

Abstract
Objectives
Little is known about mechanisms of efficacy of methotrexate (MTX) in childhood arthritis, or genetic influences upon response to MTX. The aims of this study were to use gene expression profiling to identify novel pathways/genes altered by MTX and then investigate these genes for genotype associations with response to MTX treatment.
Methods
Gene expression profiling before and after MTX treatment was performed on 11 children with juvenile idiopathic arthritis (JIA) treated with MTX, in whom response at 6 months of treatment was defined. Genes showing the most differential gene expression after treatment were selected for SNP genotyping. Genotype frequencies were compared between non-responders and responders (ACR-Ped70). An independent cohort was available for validation.
Results
Gene expression profiling before and after MTX treatment revealed 1222 differentially expressed probes sets (fold change >1.7, p< 0.05) and 1065 when restricted to full responder cases only. Six highly differentially expressed genes were analysed for genetic association to response to MTX. Three SNPs in the SLC16A7 gene showed significant association with MTX response. One SNP showed validated association in an independent cohort.
Conclusions
This study is the first, to our knowledge, to evaluate gene expression profiles in children with JIA before and after MTX, and to analyse genetic variation in differentially expressed genes. We have identified a gene which may contribute to genetic variability in MTX response in JIA, and established as proof of principle that genes which are differentially expressed at mRNA level after drug administration may also be good candidates for genetic analysis.
Keywords: Methotrexate (MTX), pharmacogenomics, treatment, juvenile idiopathic arthritis (JIA), autoimmunity, transcriptional profiling, microarray
Introduction
Methotrexate (MTX) is a major first line disease-modifying drug in the treatment of inflammatory arthritis[1, 2]. One in 1000 children has inflammatory arthritis known as juvenile idiopathic arthritis (JIA), which frequently requires disease-modifying drug treatment. 65-70 % of children with JIA will respond to MTX, either partially or fully, and the disease can enter prolonged remission on this medication[3-5]. The majority of children who respond to MTX experience an improved quality of life[6]. However for the ∼35% of children who respond poorly, or not at all, the time taken before switching to alternative treatments, such as biologic agents, means loss of quality of life due to active arthritis and drug side effects, with consequent accrual of disability which may then be difficult to reverse[7, 8]. Variation in individual response to MTX is likely influenced by genetic, environmental and psychological factors. At present it is not possible to predict with accuracy which patients will respond to MTX: predictors of response to MTX would be highly valuable, both in JIA and also in adult arthritis.
Methotrexate is a folate analogue, which blocks dihydrofolate reductase, inhibiting purine metabolism and amino acid conversion; it enters cells via the reduced folate carrier, RFC1 (or solute carrier SLC19A1) transporter[9, 10]. Inside the cell MTX is polyglutamated and inhibits several enzymes such as ATIC (5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase), TYMS (thymidylate synthase) and MTHFR (methylenetetrahydrofolate reductase). However the specific mode of action of MTX in both adult and childhood arthritis is unclear, in particular since administration of oral folate or folinic acid do not inhibit efficacy[11-13]. Considerable evidence suggests that MTX may act at least in part by raising extracellular levels of adenosine[11]. Studies in adult arthritis have indicated genetic variants which influence response to MTX within genes that affect MTX transport and metabolism, enzymes influenced by MTX, and adenosine pathways[14, 15]. Recent studies have tried to combine such genetic data with clinical factors to create predictive algorithms which may be applicable to clinical care[16]. However we cannot readily extrapolate such adult data to paediatric use, due to differences in disease processes, susceptibility loci, pharmacokinetics or side effects[17]. Small studies in JIA have suggested SNP variants which may influence response to MTX,[18] and a recent study has attempted to model genetic and clinical factors in combination[19]. However given that the mechanisms of action of MTX in JIA are unknown, we reasoned that this candidate gene approach of known pathways may miss key molecules in pathways that are influenced by MTX.
The SPARKS-CHARM (CHildhood Arthritis Response to Medication) study (hereafter referred to as CHARMS) is focused upon three aspects (genetic, environmental and psychological) influencing response to MTX in JIA. Its ultimate aim is to define a multi- factorial model of response outcomes. We have adopted a gene expression profiling approach to identify genes whose mRNA expression levels in peripheral blood mononuclear cells (PBMC) are altered after MTX administration in children with JIA. We then tested the hypothesis that genetic polymorphic variants in these differentially expressed genes may be associated with differing response outcome in JIA patients by testing SNPs in both the initial cohort and a validation cohort.
Methods
Patients and samples
This work was performed as part of the SPARKS-CHARM study (CHARMS), which recruits children who fulfill ILAR criteria for JIA [20] of all subtypes, and who are about to start new disease modifying medication for active arthritis. This report is specifically of those children treated with MTX for JIA. The study has full ethical committee approval (Institute of Child Health/Great Ormond Street NHS Trust Ethics Committee) and was fully compliant with the Declaration of Helsinki. Subjects were recruited with full informed parental consent, and child assent where appropriate. Demographic and clinical data were collected at baseline, (up to 4 weeks before commencing MTX), and after 6 months of MTX. MTX was given by either oral or subcutaneous route at 10-15 mg/m2 per week (mean dose 12.40mg/ m2 per week) Data allowing assessment of clinical response to the drug were collected using the validated core set variables and the Definition of Improvement (DOI) for JIA[21]. The variables in the core set are: physicians global assessment of disease activity (scored on a visual analogue scale of 0–10 with 0=inactive and 10=most severe), parent/patient global assessment of overall well being (scored on a visual analogue scale of 0–10 with 0=very well and 10=very bad), functional ability (measured by the childhood health assessment questionnaire, CHAQ)[22], number of joints with active arthritis, number of joints with restricted range of movement, and the ESR (mm/hr). In the DOI for JIA these 6 items of data are each considered at the start of the therapeutic intervention and after a defined period of time, typically 6 months. The DOI to achieve ACR-Ped30 for JIA is when there is at least 30% improvement from baseline in 3 of any the 6 variables, with no more than 1 of the remaining variables worsening by >30%. ACR-Ped50 and -70 for JIA require improvements of 50 or 70 % in at least 3 of the variables, respectively, again with no more than 1 of the remaining variables worsening by >30%[21]. For this study we defined non-responders (NR) as failure to reach even the 30% response improvement. Venous blood samples were taken when the child required blood sampling for routine clinical care, those for gene expression profiling at 0 and 6 months of treatment, and those for DNA preparation at any time during the study. This study utilizes the first 197 Caucasian children in CHARMS on whom full core set variable clinical data and DNA samples were available.
