


Abstract
A tetracycline-controlled gene expression system provides a powerful tool to dissect the functions of gene products. However, it often appears difficult to establish cell lines or transgenic animals stably expressing tetracycline-dependent transactivators, possibly as a result of toxicity of the transactivator domains used. In order to overcome this problem, we developed a novel tetracycline-dependent transactivator that works efficiently in mammalian cells. This transactivator is a fusion of the tet reverse repressor mutant and the transcriptional activating domain of human E2F4, which is ubiquitously expressed in vivo. We demonstrate here that this tetracycline-regulated gene expression system provides a two log transcriptional activation in mammalian cells as assessed by northern blot and luciferase analyses. Combining this system with green fluorescent protein reporter systems or microarray gene expression profiling will facilitate the study of gene function.
INTRODUCTION
The use of systems in which gene expression can be induced and repressed at will would greatly facilitate the study of gene function in complex genetic environments such as mammalian cells. Such control is especially required for products that are toxic or growth inhibitory. Although several regulatory systems have been attempted, the advantages of the tetracycline-dependent gene expression system (tet system) are remarkable.
The tet system was originally developed by Gossen and Bujard (1): it permits efficient gene expression regulation in various cell types. By fusing the repressor of the tetracycline resistance operon of Escherichia coli with the transcriptional activation domain of virion protein 16 (VP16) of herpes simplex virus, a tetracycline-controlled transactivator (tTA) was generated. This tTA can bind to an array of tet-operator (tetO) sequences positioned upstream of a minimal promoter and stimulates its transcription in the absence of tetracycline. In the presence of tetracycline, the activity of this promoter is reduced to a very low level.
Subsequently, a reverse transactivator (rtTA) was developed that can bind tetO sequences efficiently, but only in the presence of tetracycline derivatives (e.g. doxycycline) (2). Furthermore, in these systems the expression level of the target gene is controllable by the concentration of tetracycline.
One of the problems encountered with this system is the difficulty in obtaining cell lines or transgenic animals that stably express the tTA or rtTA; this is probably due to the toxicity of the VP16 protein domain, since overexpression of VP16 results in squelching. Actually, VP16 interacts with a variety of essential components of transcriptional machinery. Furthermore, regulatory properties of this system depend on the cell lines used (3). Considering these factors, Baron et al. (4) recently developed transactivators containing the minimal activator domain of VP16 and these transactivators were tolerated at higher intracellular concentrations.
We have focused on other modifications of the transactivator. We decided to select a transactivator that is ubiquitously expressed in mammalian tissues and exhibits a relatively strong transcriptional activity because such a transactivator would probably be non-harmful in mammalian cells. E2F4 met these requirements (5). E2F4, a member of the E2F family, is a transcription factor and controls the expression of genes that are involved in cell cycle regulation.
To generate a new tet transactivator, the transcriptional activation domain of E2F4 was fused to a reverse-type tetracycline repressor. Using this novel tetracycline-controlled transactivator, we were able to induce transcription in several cell lines and monitor the expression and dynamics of mutant p53 (His273) tagged by green fluorescent protein (GFP) in culture cells. We believe this novel transactivator will be useful to understand gene function in a wide range of mammalian cells.
MATERIALS AND METHODS
Construction of plasmids
A BamHI site was introduced at the stop codon of the reverse-type tetracycline repressor gene (2) by PCR (primer: 5′-GACCTCCATAGAAGACAC-3′, 5′-CGCGGATCCACTTTCACATT-3′) and the PCR product was digested with EcoRI and BamHI. The transactivator domain (amino acids 297–413) of E2F4 was amplified by PCR (primer: 5′-TGTCTAGATCTCGGCCACTGCAGTCTTCT-3′, 5′-TGGGATCCTCAGAGGTTGAGAACAGGCACACAGACACCTTCACTCTC-3′) and digested with XbaI and BamHI. In order to prevent this transactivator from binding to the RB family of proteins, four residues (amino acids 404–407) were removed by the PCR primer used (5). These fragments were cloned into pBluescript KS and verified by DNA sequencing. The transactivator domain of E2F4, digested with BglII and BamHI, was inserted into BamHI site at the 3′-end of the reverse-type tetracycline repressor. This novel transactivator replaced the original one in pUHD172-1neo (2), which is driven from the CMV promoter (prTE4d38-neo).
