transcriptase (Invitrogen Life Technologies) and an oligo(dT) primer. Synthesized cDNA was stored at −20°C until it was used. The expression of PD-L1 real-time PCR was performed as described previously [19]. In brief, cDNA (2 µl) was subjected to real-time quantitative RT–PCR using the Opticon system with SYBR green I (Molecular Probes, Eugene, OR, USA) as a fluorescent reporter. PD-L1 and β-actin cDNAs were amplified in separate reactions. The primers used for PD-L1 were 5′-GGTGAGGATGGTTCTACACAG-3′ (sense) and 5′-GAGAACTGCATGAGGTTGC-3′ (anti-sense). The threshold cycle number of duplicate reactions was determined using the Opticon software, and levels of PD-L1 mRNA expression were normalized to β-actin levels using the formula 2(Rt–Et), where Rt is the mean threshold cycle for the reference gene (β-actin) and Et is the mean threshold cycle for the experimental gene. Data are expressed in arbitrary units.
Analysis of PD-L1 expression by FACS analysis
Surface expression of PD-L1 was analysed by flow cytometry using phycoerythrin (PE)-conjugated anti-PD-L1 (clone MIH1) antibodies (eBioscience). After washing with PBS containing 10% fetal calf serum (FCS), cells were incubated with anti-PD-L1 or matching isotype control antibodies for 30 min, washed, and fixed in 1% paraformaldehyde. A total of 10 000 gated events were analysed using a FACScan (Becton Dickinson).
Apoptosis assays
To determine PD-L1-mediated cytotoxicity, co-cultures of 200 000 Jurkat T cells (or primary T cells) were established with 200 000 AGS cells (or primary gastric epithelial cells) for 24 h. T cells were added either directly to the gastric epithelial cells or placed into Transwell inserts (0·2 mm pore size; Nunc Tissue Culture, Nunc, Roskilde, Denmark). To analyse functional activity, blocking anti-PD-L1 antibodies (clone MIH5; eBioscience) were incubated with the gastric epithelial cells, and unbound antibody was removed by washing prior to addition of T cells. For detection of apoptosis, cells were stained simultaneously with fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide (PI) (both from BD PharMingen, La Jolla, San Diego, CA, USA), according to the manufacturer's instructions. Total numbers of apoptotic cells were determined by calculation of annexin V+ and PI- cells together with annexin V+ and PI+ cells. For positive controls, cells were permeabilized with saponin 0·1% for 1 h.
Western blot analysis
Proteins were separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a nitrocellulose membrane (Bio-Rad), and blocked in TBST (20 mM Tris-HCl/500 mM NaCl (pH 7·5) with 0·05% Tween 20) containing 5% dry milk. The membranes were subsequently probed with respective primary antibody in TBST containing 5% dry milk. The binding of primary antibodies was detected with horseradish peroxidase (HRP)-conjugated secondary antibody (Amersham Biosciences, Arlington Heights, IL, USA). Subsequently, membranes were washed and incubated in ECL reagent (Amersham Biosciences).
Statistical analysis
Results are expressed as means ± standard deviation (s.d.) (range) or percentage. Comparison between groups was made using Student's unpaired t-test. Welch's correction was performed when required. A P-value < 0·05 was considered statistically significant. All calculations were performed using the statistical computer package GraphPad Prism version 4·00 for Windows (GraphPad Software, San Diego, CA, USA).
Results
Expression of PD-L1 in human gastric epithelial cells and gastric biopsies
Initially, we analysed PD-L1 protein expression in primary gastric epithelial cells, AGS and gastric biopsies. In accordance with previous studies analysing human primary gastric epithelial cells [19], we detected PD-L1 expression in human primary gastric epithelial cells, AGS and gastric biopsy samples from patients negative to H. pylori infection at only low levels. In addition, PD-L1 expression was clearly detectable in gastric biopsy samples from H. pylori-infected patients (Fig. 1a). Eleven preparations of primary gastric epithelial cells were tested for PD-L1 expression using Western blot. No significant differences were found in the levels of PD-L1 expression between primary gastric epithelial cells from 11 different human cases. A total of 22 gastric biopsy samples were analysed from 12 H. pylori-positive and 10 H. pylori-negative patients. H. pylori-positive samples showed more PD-L1 expression than H. pylori-negative samples (Fig. 1b).