For the genetic studies, DNA from a replication cohort of 210 Caucasian children with polyarticular or oligoarticular JIA from Cincinnati Children's Hospital Medical Centre, who had also been treated with MTX and analysed pre treatment and at 6 months, were available. The study had full ethical committee approval (CCHMC IRB) and was fully compliant with the Declaration of Helsinki. In this patient cohort improvement of arthritis was defined using active joint count alone. Levels of improvement at 30, 50 and 70% were defined, and non-responders (NR) were those who did not reach a 30% improvement in active joint count.
Gene expression profiling
Peripheral blood mononuclear cells (PBMC) from the first 11 children who had paired samples available from pre treatment, and at 6 months of MTX treatment, were analysed. PBMC were prepared from venous blood by standard density centrifugation. 3 × 106 cells were lysed into Trizol reagent (Invitrogen, UK). In accordance with our previous work, strict protocols were followed for time from sample collection to storage of sample in Trizol at -80C, which we have previously shown is ideally performed within 4 hours [23, 24]; all samples in this study were processed and stored at -80C within 3 hours. Total RNA was prepared by standard methods. High quality and purity of RNA were confirmed using the Bioanalyser 2100 (Agilent Technologies, Palo Alto, CA). cDNA and subsequent cRNA synthesis was performed as described. Purified cRNA transcripts were fragmented and hybridized to human U133 plus 2.0 GeneChips according to Affymetrix standard protocols (http://www.affymetrix.com). Complete gene expression data are available on the GEO database (www.ncbi.nlm.nih.gov/geo).
Gene expression data analysis
Signal values were calculated using MAS 5.0, scaled to 100 and normalized to the median prior to analysis with GeneSpring GX 7.3 software (Agilent Technologies). Probesets were excluded if the signal strength did not significantly exceed background values and if expression did not reach a threshold value for reliable detection (defined as in 4 of 22 samples). A list of probesets where the difference between mean expression levels after MTX therapy, compared to prior to treatment, was greater than 1.7 fold was statistically analyzed by the Student's t test with Welch's correction and a p value cut-off of 0.05 was applied. Functional categories for corresponding genes were assigned using gene ontology terms from the Gene Ontology Consortium (www.geneontology.com) and Ingenuity Pathway Analysis (IPA, Ingenuity Systems) and confirmed via Entrez. Differentially expressed probesets were corrected for multiple comparisons using the Benjamini Hochberg correction at a false discovery rate (FDR) of 5%. Genes were ranked on p-value in Genespring and are defined here by excluding probesets without a gene symbol, or those annotated as hypothetical proteins. To list unique genes where multiple probesets were differentially expressed, fold-change and p-value are given for the probe with the smallest p-value. Gene lists were further analysed to identify pathways or networks, which showed differential expression after treatment with MTX using IPA.
Real-time quantitative RT-PCR
Total RNA was prepared from PBMC as above and first strand cDNAs were prepared using the SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen Corp., Carlsbad, CA) with priming using random hexamers. Gene expression was measured by PCR amplification on a Bio-Rad iCycler Optical System using iQ™ SYBR green real-time PCR kit (Bio-Rad Laboratories, Inc.) according to manufacturer's instructions.
Primers were designed by using PrimerSelect version 2.0 and sequences are as follows: B2M: forward 5′-AGCGTACTCCAAAGATTCAGGTT, reverse 5′-CACATGGTTCACACGGCAGG; SLC16A7: forward 5′-CAGCCGGCCGGTGGTGATAG, reverse 5′-GTTAAGGCGGGTTGCAGGTTGAAG; BAT1: forward 5′-TCGGGAGTTGGCTTTTCAG, reverse 5′-GATATGCGG GCAGTTCTTCTTCAG; MALAT1: forward 5′-TCACCAGCGGAAAACTCAAG, reverse 5′- GCTGTAGGCCCCAAAGATGTC; ZEB1(TCF8): forward 5′-TTCACAGTGGAGAGAAGC, reverse 5′-AGAA CACTGAGATGTCTTG; THAP6: forward 5′-TGACATTTCACGTATTCCC, reverse 5′-TGGAGCACTTCTGTCAA; and NFATC2IP: forward 5′-GAAGTCGGAGCCCCTGCAGAGTG, reverse 5′- TGGGAGTGGCAGTAGGTGATAGC. Data were normalized to reference gene B2M.
Genetic studies
SNP selection
Genes for SNP genotyping were selected from genes which were significantly differentially expressed at mRNA level in ACR-Ped70 patients (n=7) pre versus post MTX, with Benjamini-Hochberg correction applied at FDR of 5%. The final selection was based on fold change and biological relevance using GO functional categories and published literature. Pair-wise tagging SNPs for each gene were selected using HapMap release 22 (www.hapmap.org) and the tagger function in Haploview version 4.1 [25] (http://www.broadinstitute.org/haploview/haploview), using an r2 cutoff ≥ 0.8 and MAF ≥ 0.05 within 10kb up and down stream of each gene.
Genotyping
Genomic DNA was prepared from venous blood by standard methods. SNP genotyping was performed using the Sequenom iPlex® MassARRAY platform according to manufacturer's instructions (Sequenom, San Diego, CA. http://www.sequenom.com/). Only samples and SNPs exceeding a 90% success rate were used in the analysis. For the US cohort, the genotypes provided were part of a larger genome-wide association study. The genotyping was done at Affymetrix Service Center (South San Francisco, CA) and used the Affymetrix Genome-Wide Human SNP Array 6.0. The Birdseed calling algorithm was used and the overall call rate was 98.97%.