To create a mutant p53–GFP fusion gene, an XbaI restriction site was introduced at the stop codon of mutant p53 (mtp53) by PCR (primer: 5′-AGGAATTCCTTCCGGGTCACTGCCA-3′, 5′-TATCTAGAGTCTGAGTCAGGCCCTT-3′). GFP was fused to the C-terminus of mutant p53 (His273). It was placed under the transcriptional control of the tetO (pTO–mtp53–GFP).
Cell culture and transient transfections
NIH 3T3, COS-7, SaOs-2 and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% tet system-approved fetal bovine serum (Clontech). Transfections by LipofectAMINE 2000 reagent (Gibco) were performed according to the manufacturer’s instructions. pUHC13-3 (100 ng), which contains the luciferase cDNA under the control of a tTA-dependent minimal promoter (1), and 20 ng of pRL-CMV (Renilla luciferase expression vector; Promega) were co-transfected with 700 ng of pUHD172-1 or prTE4d38 into 1–3 × 105 cells grown in 24-well plates. After 12 h, doxycycline (final concentration 1 µg/ml) was administered and cells were cultured for 24 h. The pRL-CMV vector, containing the Renilla luciferase cDNA expressed from a CMV enhancer and promoter, was used to standardize the transfection efficiency between individual samples. Standardized basal levels of luciferase activities and standardized induced levels of luciferase activities were used to estimate induction ratios.
Generation of stably transfected cell lines
NIH 3T3, COS-7, SaOs-2 and HeLa cells were maintained in 60 mm dishes and transfected with 3 µg circular plasmid DNA pUHD172-1neo and prTE4d38-neo using LipofectAMINE 2000 reagent (Gibco). After 48 h, cells were transferred to 10 cm dishes. Selection of stable cell clones was carried out in the presence of 400–500 µg/ml G418. Resistant clones were picked up after 2–3 weeks and expanded separately. These selected clones were plated at 1–3 × 105 cells per well in 24-well plates and transiently transfected with 100 ng of pUHC13-3 plasmid DNA by LipofectAMINE 2000 reagent. Doxycycline (final concentration 1 µg/ml) was administered as described above and luciferase activities were measured.
pUHC13-3 and pTO–mtp53–GFP were transfected with pgk-hyg plasmid into SaOs-2 cell clones expressing novel tetracycline-dependent transactivator. Clones resistant to Hygromycin (200 µg/ml) were isolated and expanded. Cells containing chromosomally integrated pTO–mtp53–GFP were cultured with medium containing doxycycline (1 µg/ml) for 24 h and examined by fluorescent microscopy.
Luciferase assay and northern blot analysis
After transfection, cells were washed twice with PBS and harvested at 36 h. Luciferase assays were performed using Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
Total RNA was isolated using the RNeasy kit (Qiagen). Total RNA (10 µg) was denatured, electrophoresed on a 1% agarose formaldehyde gel and transferred onto Hybond nylon membrane (Amersham). GAPDH and GFP genes were used as probes. The band intensities were quantified with a STORM830 (Molecular Dynamics) and standardized with GAPDH.
RESULTS AND DISCUSSION
Construction and initial testing of the novel transactivator
We used the transcriptional activation domain of E2F4 to generate a novel tetracycline-dependent transactivator. E2F4 is expressed in all tissues (heart, brain, placenta, lung, liver, skeletal muscle, kidney and pancreas) and is present throughout the cell cycle (5). In addition, the transcriptional activity of E2F4 is stronger than E2F1 (6). We therefore thought that the E2F4 transactivator domain would be suitable to replace the VP16 of rtTA or tTA. In order to prevent the RB family of proteins from binding to E2F4, four residues of the pocket protein binding domain of E2F4 were deleted. The transactivator domain (amino acids 297–413) of E2F4 was then fused to a reverse-type tetracycline repressor (Fig. 1).
Figure 1.

The structure of the novel transactivator. The transactivator domain (amino acids 297–413) of E2F4 was fused to reverse type tetracycline repressor. In order to prevent the RB family proteins from binding to E2F4, four residues (amino acids 404–407) of the pocket protein binding domain of E2F4 were deleted. This transactivator is driven by the CMV promoter (prTE4d38).