Fig. 1.
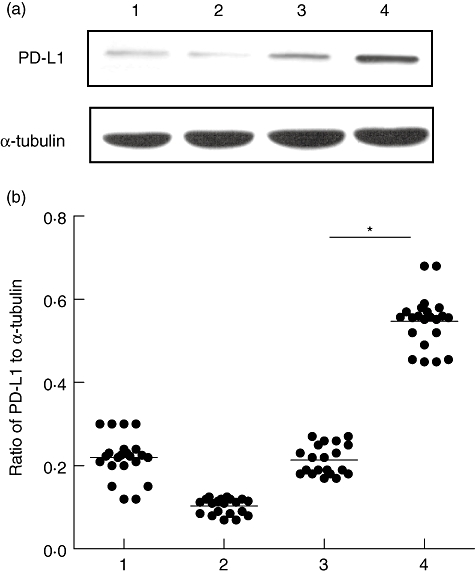
Western blot of programmed death-ligand-1 (PD-L1) expression using mouse anti-human PD-L1 monoclonal antibody (mAb). (a) Lane 1, primary gastric epithelial cell; lane 2, human gastric adenocarcinoma (AGS); lane 3, gastric biopsy samples from patient negative to Helicobacter pylori infection; lane 4, gastric biopsy samples from H. pylori-infected patient. α-Tubulin was used as an internal control. (b) Densitometric scan was performed using BioRad's Quantity One software. Percentage of adjacent volume was determined for each band of PD-L1 and α-tubulin. Ratio of percentage of adjacent volume was plotted. *P < 0·05 compared to control.
Subsequently, we wanted to investigate whether PD-L1 expression in gastric epithelial cells may be affected directly by H. pylori infection. First, we analysed AGS cells 24 h and 48 h after infection with H. pylori. PD-L1 mRNA expression was induced significantly in AGS cells by H. pylori infection compared to uninfected controls [12·5-fold (P < 0·05) 24 h and 18·7-fold (P < 0·05) 48 h after infection; Fig. 2a)]. Secondly, we analysed primary gastric epithelial cells 24 h and 48 h after infection with H. pylori. PD-L1 mRNA expression was induced significantly in primary gastric epithelial cells by H. pylori infection compared to uninfected controls [3·6-fold (P < 0·05) 24 h and 7·8-fold (P < 0·05) 48 h after infection; Fig. 2b], demonstrating that H. pylori induced elevated levels of PD-L1.
Fig. 2.
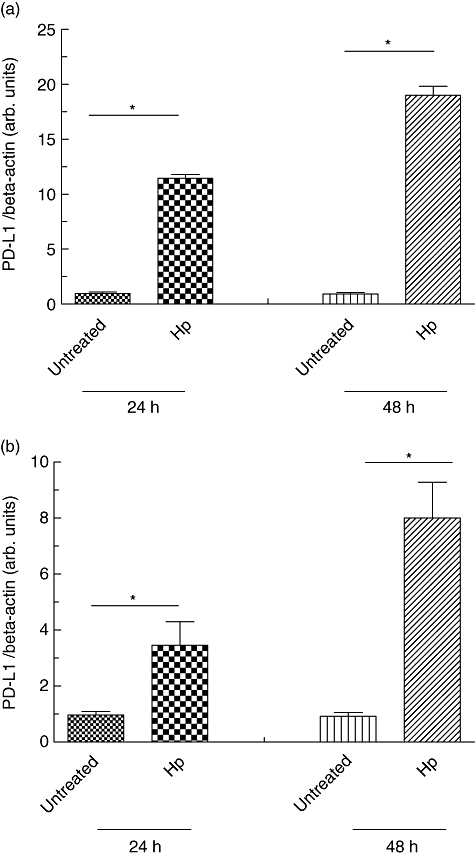
Programmed death-ligand-1 (PD-L1) expression of human gastric epithelial cells after Helicobacter pylori infection. (a) Human gastric adenocarcinoma (AGS) cells were infected with H. pylori and RNA was isolated 24 and 48 h after infection. Non-infected AGS cells served as controls. (b) Primary human gastric epithelial cells were infected with H. pylori and RNA was isolated 24 and 48 h after infection. Non-infected primary human gastric epithelial cells served as controls. PD-L1 mRNA expression was quantified in relation to β-actin expression by real-time polymerase chain reaction. *P < 0·05 compared to control.