Statistical analysis
Genotype frequencies were compared between non-responders and responders (ACR-Ped70) using the trend test and allelic odds ratios with 95% confidence intervals were calculated using PLINK [26] (http://pngu.mgh.harvard.edu/∼purcell/plink/index.shtml). Haplotype analysis and linkage disequilibrium measures were performed using Haploview version 4.1 [25] (http://www.broadinstitute.org/haploview/haploview). Linear regression analysis of SNP genotype data and expression data was performed using STATA SE9.2.
Results
Subjects
We present results from the first 197 Caucasian children in CHARMS on whom full core set variable clinical data and DNA samples were available, with replication of genetic findings in the Cincinnati cohort (210 Caucasian children). The demographic data on the two cohorts are summarised in Table 1. Subtype distribution and disease activity was as expected for use of MTX in JIA and in line with previous studies[4]. As expected, there was a higher prevalence in females, in both cohorts. Disease duration has been recently reported as linked with MTX response[27]: the median time of disease duration prior to starting MTX in these two cohorts was 1 year (Table 1).
Table 1. Demographics of UK and US JIA patient cohorts.
| UK cohort | US cohort | |
|---|---|---|
| Number of cases | 197 | 210 |
| JIA Subtype at time of starting MTX (in %) : | ||
| Oligoarticular Persistent | 9.1 | 8.6 |
| Oligoarticular Extended | 26.4 | 20.9 |
| Polyarticular RF- | 37.6 | 67.6 |
| Polyarticular RF+ | 5.6 | 1.9 |
| Enthesitis related arthritis | 6.0 | 0 |
| Psoriatic | 2.5 | 0.95 |
| Systemic | 10.7 | 0 |
| Unclassified | 2.0 | 0 |
| Female (%) | 132 (67%) | 164 (78%) |
| Median and range of disease duration at starting MTX (decimal years) | 1.00, 11.53 | 1.08, 15.8 |
| Median and range of age at starting MTX (decimal years) | 7.93, 14.97 | 7.25, 18.91 |
| Median and range of active joints at start of MTX | 6, 34 | 8, 61 |
| Median and range of restricted joints at start of MTX | 4, 41 | 4, 44 |
In the UK cohort, where the six ‘core set’ data items, defined at baseline (before MTX), and 6 months of treatment, were used to define response to MTX, 141 (71.5%), 119 (60.4%) and 84 (42.6%) cases reached ACR-Ped30, 50 and 70 respectively at 6 months of treatment. The 56 (28.4%) cases that failed to reach ACR-Ped30 were defined as non responders (NR). Note that all children who reach ACR-Ped70 automatically also reach ACR-30 and 50, while those who achieve ACR-Ped50 also achieve ACR-Ped30. In the US cohort, where response was defined using joint count alone, 131 (63%), 107 (51.7%) and 67 (32.4%) children reached the 30, 50, and 70 % levels of response respectively, while 76 (36.7%) were NR at 6 months of treatment and 3 children had missing data.
There were no significant differences between responders (ACR70) and non-responders in the mean dose or route of MTX, nor in the co-administration of steroids at the start of MTX therapy (intra-articular, oral or pulsed iv), or the rates of gastrointestinal toxicity as assessed by child/parent report of vomiting (Supplementary data, Table S1 http://links.lww.com/FPC/A204).
Gene expression profiling and analysis
To identify differentially expressed genes after administration of MTX, we performed gene expression profiling using RNA from total PBMC obtained prior to, and at 6 months of MTX therapy, on the first 11 cases from whom paired samples were prepared. Of these, 7 cases reached ACR-Ped70 response and were analysed further as ‘best responders’ to MTX. Flow cytometric analysis of PBMC from the same patients at the same time points showed that T, B cell and monocyte populations were all within normal limits, and were not significantly altered by MTX treatment (data not shown). Genes whose expression was significantly higher or lower after MTX compared to prior to MTX, with a cut off of fold change of 1.7 and p value of ≤0.05 were considered for further analysis. The raw data of these arrays are all available at the GEO website (www.ncbi.nlm.nih.gov/geo). Analysis of gene expression from the 11 subjects (22 sets of data) using a serial sample approach, yielded a total of 1222 probe sets, that were either up-regulated (n=823) or down- regulated (n=399) after MTX compared to before MTX. Pathway analysis performed using IPA on the 1222 probe sets revealed 204 implicated pathways altered by MTX, of which 28 pathways reached overall significance of p<0.05, (Table 2).
Table 2.
Canonical Pathways identified by IPA from the probe sets (1222) that were differentially expressed after MTX compared to before MTX, (n=11 cases, 22 datasets). P values are for the pathway as a whole.