To estimate the transcriptional activity of this novel transactivator, prTE4d38 or pUHD172-1 was transiently transfected with pUHC13-3 (tetO–luciferase plasmid) into several mammalian cell lines (NIH 3T3, COS-7, SaOs-2 and HeLa) from various origins. These cells were cultured in the presence of doxycycline for 24 h and harvested for the luciferase assay. As shown in Table 1, a 28–52-fold increase in activation was observed upon transient transfection of prTE4d38. When comparing regulatory properties of prTE4d38 with those of pUHD172-1, pUHD172-1 showed a 1.5–4.0-fold higher induction than prTE4d38. However, cells transfected with prTE4d38 displayed lower basal luciferase level than cells transfected with pUHD172-1, probably due to the property of transactivator domains used.
Table 1. Transcriptional activation by transiently transfected tetracycline-dependent transactivators.
| Cell line | Plasmid transfected with | std. RLU | x-fold induction | |
| |
pUHC13-3 |
Doxy(–) |
Doxy(+) |
|
| HeLa | pUHD172-1neo | 2.72 | 335.31 | 123.2 |
| prTE 4d38-neo | 1.22 | 38.25 | 31.4 | |
| Cos-7 | pUHD172-1neo | 29.4 | 1226.7 | 41.7 |
| prTE 4d38-neo | 20.0 | 565.9 | 28.3 | |
| NIH 3T3 | pUHD172-1neo | 8.75 | 687.8 | 78.6 |
| prTE 4d38-neo | 2.52 | 132.1 | 52.4 | |
pUHC13-3 and pRL-CMV (Renilla luciferase expression vector) were co-transfected with pUHD172-1 or prTE4d38 into 1–3 × 105 cells. Cultures were incubated in the presence or absence of doxycycline for 24 h before luciferase activity was measured and standardized to Renilla luciferase activity. Induction ratios were estimated by standardized basal levels of luciferase activities [std. RLU, Doxy(–)] and standardized induced levels of luciferase activities [std. RLU, Doxy(+)]. Doxy(+) or (–), presence or absence of doxycycline, respectively. Results were the average from three experiments.
Analysis of stable cell lines expressing tet-transactivator
To compare the novel and original tet-transactivators under stable expression conditions, prTE4d38-neo and pUHD172-1neo were transferred into several mammalian cell lines (NIH 3T3, COS-7, SaOs-2 and HeLa). G418 resistant clones were obtained and screened for expression of a functional transactivator by transient transfection with pUHC13-3. About 50% of resistant clones from prTE4d38-neo showed inducible luciferase activity on addition of doxycycline to the culture medium, while inducible luciferase activity was observed only in HeLa cell clones but not other cell clones when pUHD172-1neo was transfected (Table 2). The cells transfected with rTA or rtTA frequently showed growth retardation. The E2F4 transactivator appears to be tolerated in a wide range of mammalian cells.
Table 2. Frequency of stable clones expressing the tet-transactivator.
| Cell line |
pUHD172-1neo |
prTE4d38-neo |
| HeLa | 14/18 | 10/16 |
| COS-7 | 0/14 | 14/25 |
| NIH 3T3 | 0/12 | 6/16 |
| SaOS-2 | 0/15 | 14/22 |
The ratios in the table indicate the number of expression-positive clones/total number of clones analyzed.
Kinetics of doxycycline-mediated luciferase activity
pUHC13-3 was transfected with pgk-hyg plasmid into one of SaOs-2 cell lines, ST-7, which constitutively produces novel tet-transactivator. The positive clone ST7L-8 was chosen to analyze the kinetics of luciferase induction by doxycycline (Fig. 2). After addition of doxycycline to the medium, >100-fold induction was achieved by 24 h.
Figure 2.

Kinetics of luciferase activity in ST7L-8 cells by doxycycline. ST7L-8 cells were plated in 24-well plates (at a density of 3 × 104 cells per well). After addition of doxycycline (final concentration 1 µg/ml), luciferase activity was measured at different time points (closed circles). After replacement to tetracycline-free medium, luciferase activity was measured at different time points (open circles).
On the other hand, after incubation of ST7L-8 cells in the presence of doxycycline (1 µg/ml) for 3 days, ST7L-8 cells were rinsed with PBS and cultured in doxycycline-free medium. Within 12 h, luciferase activity dropped to ∼30% and reached <20% of its original value after 24 h.