Induction of PD-L1 expression in gastric epithelial cells by activated T cells
Because Helicobacter pylori infections induce T cell responses, in order to analyse whether T cells might influence levels of PD-L1 expression in gastric epithelial cells co-cultures were established of AGS cells with resting or activated Jurkat T cells and naive or activated primary T cells. Quantitative PCR analysis revealed a significant up-regulation of PD-L1 expression in AGS cells co-cultured with activated Jurkat T cells or activated primary human T cells compared to AGS cells in monoculture (control) (5·1-fold P = 0·001 and 9·4-fold P = 0·007, respectively; Fig. 3a). Also, co-culture with resting T cells induced an increased PD-L1 expression (1·7- and 3·1-fold, respectively) but these differences did not reach statistical significance.
Fig. 3.
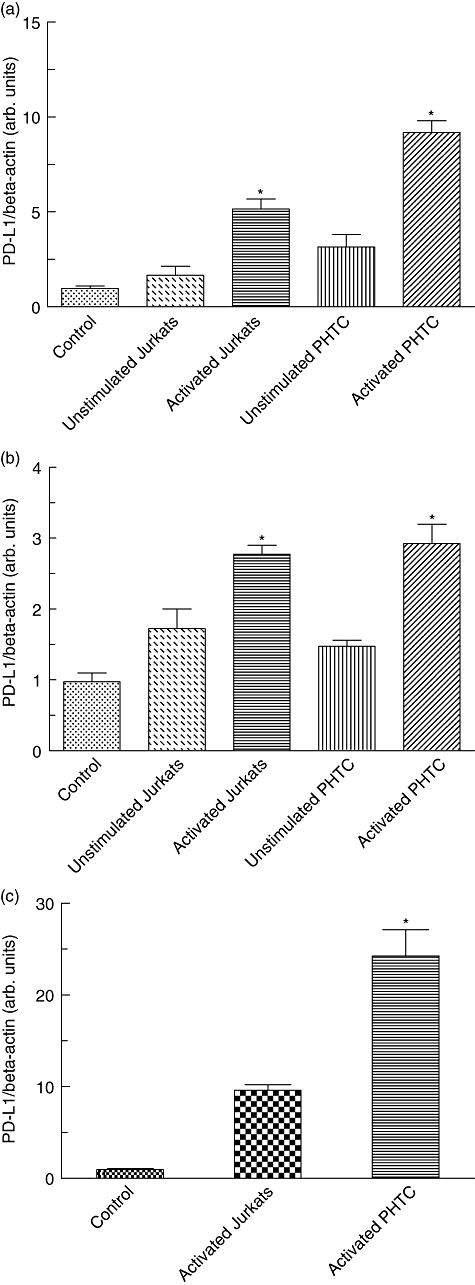
Programmed death-ligand-1 (PD-L1) expression of gastric epithelial cells after co-incubation with T cells. Human gastric adenocarcinoma (AGS) cells (a) and primary human gastric epithelial cells (b) were co-cultured with resting cells or activated Jurkat T cells as well as naive or activated primary human T cells (PHTC), respectively. Data represent PD-L1/β-actin ratio on gastric epithelial cell. Furthermore, Transwell co-cultures were established with AGS cells and activated Jurkat T cells or activated primary human T cells (PHTC), respectively (c). PD-L1 mRNA expression in AGS cells and primary human gastric epithelial cells was quantified in relation to β-actin expression by real-time polymerase chain reaction. AGS cells and primary human gastric epithelial cells in monoculture served as controls.*P < 0·05 compared to control.
Next we wanted to confirm these findings in primary gastric epithelial cells. Similar to AGS cells, co-culture with activated Jurkat T cells or primary T cells induced a significant up-regulation of PD-L1 expression (2·76-fold P = 0·036 and 2·9-fold P = 0·02, respectively; Fig. 3b).
To investigate whether direct cell-to-cell contact is important for the observed PD-L1 up-regulation, additional Transwell experiments were performed using a membrane with 0·2 µm pore size to separate activated Jurkat T cells or primary T cells from AGS cells. In this experimental setting a significant up-regulation of PD-L1 mRNA was also observed in AGS cells (23·8-fold; P = 0·02 Fig. 3c). In Transwell experiments PD-L1 up-regulation in AGS cells was even higher than observed in the conventional co-culture system (Fig. 3a and 9·4-fold), indicating that direct contact with activated T cells was not required for induction of PD-L1.