| Pathway | p-value | Molecules |
|---|---|---|
| PI3K/AKT Signaling | 0.003 | RELA, JAK1, FOXO1, SOS1, LIMS1, MAP3K8, GSK3A, ITGA3, RHEB, EIF4E, PTEN |
| Cdc42 Signaling | 0.005 | ACTR2, DIAPH1, CDC42BPA, TNK2, EXOC7, MYL10, ITGA3, IQGAP1, CLIP1, LIMK1 |
| IL-9 Signaling | 0.005 | SOCS3, RELA, JAK1, IRS2, STAT5B |
| Insulin Receptor Signaling | 0.006 | SOCS3, PPP1R3D, JAK1, CBL, PPP1R10, FOXO1, SOS1, IRS2, GSK3A, EIF4E, PTEN |
| Glycosphingolipid Biosynthesis – Lactoseries | 0.009 | FUT2, FUT6, ST3GAL3 |
| Protein Kinase A Signaling | 0.009 | RELA, CREM, PDE7A, ITPR2, GSK3A, PLCL2, TTN, ROCK1, GNB4, GNAS, PPP1R3D, NFAT5, PPP1R10, TGFB2, ADD1, LEF1, MYL10, GNG12, PRKCB |
| iCOS-iCOSL Signaling in T Helper Cells | 0.010 | PTPRC, ICOS, RELA, NFAT5, CD40, ITPR2, PLEKHA2, ICOSLG, PTEN |
| Thrombin Signaling | 0.010 | RELA, F2RL2, ITPR2, PLCL2, ROCK1, GNAS, GNB4, RHOB, GNAT2, ARHGEF16, SOS1, MYL10, GNG12, PRKCB |
| Ephrin Receptor Signaling | 0.011 | ACTR2, ITGA3, PDGFB, FGF1, LIMK1, ROCK1, GNAS, GNB4, GNAT2, EFNA5, ACP1, SOS1, GNG12 |
| IL-4 Signaling | 0.011 | B2M, SOCS1, IL4R, NFAT5, JAK1, IRF4, SOS1 |
| ATM Signaling | 0.011 | SMC3, GADD45A, TLK1, CCNB2, CDC2, SMC1A |
| Apoptosis Signaling | 0.012 | ROCK1, CAPN5, RELA, CASP3, CASP2, CYCS, CAPN2, CDC2 |
| Glycosphingolipid Biosynthesis | 0.015 | GCNT4, ST8SIA1, FUT2, FUT6 |
| fMLP Signaling in Neutrophils | 0.017 | NOX1, GNAS, ACTR2, RELA, GNB4, NFAT5, ITPR2, GNG12, PRKCB |
| PTEN Signaling | 0.017 | RELA, CBL, CASP3, FOXO1, SOS1, GSK3A, ITGA3, PTEN |
| Primary Immunodeficiency Signaling | 0.019 | PTPRC, ICOS, CD40, IGL, IGHG1 |
| Integrin Signaling | 0.022 | CAPN5, ACTR2, LIMS1, TNK2, ITGA3, TTN, PTEN, ROCK1, ARF1, RHOB, SOS1, CAV1, CAPN2 |
| Molecular Mechanisms of Cancer | 0.024 | RELA, TCF4, JAK1, CASP3, GSK3A, AURKA, TCF3, GNAS, CBL, RHOB, FOXO1, ARHGEF16, GNAT2, SOS1, TGFB2, CYCS, LEF1, HIPK2, MAP3K7IP1, PRKCB |
| Erythropoietin Signaling | 0.035 | SOCS3, RELA, CBL, SOS1, STAT5B, PRKCB |
| Angiopoietin Signaling | 0.035 | RELA, FOXO1, SOS1, STAT5B, BIRC5, TIE1 |
| Cardiac Hypertrophy Signaling | 0.035 | CACNA1D, PLCL2, EIF4E, ROCK1, GNAS, GNB4, RHOB, GNAT2, SOS1, TGFB2, MAP3K8, MYL10, MAP3K7IP1, GNG12 |
| DNA Methylation & Transcriptional Repression | 0.037 | CHD4, ARID4B, RBBP4 |
| Oncostatin M Signaling | 0.037 | JAK1, SOS1, PLAU, STAT5B |
| CD27 Signaling in Lymphocytes | 0.042 | RELA, CASP3, CYCS (includes EG:54205), MAP3K8, MAP2K5 |
| PDGF Signaling | 0.042 | JAK1, ACP1, SOS1, CAV1, PDGFB, PRKCB |
| Actin Cytoskeleton Signaling | 0.043 | ACTR2, ITGA3, IQGAP1, PDGFB, TTN, FGF1, LIMK1, ROCK1, DIAPH1, SOS1, WASF2, MYL10, GNG12 |
| RhoA Signaling | 0.044 | ROCK1, ACTR2, RHPN1, SEPT7, MYL10, SEPT2, TTN, LIMK1 |
| One Carbon Pool by Folate | 0.048 | TYMS, MTHFD1L, GART |
Interestingly several of the differentially expressed genes identified have been implicated as being involved in pathways influenced by MTX and the response to MTX, in adult arthritis. Specifically, thymidylate synthase (TYMS) and Serine hydroxymethyltransferase 1 (SHMT1) [28, 29] mRNA levels were down regulated in children with JIA after MTX administration. In addition, IPA analysis implicated the one carbon folate pathway along with many pathways which play a role in the immune system. These included signaling pathways that are involved in lymphocyte and immune function such as PI3 kinase/AKT, PTEN, iCOS and protein kinase pathways, interleukin signaling pathways including IL-4 and IL-9, CD27 as well as integrin signalling. The increased expression of PTEN is interesting since PTEN is a major inhibitor of T cell activation, in particular by inhibition of the AKT pathway, and factors affecting the PI3K/AKT pathway are critical to control of regulatory T cells [30, 31]. In addition several genes known to affect DNA methylation and epigenetic control were indicated including ARID4B, RBBP4 [32].
Narrowing of targets for further genetic analysis from over 1000 genes is challenging. To stratify genes that were highly differentially expressed in patients who had responded clinically to MTX, we next performed a more stringent analysis on the ‘best responder’ (ACR-Ped70) group, (n=7). Analysis of gene expression in this group, after application of the stringent Benjamini-Hochberg correction for multiple testing, to control for false discovery rate (pre correction probeset included 1065 probes), yielded a total of 87 gene probes that were significantly altered post MTX, Figure 1 and Supplementary information, Table S2 http://links.lww.com/FPC/A205). These 87 probe sets represented 62 genes of which the top 28 ranked genes are shown in Table 3.
Figure 1. Gene expression changes in JIA patients with ‘best response’ after MTX, each compared to before starting MTX.

Heat map contains probe sets (n=87) that are 1.7-fold differentially expressed at a significance of p < 0.05 by the Student's t-test with Welch's correction and after Benjamini-Hochberg correction. Each row represents a probeset and each column represents a patient, either before or after MTX as shown. The normalized expression level for each probe set is indicated by colour with red and blue reflecting higher or lower expression levels as shown.