Regulation of mutant p53 gene expression by tetracycline
To illustrate the utility of this system, a construct was made encoding GFP fused to the C-terminus of mutant p53 (His273), under the transcriptional control of the tetO (pTO–mtp53–GFP). This construct was transfected with pgk-hyg plasmid into SaOs-2 cell clones stably expressing the novel transactivator. Colonies containing both prTE4d38 and pTO–mtp53–GFP were obtained. Doxycycline-dependent mtp53–GFP gene expression was estimated by northern blot and/or fluorescent microscopy. A representative result obtained with one clone (D28 clone) is shown in Figures 3 and 4.
Figure 3.
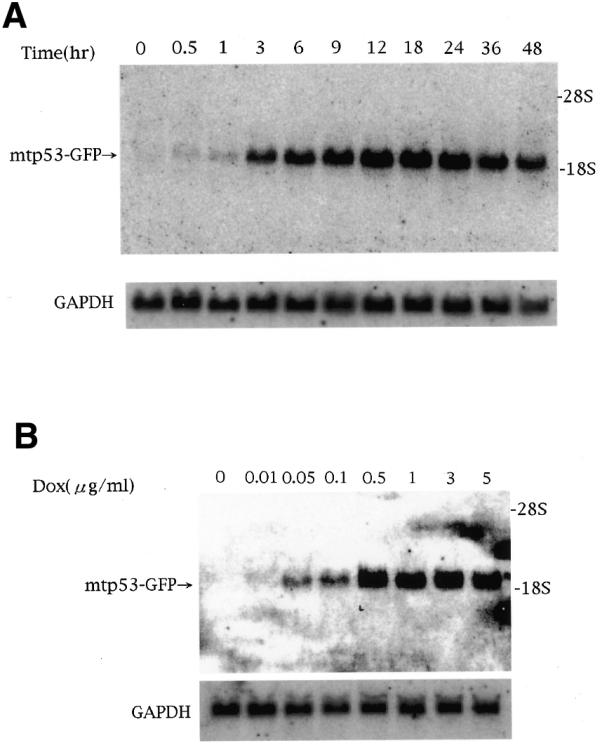
Northern blot analysis of doxycycline-dependent mtp53–GFP expression in D28 cells. Total RNA (10 µg) was loaded into each lane. The blot was hybridized with a DNA probe for the GFP gene and, as a control for loading, with a GAPDH probe. (A) Kinetics of mtp53–GFP induction in D28 cell. After administration of doxycycline (final concentration 1 µg/ml), cells were harvested at different time points (0, 0.5, 1, 3, 6, 9, 12, 18, 24, 36 and 48 h). (B) Dose–response analysis of doxycycline on the D28 clone. Cells were harvested after 24 h in the presence of doxycycline.
Figure 4.
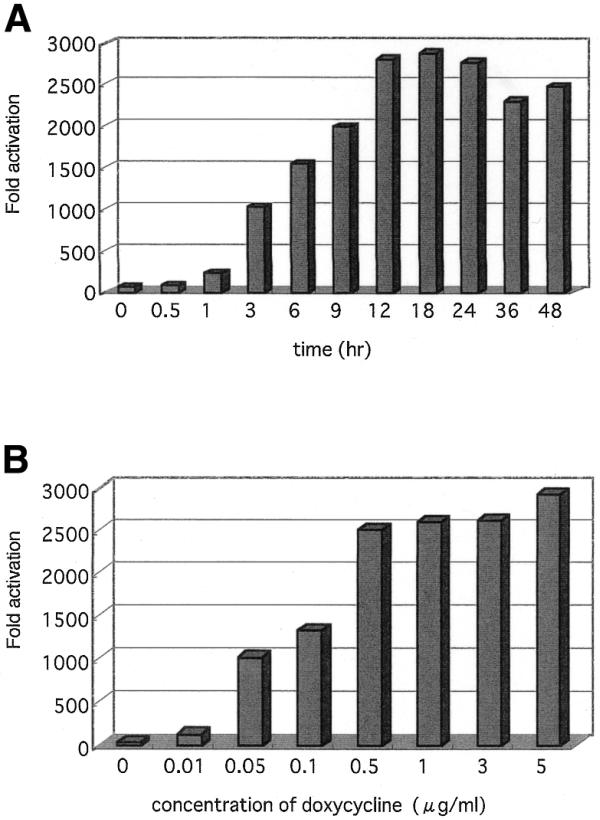
Relative activity of induced gene expression. The expression of mtp53–GFP was standardized with GAPDH. (A) Kinetics of mtp53–GFP induction in D28 cell after administration of doxycycline. (B) Dose–response analysis of doxycycline on the D28 clone.