Induction of PD-L1 expression by Th1 cytokines IFN-γ and TNF-α
To test whether Th1 cytokines IFN-γ and TNF-α might modulate PD-L1 expression in gastric epithelial cells, we performed dose–response and time–course experiments initially using AGS cells. As determined by quantitative PCR, expression of PD-L1 mRNA was directly dependent upon the IFN-γ concentrations. PD-L1 expression was induced at concentrations as low as 10 U/ml and reached a plateau at concentrations higher than 1000 U/ml (Fig. 4a). Applying 100 U/ml IFN-γ, a significant increase of PD-L1 expression was observed already 6 h after stimulation. Maximal PD-L1 expression was observed 24 h after stimulation with IFN-γ, and expression levels were maintained for 48 h (Fig. 4b). Next we performed dose–response and time–course experiments applying TNF-α. PD-L1 expression was induced at concentrations as low as 10 ng/ml TNF-α, and increased significantly with increasing doses of TNF-α (tested up to 60 ng/ml) (Fig. 4c). The time–course of TNF-α-mediated PD-L1 mRNA induction (tested at a concentration of 40 ng/ml) was similar to that seen for IFN-γ (Fig. 4d). Based upon the results obtained from dose–response and time–course experiments with AGS cells, we investigated the effect of both Th1 cytokines on PD-L1 expression in human primary gastric epithelial cells. Similar to that seen in AGS cells, 24 h stimulation with both TNF-α (40 ng/ml) or IFN-γ (100 U/ml) induced a significant up-regulation of PD-L1 mRNA expression in human primary gastric epithelial cells [35-fold (P < 0·005) and 27-fold (P < 0·005), respectively] (Fig. 5a). Next, we wanted to confirm our finding on the protein level. Here, as in subsequent analysis, we focused upon human primary gastric epithelial cells. FACS analysis revealed only minimal expression of PD-L1 on the surface of human primary gastric epithelial cells, suggesting that constitutive PD-L1 expression in human primary gastric epithelial cells occurred at only low levels (MFI 18·5; isotype control MFI 10·2) (Fig. 5b). FACS analysis was used to confirm these results at the protein level and revealed a strong up-regulation of PD-L1 on the cell surface of AGS cells as well as human primary gastric epithelial cells following stimulation with either IFN-γ (MFI 587·6) or TNF-α (MFI 557·4) (Fig. 5b).
Fig. 4.
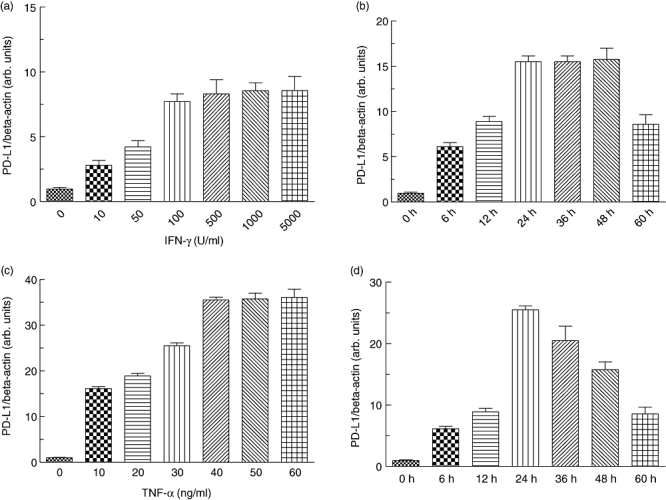
Effects of interferon (IFN)-γ and tumour necrosis factor (TNF)-α on programmed death-ligand-1 (PD-L1) expression. Human gastric adenocarcinoma (AGS) were stimulated with the indicated concentrations of IFN-γ (a) and TNF-α (c) for 24 h, and PD-L1 expression was analysed. Time–course of IFN-γ (b) and TNF-α (d) induced PD-L1 mRNA-expression. At the indicated time intervals after stimulation RNA was isolated and PD-L1 mRNA expression was quantified in relation to β-actin expression by real-time polymerase chain reaction. Data represent means ± standard deviation from three separate experiments.