Table 3. Top 28 Differentially expressed genes in ACR-Ped70 patients after MTX, Benjamini-Hochberg correction applied.
| P-value | Fold Change | Gene Symbol | Entrez Gene Name | Location | Annotation | |
|---|---|---|---|---|---|---|
| 1 | 0.004 | 12.8 | MALAT1 | metastasis associated lung adenocarcinoma transcript 1 (non-coding RNA) | Unknown | Other |
| 2 | 0.004 | 6.7 | CHD9 | chromodomain helicase DNA/binding protein 9 | Cytoplasm | DNA repair |
| 3 | 0.006 | 6 | OSBPL8 | oxysterol binding protein-like 8 | Cytoplasm | Transporter |
| 4 | 0.006 | 3.3 | FBXO42 | F-box protein 42 | Unknown | Ubiquitin cycle |
| 5 | 0.006 | 3.3 | KLF3 | Kruppel-like factor 3 (basic) | Unknown | Transcription Regulator |
| 6 | 0.008 | 3.8 | THAP6 | THAP domain containing 6 | Unknown | Other |
| 7 | 0.008 | 3.5 | ZNF539 | Zinc finger protein 254 | Nucleus | Transcription Regulator |
| 8 | 0.008 | 3.8 | CAP350 | centrosome-associated protein 350 | Cytoplasm | Microtubule Organisation |
| 9 | 0.008 | 5.6 | SYNJ2BP | Synaptojanin 2 binding protein | Cytoplasm | Other |
| 10 | 0.0106 | 3.2 | SPAG9 | sperm associated antigen 9 | Plasma Membrane | Other |
| 11 | 0.0131 | 4.3 | SLC16A7 | solute carrier family 16 (monocarboxylic acid transporters), member 7 | Plasma Membrane | Transporter |
| 12 | 0.0139 | 5.3 | TCF8 (ZEB1) | transcription factor 8 (zinc finger enhancer protein 1) | Nucleus | Transcription Regulator |
| 13 | 0.0216 | 11.5 | C20orf94 | chromosome 20 open reading frame 94 | Unknown | Other |
| 14 | 0.0216 | 3.3 | PTPRC | protein tyrosine phosphatase, receptor type, C | Plasma Membrane | Signal Transduction |
| 15 | 0.0218 | 2.6 | NFATC2IP | nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 2 interacting protein | Nucleus | Protein Modification |
| 16 | 0.0218 | 4.7 | ATRX | alpha thalassemia/mental retardation syndrome X-linked | Nucleus | Transcription Regulator |
| 17 | 0.0218 | 4 | ABLIM1 | actin binding LIM protein 1 | Cytoplasm | Cytoskeleton Organization |
| 18 | 0.0218 | 4.6 | EXT1 | Exostoses (multiple) 1 | Unknown | Signal Transduction |
| 19 | 0.0218 | 3.1 | HECTD1 | HECT domain containing 1 | Unknown | Enzyme |
| 20 | 0.0218 | 5.8 | IQGAP1 | IQ motif containing GTPase activating protein 1 | Cytoplasm | Signal Transduction |
| 21 | 0.0218 | 10.8 | OTUD4 | OTU domain containing 4 | Unknown | Other |
| 22 | 0.0222 | -1.8 | EIF4G3 | Eukaryotic translation initiation factor 4 gamma, 3 | Unknown | Protein Biosynthesis |
| 23 | 0.0222 | -1.8 | LOC134492 | NudC domain containing 2 | Unknown | Other |
| 24 | 0.0222 | 2 | SYNCOILIN | Syncoilin, intermediate filament 1 | Unknown | Other |
| 25 | 0.0222 | 2 | ZNF567 | zinc finger protein 567 | Nucleus | Transcription Regulator |
| 26 | 0.0222 | 3.7 | ZNF587 | Zinc finger protein 587 | Unknown | Transcription Regulator |
| 27 | 0.0224 | 4.7 | HNRPD | Heterogeneous nuclear ribonucleoprotein D | Unknown | Other |
| 28 | 0.0237 | 3.8 | BAT1 | HLA-B associated transcript 1 | Nucleus | Enzyme |
Genes in bold are SNP genotyped
Gene selection for genotyping
From these 62 genes that were highly differentially expressed after MTX treatment in best responders, six genes were selected for SNP genotyping based on both expression level fold-change and potential biological relevance. Gene expression pre and post MTX for these 6 genes were confirmed by RT-PCR (Figure 2). These 6 genes were selected for genetic study. Metastasis associated lung adenocarcinoma transcript 1 (MALAT1), which had the highest fold change after MTX treatment. This gene has been previously shown to be misregulated in tumour cells[33, 34] and its expression levels are upregulated in cell lines treated by several chemotherapeutic drugs including MTX [35]. Solute carrier family 16, member 7 (SLC16A7), also known as monocarboxylate transporter 2 (MCT2), is a widely expressed transporter that imports and exports lactate and pyruvate. Interestingly, a recent meta analysis of GWAS studies has implicated SLC16A7 as associated with autoimmune inflammatory bowel disease[36], and a related SLC family member SLC19A1 has been associated with response to MTX in rheumatoid arthritis[37, 38]. The zinc finger –enhancer protein 1, ZEB1 (also known as transcription factor 8, TCF8) is a zinc finger protein which interacts with SMAD proteins and enhances signaling of the immune suppressive factor TGFβ[39] and represses IL-2 expression[40]. Polymorphisms in the promoter of HLA-B associated transcript-1 (BAT1) have been associated with susceptibility to rheumatoid arthritis[41] and BAT1 is thought to be involved in regulation of TNF and other inflammatory cytokines[42, 43]. Nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 2 interacting protein (NFATC2IP) alters expression of cytokine genes in T-cells[44, 45]. THAP domain containing 6 (THAP6) annotations indicate that it plays a role in nucleic acid binding.
Figure 2. mRNA expression by QPCR of the 6 genes selected for genotyping in ‘best’ responder JIA patients, comparing before and after MTX treatment.

Bars show mean expression levels relative to beta2 microglobulin (B2M) gene expression, pre- (white bars) and post- (black bars) MTX; error bars indicate 1 standard error of the mean. Significant differences (p < 0.05) between pre- and post- MTX are indicated by *.