D28 cells were plated at the same density in 6 cm dishes and cultured in the absence of doxycycline. After administration of doxycycline (final concentration 1 µg/ml), cells were harvested at different time points (0, 0.5, 1, 3, 6, 9, 12, 18, 24, 36 and 48 h). To monitor the time course of doxycycline-dependent gene expression, 10 µg of total RNA was analyzed by northern blotting. mtp53–GFP expression was detected after 0.5 h and had increased induction of gene expression by ∼100-fold after 12 h. After 24 h, the mtp53–GFP expression was slightly reduced, probably due to degradation of doxycycline (Figs 3A and 4A). Doxycycline concentration-dependent gene expression was also monitored with maximal activation achieved at 5 µg/ml (Figs 3B and 4B). The mtp53–GFP was detected in the nuclei of living cells by fluorescent microscopy. The various mtp53–GFP expression levels did not affect cell growth or morphology of SaOs-2 cells.
Taken together, our data show that this transactivator is not deleterious to the mammalian cells used and appears suitable for the generation of conditional transgenic animals as well as the Cre/loxP system (7).
Recently, two new tet regulatable systems have been developed. One is the regulatory system that permits reversible control of two genes in a mutually exclusive fashion (8), the other is a tetracycline-dependent transrepressor, which was generated to reduce the background (9). These new tet systems represent significant improvements over the previous systems. Indeed, the combination of these improved systems and the GFP reporter system and/or microarray technology for gene expression profiling will facilitate the study of gene function.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Prof. H. Bujard for the gift of pUHD15-1, pUHD172-1 and pUHDC13-3 and Dr R.L. Beijersbergen for the gift of the E2F4 gene. prTE4d38 is available on request. This work was supported by HFSP long-term fellowship LT-204/94.
References
- 1.Gossen M. and Bujard,H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl Acad. Sci. USA, 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gossen M., Freundlieb,S., Bender,G., Muller,G., Hillen,W. and Bujard,H. (1995) Transcriptional activation by tetracyclines in mammalian cells. Science, 268, 1766–1769. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann A., Villalba,M., Journot,L. and Spengler,D. (1997) A novel tetracycline-dependent expression vector with low basal expression and potent regulatory properties in various mammalian cell lines. Nucleic Acids Res., 25, 1078–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baron U., Gossen,M. and Bujard,H. (1997) Tetracycline-controlled transcription in eukaryotes: novel transactivators with graded transactivation potential. Nucleic Acids Res., 25, 2723–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginsberg D., Vairo,G., Chittenden,T., Xiao,Z.-X., Xu,G., Wydner,K.L., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) E2F-4, a new member of the of the E2F transcription factor family, interacts with p107. Genes Dev., 8, 2665–2679. [DOI] [PubMed] [Google Scholar]
- 6.Hijmans E.M., Voorhoeve,P.M., Beijersbergen,R.L., Veer,L,J. and Bernards,R. (1995) E2F-5, a new E2F family member that interacts with p130 in vivo. Mol. Cell. Biol., 15, 3082–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akagi K., Sandig,V., Vooijs,M., van der Valk,M., Giovannini,M., Strauss,M. and Berns,A. (1997) Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res., 25, 1766–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baron U., Schnappinger,D., Helbl,V., Gossen,M., Hillen,W. and Bujard,H. (1999) Generation of conditional mutants in higher eukaryotes by switching between the expression of two genes. Proc. Natl Acad. Sci. USA, 96, 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossi F.M.V., Guicherit,O.M., Spicher,A., Kringstein,A.M., Fatyol,K., Blakely,B.T. and Blau,H.M. (1998) Tetracycline-regulatable factors with distinct dimerization domains allow reversible growth inhibition by p16. Nature Genet., 20, 389–393. [DOI] [PubMed] [Google Scholar]