Fig. 5.
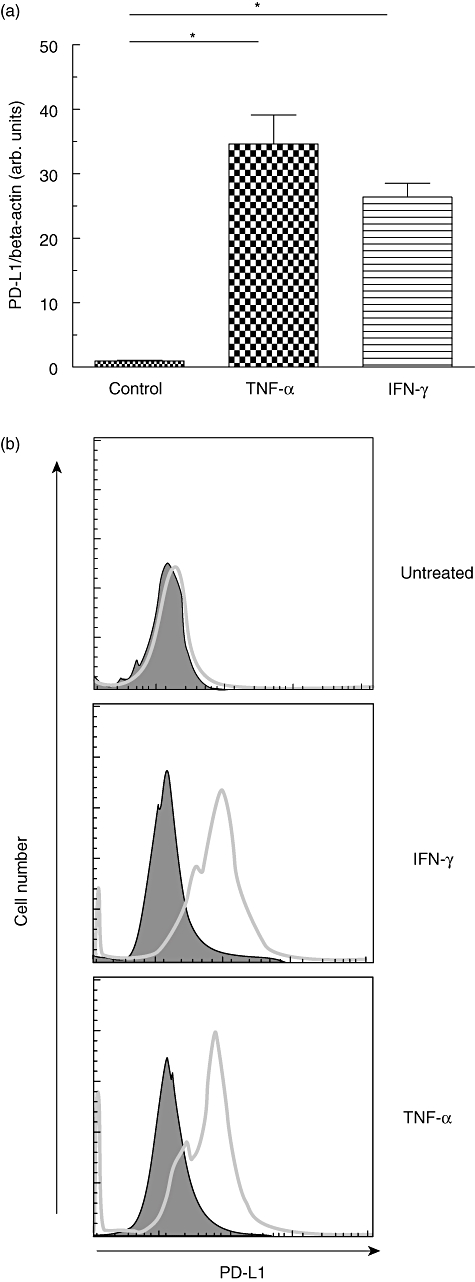
Effects of interferon (IFN)-γ and tumour necrosis factor (TNF)-α on programmed death-ligand-1 (PD-L1) expression in primary human gastric epithelial cells. Primary human gastric epithelial cells were stimulated with IFN-γ (100 U/ml) and/or TNF-α (40 ng/ml) for 24 h (for PD-L1 mRNA analysis) or 48 h (for analysis of PD-L1 surface expression), respectively. (a) PD-L1 mRNA expression was quantified in relation to β-actin expression by real-time polymerase chain reaction. (b) Expression of cell surface PD-L1 determined by fluorescence activated cell sorter (FACS) analysis (open profiles, anti-PD-L1 antibody; filled profiles, isotype control). *P < 0·05 compared to control.
Expression of PD-L1 in gastric epithelial cell-mediated T cell apoptosis
It has been demonstrated that PD-L1 expressed by tumour cells is able to induce apoptosis in T cells. With regard to these observations, we wanted to evaluate whether T cell apoptosis might be regulated by PD-L1 expressed in gastric epithelial cells. Therefore, AGS cells were cultured in the presence or absence of IFN-γ or TNF-α and blocking antibodies to PD-L1, respectively. Unbound antibody was removed prior to establishment of co-cultures with Jurkat T cells to ensure that the effect of the antibodies was due to blockade of PD-L1 on the gastric epithelial cells and not on T cells, as PD-L1 can be expressed on activated human T cells [23]. After 8 h of co-culture, apoptosis in Jurkat T cells was examined by FACS analysis. Numbers of apoptotic Jurkat T cells were increased significantly in co-cultures with AGS cells activated by IFN-γ or TNF-α compared to co-cultures with untreated control cells (2·39 ± 0·9-fold, P = 0·013 and 2·05 ± 0·7-fold, P = 0·006, respectively). In the presence of blocking antibodies to PD-L1, apoptosis in Jurkat T cells was reduced significantly in co-cultures with IFN-γ- and TNF-α-activated AGS cells (0·59 ± 0·2, P = 0·0003 and 0·65 ± 0·1, P = 0·004, respectively), demonstrating that PD-L1 expression on the surface of gastric epithelial cells mediates T cell apoptosis. A representative FACS result is depicted in Fig. 6. Similar results were obtained in co-culture experiments with primary human gastric epithelial cells and T cells, respectively (data not shown).