SNP genotyping
45 SNPs were genotyped across the 6 genes, with a per gene coverage of 95% after QC. From 197 children with available core set variable data in the CHARMS study, 175 DNA samples achieved >90% success in SNP genotyping. Of these 175 cases, patients were included in the statistical analysis if they were either ACR-Ped70 (n=72) or non-responders (n=51) thus comparing ‘extremes’ of the response phenotype. Three SNPs were significant with trend p-value <0.05, all of which lie within the SLC16A7 gene (Table 4). Results for all SNPs, and the allele frequencies for the three significant SNPs represented graphically, can be found in supplementary data, in Table S3 http://links.lww.com/FPC/A206) and Figure S1 http://links.lww.com/FPC/A207) respectively. For two SNPs in the SLC16A7 gene, rs2711655 and rs3763980, the minor alleles G and A respectively, are associated with increased risk of not responding to MTX and for the other SNP, rs10877333, the major allele T is associated with increased risk of not responding to MTX.
Table 4.
Association analysis between non-responders and responders to MTX, across the SLC16A7 gene, in UK and US dataset.
| SNP | Gene | CHR | Position | Minor allele | Major allele | UK MAF NR (n=51) | UK MAF ACR-Ped70 (n=72) | OR (95% CI) | P trend | US MAF NR (n=76) | US MAF Joint count 70 (n=67) | OR (95% CI) | P trend |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs17122830 | SLC16A7 | 12 | 58359507 | G | A | 0.07 | 0.06 | 1.11 (0.40-3.07) | 0.84 | 0.06 | 0.05 | 1.16 (0.42-3.2) | 0.79 |
| rs1497474 | SLC16A7 | 12 | 58360056 | T | C | 0.27 | 0.27 | 0.99 (0.56-1.77) | 0.98 | 0.22 | 0.28 | 0.76 (0.44-1.29) | 0.3 |
| rs10877327 | SLC16A7 | 12 | 58360452 | T | G | 0.15 | 0.13 | 1.21 (0.58-2.52) | 0.64 | NT | NT | ||
| rs7976956 | SLC16A7 | 12 | 58365097 | A | G | 0.25 | 0.23 | 1.12 (0.61-2.03) | 0.73 | NT | NT | ||
| rs1000708 | SLC16A7 | 12 | 58369755 | G | A | 0.41 | 0.48 | 0.75 (0.44-1.29) | 0.25 | NT | NT | ||
| rs7971953 | SLC16A7 | 12 | 58376474 | C | G | 0.20 | 0.26 | 0.70 (0.37-1.35) | 0.28 | 0.25 | 0.28 | 0.87 (0.52-1.48) | 0.62 |
| rs12231740 | SLC16A7 | 12 | 58390307 | T | C | 0.38 | 0.28 | 1.61 (0.94-2.76) | 0.07 | 0.28 | 0.20 | 1.56 (0.9-2.71) | 0.12 |
| rs2711669 | SLC16A7 | 12 | 58415239 | T | C | 0.09 | 0.07 | 1.45 (0.55-3.80) | 0.43 | 0.07 | 0.04 | 1.5 (0.53-4.25) | 0.46 |
| rs2706301 | SLC16A7 | 12 | 58421669 | C | G | 0.38 | 0.44 | 0.79 (0.46-1.36) | 0.36 | NT | NT | ||
| rs10877333 | SLC16A7 | 12 | 58442313 | G | T | 0.12 | 0.22 | 0.48 (0.23-0.98) | 0.03 | 0.21 | 0.25 | 0.83 (0.48-1.45) | 0.51 |
| rs2711655 | SLC16A7 | 12 | 58452688 | G | A | 0.50 | 0.35 | 1.84 (1.10-3.09) | 0.02 | NT | NT | ||
| rs12718000 | SLC16A7 | 12 | 58455532 | A | G | 0.09 | 0.09 | 0.91 (0.36-2.33) | 0.84 | NT | NT | ||
| rs3763980 | SLC16A7 | 12 | 58459623 | A | T | 0.36 | 0.23 | 1.96 (1.11-3.44) | 0.02 | 0.28 | 0.17 | 1.83 (1.01-3.31) | 0.04 |
| rs10784000 | SLC16A7 | 12 | 58467231 | G | A | 0.28 | 0.28 | 1.00 (0.57-1.75) | 0.99 | NT | NT |
NOTES : CHR=chromosome, MAF= minor allele, NR=non-responder; NT- not tested; significantly associated SNPs (p<0.05) in bold
Analysis of all the SNPs within the SLC16A7 gene in Haploview found that the 3 associated SNPs lie in a block of LD, spanning 45kb (Figure 3). Haplotype analysis showed that the most common haplotype (Haplotype 1), carrying the ‘risk’ alleles of the 3 associated SNPs (T, G and A), is associated with increased risk of non-response to MTX (Table 5). The results suggest that it is the A allele of rs3763980 that is driving the association of haplotype 1. The SNP rs3763980 is a non-synonymous coding SNP causing an amino acid change from Threonine to Serine at position 445. In contrast, haplotype 3, carrying the ‘protective’ alleles of the 3 SNPs (G, A and T) is associated with a good response to MTX. The minor G allele of rs10877333 is driving the association of haplotype 3.
Figure 3. Linkage disequilibrium plot for SNPs across the SLC16A7 gene using Haploview version 4.1, showing LD block around the associated SNPs.

Significant SNPs highlighted in black boxes. Pairwise LD calculated using D', with the coloured squares showing the strength of LD, with red denoting high LD, blue moderate LD and white low LD. The numbers in the block denotes LD calculated using r2.
Table 5.