Fig. 6.
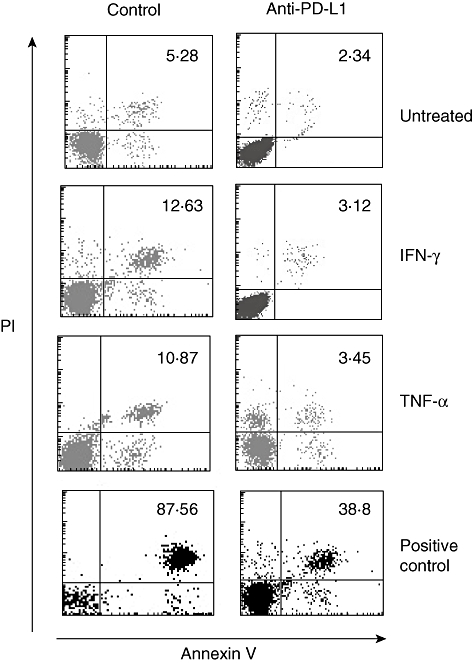
Expression of programmed death-ligand-1 (PD-L1) in gastric epithelial cell-mediated T cell apoptosis. Human gastric adenocarcinoma (AGS) cells (1 × 106) were activated by addition of interferon (IFN)-γ (100 U/ml) and/or tumour necrosis factor (TNF)-α (40 ng/ml) in the presence or absence of blocking antibodies to PD-L1 (10 µg/ml). Unbound antibody was removed prior to establishment of co-cultures with Jurkat T cells (5 × 106). After 8 h of co-culture, apoptosis in Jurkat T cells was examined by fluorescence activated cell sorter (FACS) analysis using annexin V/PI-staining. The percentages of apoptotic T cells (annexin V+/PI+) are indicated. One of three independent experiments is depicted. For positive controls, cells were permeabilized with saponin 0·1% for 1 h.
Discussion
We have demonstrated for the first time that primary human gastric epithelial cells express constitutively low levels of PD-L1, while its expression is enhanced strongly by activated T cells and H. pylori infection, and augmented markedly by further stimulation with IFN-γ or TNF-α. Moreover, PD-L1 expression on primary human gastric epithelial cells induces apoptosis in T cells.
H. pylori infection is one of the most common infections of humans because 50% of the global population carries this bacterium in the stomach, and in some individuals infection leads to serious diseases. Despite the inflammatory and immune responses that are elicited by H. pylori infection, it remains chronic. Various studies have suggested that T cell functions are affected during infection with H. pylori. Early studies to assess T cell function in response to H. pylori suggested that T cells in infected individuals were hyporesponsive compared with non-infected individuals. Gastric T cells from infected individuals were suppressed in their proliferation and IFN-γ production [24]. The mechanisms responsible for the low response were not clear.
Basic expression but even more inducible induction of PD-L1 on gastric epithelial cells may have several implications. IFN-γ is a key proinflammatory molecule released by activated T cells, up-regulating MHC expression and antigen presentation pathways. However, IFN-γ can also dampen the extent of T cell activation and limit T cell-mediated tissue damage [17,25–27]. The observed profound up-regulation of PD-L1 expression on gastric epithelial cells may serve as a part of a tissue protective negative feedback mechanism in T cell-mediated inflammatory events. PD-L1 delivers an inhibitory signal to T cell activity; our finding of induced PD-L1 expression on human gastric epithelial cells in response to TNF-α is of particular interest. One might speculate that TNF-α therapy affects both anti-bacterial but, via PD-L1, also immunosuppressive and anti-inflammatory mechanisms, eventually favouring bacterial persistence. We would like to hypothesize that TNF-α-induced PD-L1 on human gastric epithelial cells accounts in part for some of these reported gastric protective effects.
Interestingly, we observed increased levels of PD-L1 in human primary gastric epithelial cells after H. pylori infection. Similarly, previous studies found an up-regulation of PD-L1 expression in gastric epithelial cell lines in response to H. pylori[28] and in lymphocytes in response to H. pylori[29]. Taking into account that PD-L1 may have receptors other than PD-1 on activated T cells [30–32], we used anti-PD-L1 mAb instead of anti-PD-1 mAb or fusion protein and could partially reverse the proapoptotic effect of human gastric epithelial cells on lymphocytes. At this time, however, we do not know whether this is mediated through ligation of PD-1. Conversely, it may be speculated that the observed PD-L1 up-regulation in human gastric epithelial cells by activated lymphocytes is mediated exclusively via substantial IFN-γ or TNF-α expression compared to resting cells. In any case, PD-L1 up-regulation by activated lymphocytes has been observed in activated hepatic stellate cells [32]. Furthermore, IFN-γ has been shown to up-regulate PD-L1 expression in dermal fibroblasts [33]. Interestingly, also in vivo substantially increased expression of B7-H1 on keratinocytes was found close to numerous T cell infiltrates in the subepithelium of patients with chronic inflammatory mucocutaneous disease [12]. In addition, in Transwell experiments, the level of PD-L1 expression was even higher in the Transwell cultures, indicating that perhaps cell–cell contact reduced the soluble factors released by activated T cells.
Our results suggest a bidirectional interaction between human gastric epithelial cells and lymphocytes. Primary human gastric epithelial cells isolated from human stomach express low levels of PD-L1. Exposure to IFN-γ or TNF-α, an inflammatory cytokine released by activated T cells, results in up-regulation of PD-L1 on gastric epithelial cells that in turn induces apoptosis in lymphocytes. Herewith, gastric epithelial cells may contribute to gastric tolerance induction by deletion of activated T cells through induction of apoptosis. Currently, the cellular mechanism by which, in certain in vivo settings, PD-L1 co-stimulates T cell responses, and in other circumstances these responses are inhibited, remains elusive. Our study may provide a first step for the elucidation of this mechanism in the stomach, an organ with unique immunological properties.
Acknowledgments
This work was supported by grants from the National Science Council, Taiwan (NSC97-2320-B-039-045; NSC 98-2320-B-039 -012 -MY3) and China Medical University, Taiwan (CMU96-266; CMU97-299). We would like to thank Dr Chin-Tung Hsieh and Mr Jeffrey Conrad for their valuable technique assistance and critical review of the manuscripts.
Disclosure
No potential conflicts of interest were disclosed.
References
- 1.Parsonnet J. Molecular mechanisms for inflammation-promoted pathogenesis of cancer – the Sixteenth International Symposium of the Sapporo Cancer Seminar. Cancer Res. 1997;57:3620–4. [PubMed] [Google Scholar]
- 2.Parsonnet J, Friedman GD, Vandersteen DP, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–31. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 3.Bamford KB, Fan X, Crowe SE, et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology. 1998;114:482–92. doi: 10.1016/s0016-5085(98)70531-1. [DOI] [PubMed] [Google Scholar]
- 4.D'Elios MM, Manghetti M, De Carli M, et al. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J Immunol. 1997;158:962–7. [PubMed] [Google Scholar]
- 5.Lindholm C, Quiding-Jarbrink M, Lonroth H, Hamlet A, Svennerholm AM. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–71. doi: 10.1128/iai.66.12.5964-5971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sommer F, Faller G, Konturek P, et al. Antrum- and corpus mucosa-infiltrating CD4(+) lymphocytes in Helicobacter pylori gastritis display a Th1 phenotype. Infect Immun. 1998;66:5543–6. doi: 10.1128/iai.66.11.5543-5546.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner S, Beil W, Westermann J, et al. Regulation of gastric epithelial cell growth by Helicobacter pylori: evidence for a major role of apoptosis. Gastroenterology. 1997;113:1836–47. doi: 10.1016/s0016-5085(97)70003-9. [DOI] [PubMed] [Google Scholar]