Haplotype analysis across the LD block within SLC16A7.
| Haplotype | rs2706301 | rs10877333* | rs2711655* | rs12718000 | rs3763980* | rs10784000 | Haplotype Frequency. | Case,Control Frequencies | Chi Square | P Value |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | G | T | G | G | A | A | 0.28 | 0.36, 0.22 | 5.8 | 0.01 |
| 2 | C | T | A | G | T | G | 0.28 | 0.27,0.28 | 0.03 | 0.86 |
| 3 | G | G | A | G | T | A | 0.18 | 0.12,0.22 | 4.5 | 0.03 |
| 4 | C | T | A | G | T | A | 0.12 | 0.11,0.14 | 0.49 | 0.48 |
| 5 | G | T | G | A | T | A | 0.09 | 0.1,0.09 | 0.09 | 0.77 |
| 6 | G | T | G | G | T | A | 0.04 | 0.03,0.04 | 0.39 | 0.53 |
significantly associated SNPs (p<0.05); Haplotype 1 = ‘risk’ haplotype (associated with increased risk of non-response to MTX), and Haplotype 3 = ‘protective’ haplotype (associated with a good response to MTX).
In the validation cohort (US cases) genotype data were available for 7 of the 14 SNPs in the SLC16A7 gene which had also been genotyped in the UK CHARMS study including 2 of the 3 SNPS that had been found to be significant. Of the 210 US individuals with genotype data available, patients were included in the statistical analysis if they had improvement of >70% in active joint count, responders (n=67) or <30%, non responders (n=76). Despite the fact that a different measurement of clinical response to MTX was used in the US cohort, association analysis again showed that the minor A allele of the SNP rs3763980 was associated with increased risk of not responding to MTX. Meta-analysis of the two studies strengthened the evidence of an association of the SNP rs3763980, with non-response, with a combined p-value of 0.002 (Figure 4). The combined odds ratio (OR) of ‘risk’ of being a non-responder for patients carrying the minor (A) allele was 1.89 when both data sets were considered together. There was no significant evidence for heterogeneity between the studies (p=0.87).
Figure 4. Meta-analysis of rs3763980 in UK CHARMS dataset and US dataset.
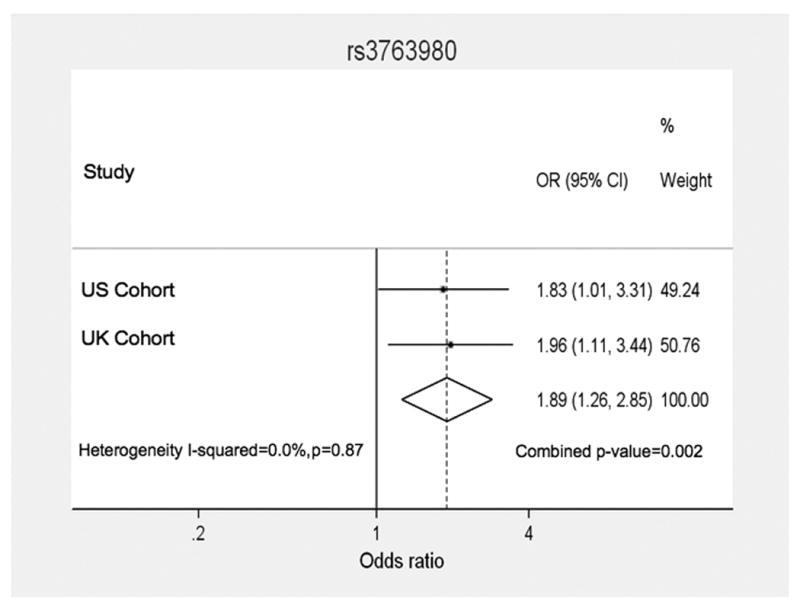
Forest plot displaying odds ratios (OR) and 95% confidence intervals for each of the two studies (US, UK) and the weighting for each study according to sample size. The combined OR (1.89) from the two studies is displayed as a diamond with the peaks denoting the upper and lower limits of the confidence intervals (1.26 – 2.85). The combined p-value for association of the A allele with non-responder status was 0.002. The Breslow-Day test was performed to test for heterogeneity between the two studies, and showed no evidence of heterogeneity (p=0.87).
To assess the functional role of this SNP, we also investigated a gene expression dataset generated from transformed B-cell lines from a large number of unrelated HapMap individuals[46]. Linear regression analysis found a significant correlation between genotype at rs3763980 and expression of SLC16A7 (p=2.9 × 10-7) (Figure 5).
Figure 5. SLC16A7 gene expression by genotype of rs3763980.
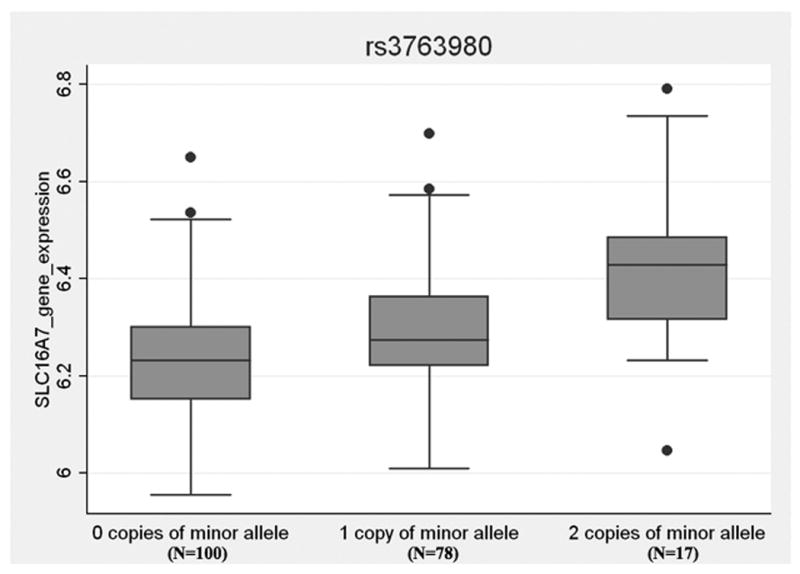
Gene expression levels in B cells from 195 healthy individuals was plotted by genotype at the rs3763980 SNP in the SLC16A7 gene. Data obtained from reference 46. The box and whisker plot shows the correlation of genotype at rs3763980 with normalized log2 quantitative gene expression of SLC16A7.