- 8.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–92. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 9.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 10.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 12.Youngnak-Piboonratanakit P, Tsushima F, Otsuki N, et al. The expression of B7-H1 on keratinocytes in chronic inflammatory mucocutaneous disease and its regulatory role. Immunol Lett. 2004;94:215–22. doi: 10.1016/j.imlet.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Ruddle NH, Picarella D, Kratz A, Li CB, Flavell RA. Probing the mechanism of TNF-alpha(cachectin)- and TNF-beta(lymphotoxin)-induced pancreatic inflammation with transgenic mice. Res Immunol. 1993;144:336–42. doi: 10.1016/s0923-2494(93)80077-c. [DOI] [PubMed] [Google Scholar]
- 14.Guerder S, Picarella DE, Linsley PS, Flavell RA. Costimulator B7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc Natl Acad Sci USA. 1994;91:5138–42. doi: 10.1073/pnas.91.11.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuff CA, Sacca R, Ruddle NH. Differential induction of adhesion molecule and chemokine expression by LTalpha3 and LTalphabeta in inflammation elucidates potential mechanisms of mesenteric and peripheral lymph node development. J Immunol. 1999;162:5965–72. [PubMed] [Google Scholar]
- 16.Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–81. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- 17.Oei E, Kalb T, Beuria P, et al. Accessory cell function of airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L318–31. doi: 10.1152/ajplung.00174.2003. [DOI] [PubMed] [Google Scholar]
- 18.Subudhi SK, Alegre ML, Fu YX. The balance of immune responses: costimulation versus coinhibition. J Mol Med. 2005;83:193–202. doi: 10.1007/s00109-004-0617-1. [DOI] [PubMed] [Google Scholar]
- 19.Wu YY, Tsai HF, Lin WC, et al. Upregulation of CCL20 and recruitment of CCR6+ gastric infiltrating lymphocytes in Helicobacter pylori gastritis. Infect Immun. 2007;75:4357–63. doi: 10.1128/IAI.01660-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu YY, Tsai HF, Lin WC, et al. Helicobacter pylori enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in human gastric epithelial cells. World J Gastroenterol. 2004;10:2334–9. doi: 10.3748/wjg.v10.i16.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smoot DT, Sewchand J, Young K, Desbordes BC, Allen CR, Naab T. A method for establishing primary cultures of human gastric epithelial cells. Methods Cell Sci. 2000;22:133–6. doi: 10.1023/a:1009846624044. [DOI] [PubMed] [Google Scholar]
- 22.Tsai HF, Lai JJ, Chou AH, Wang TF, Wu CS, Hsu PN. Induction of costimulation of human CD4 T cells by tumor necrosis factor-related apoptosis-inducing ligand: possible role in T cell activation in systemic lupus erythematosus. Arthritis Rheum. 2004;50:629–39. doi: 10.1002/art.20038. [DOI] [PubMed] [Google Scholar]
- 23.Brown JA, Dorfman DM, Ma FR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–66. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 24.Fan XJ, Chua A, Shahi CN, McDevitt J, Keeling PW, Kelleher D. Gastric T lymphocyte responses to Helicobacter pylori in patients with H. pylori colonisation. Gut. 1994;35:1379–84. doi: 10.1136/gut.35.10.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eriksson U, Kurrer MO, Sebald W, Brombacher F, Kopf M. Dual role of the IL-12/IFN-gamma axis in the development of autoimmune myocarditis: induction by IL-12 and protection by IFN-gamma. J Immunol. 2001;167:5464–9. doi: 10.4049/jimmunol.167.9.5464. [DOI] [PubMed] [Google Scholar]
- 26.Nagano H, Mitchell RN, Taylor MK, Hasegawa S, Tilney NL, Libby P. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J Clin Invest. 1997;100:550–7. doi: 10.1172/JCI119564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pewe L, Haring J, Perlman S. CD4 T-cell-mediated demyelination is increased in the absence of gamma interferon in mice infected with mouse hepatitis virus. J Virol. 2002;76:7329–33. doi: 10.1128/JVI.76.14.7329-7333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol. 2006;176:3000–9. doi: 10.4049/jimmunol.176.5.3000. [DOI] [PubMed] [Google Scholar]
- 29.Beswick EJ, Pinchuk IV, Das S, Powell DW, Reyes VE. Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect Immun. 2007;75:4334–41. doi: 10.1128/IAI.00553-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med. 2003;197:1083–91. doi: 10.1084/jem.20021752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tseng SY, Otsuji M, Gorski K, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–46. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Gao JX, Wen J, et al. B7DC/PDL2 promotes tumor immunity by a PD-1-independent mechanism. J Exp Med. 2003;197:1721–30. doi: 10.1084/jem.20022089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SK, Seo SH, Kim BS, et al. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. 2005;40:95–103. doi: 10.1016/j.jdermsci.2005.06.008. [DOI] [PubMed] [Google Scholar]