Discussion
Although MTX has been used for many years to treat juvenile arthritis, there are still no reliable biomarkers with which to predict good or poor response to MTX, and the mechanisms of MTX and its interactions with inflammatory pathways are still unclear. Several recent studies have suggested that genes which are differentially expressed in a pathological process are also more likely to harbour disease-associated genetic variants for a specific disease. This has been demonstrated in prostate cancer and type I diabetes: such SNPs have been labeled ‘functionally interpolating SNPs or FitSNPs”[47, 48]. In addition there are clearly genetic influences upon gene expression levels, and so called ‘very important pharmacogenes (VIPs)’ have been previously shown to have expression patterns that are associated with genetic variation[49]. Other gene expression profiling studies, in cancer, have identified transcripts whose early expression correlates with good or poor response to MTX[50]. In this study we have generated transcriptional profiling data from children given MTX for treatment of JIA, in order to identify pathways which are altered at a transcriptional level after MTX therapy, and then tested whether variation in highly differentially expressed genes makes a contribution to the genetic component of drug response. To our knowledge this is the first pharmacogenomic study in which this ‘dual’ approach has been applied.
Using analysis of genes differentially expressed in peripheral blood cells after MTX treatment in a group of 11 children with JIA, we first identified genes which had a fold change of 1.7 and reached significance of p<0.05. From the differentially expressed probes (n=1222) after MTX treatment, we used the IPA database to identify canonical pathways that were implicated by the differentially expressed genes in the dataset. 28 pathways reached a significance level of p<0.05. Many of these pathways were related to immune cell functioning, signalling and cytokines, while others indicated involvement of the one-carbon folate, purine and pyrimidine synthesis pathways, as expected. The top pathway, PI3K/AKT signalling, is known to be of functional importance in regulatory T cells, and the negative regulator of IL-2 signalling PTEN (increased 2.6 fold post MTX) is critical to maintaining tolerance and maintains the anergic phenotype of regulatory cells[31, 51, 52]. Several other immunosuppressive pathways including TGFß signalling and the PPAR (Peroxisome proliferator-activated receptors) pathway were identified, although at a lower significance level. In addition, genes known to be involved in MTX metabolism including thymidylate synthase (TYMS) and Serine hydroxymethyltransferase 1 (SHMT1) were found to have altered gene expression after 6 months of MTX therapy. TYMS is involved in generation of dihydrofolate and is inhibited by MTX polyglutamates, while SHMT1 is a pivotal enzyme in the folate pathway that synthesizes 5, 10-methylene tetrahydrofolate [26,27]. These genes are being investigated further.
One possible caveat relating to our strategy, would be that genes down regulated after successful MTX treatment may reflect the gene signature of active inflammation in JIA, which would be predicted to fall in expression after treatment. To address this issue, direct comparison of our data with a study of gene expression in JIA was performed. This study by Griffin et al used gene expression profiling of PBMC to identify distinct gene signatures in active JIA[23]. Comparison of the top 75 genes up-regulated in these signatures with our initial 1222 probe sets, showed overlap of only 6 genes (DUSP4, CREM, C9orf3, TcnRNA MAD1L1, and ACPP); of these, 4 were upregulated in the Griffin study (active JIA), and higher pre-treatment in our study. However the majority of genes in our differentially expressed list were not in the signature of ‘active JIA’ suggesting that our data do not simply represent a ‘downregulation of disease’ signature.
One previous study in adult RA used gene expression profiling from whole blood before and after MTX or anti-TNF treatment, but considered only genes that are involved in the NFκB pathway and did not identify genes specifically altered by MTX[53]. In the cancer field, where MTX doses administered are higher than those used to treat arthritis, differential gene expression in leukaemic cells has been used to screen for genes whose expression may predict a poor response to MTX[50]. That study did not attempt to relate genetic variation to the expression findings. A caveat of our study is that gene expression data generated from peripheral blood, may not represent the inflamed tissue (synovium). However we adopted this approach since one longterm goal is to generate predictive biomarkers that will be clinically amenable to simple testing in a blood test. A further caveat is that from pediatric blood samples it is challenging to generate cell-specific gene expression profiling data, to indicate which cell types within the PBMC contribute most to changes demonstrated: further functional studies on sorted cells will be required to elucidate this.
To ask if the ‘top ranking genes’ of our differentially expressed gene dataset contained SNPs that could inform us about the genetic component of drug response to MTX in JIA, we selected a small number of genes from those which were differentially expressed for genotyping analysis in a large cohort of children recruited to CHARMS. We took a strategy of screening the whole gene using a tagging SNP approach, this ensured that we had >95% coverage for the genes investigated, which is a more thorough approach than some existing studies which tend to only look at only a few SNPs in candidate genes.
To identify effects, we chose to compare extreme ends of the response spectrum, comparing non-responders to those who achieved or exceeded an ACR-Ped70 response. In the SLC16A7 gene, which was highly up-regulated after administration of MTX, we found three SNPs significantly associated with response to MTX treatment. However the haplotype analysis suggested that rs2711655 lies on the same ‘risk’ haplotype as rs3763980 and that it is the latter SNP, which is driving the association. We also found evidence for a ‘protective’ haplotype and the minor allele of rs10877333 is driving this association. Interestingly the SNP rs3763980 is a non-synonymous coding SNP, resulting in a substitution at amino acid 445 threonine to serine. This is in a predicted cytoplasmic domain, with implications for phosphorylation and signaling and is therefore a potential functional SNP.
We also validated the association of the SNP rs3763980 with risk of non-response to MTX in an independent cohort of JIA patients collected in the US. In this cohort a different definition for response was used, based upon joint count alone. Again the ends of the spectrum were compared, using ‘best responders’ versus non-responders. Interestingly, when the CHARMS clinical dataset was recoded using joint count alone, there was still a trend towards association of SNP rs3763980 with non-response, although this no longer reached statistical significance (p_trend = 0.08 OR 1.63 95%CI 0.94-2.84). Meta-analysis of the cohorts strengthened the evidence for association of the SNP, rs3763980, with a combined p-value of 0.002. However, further validation of these results in large cohorts will be necessary.
Interestingly, the data also show that the SNP also influences expression of the gene. Thus the minor allele of rs3763980 is associated with increased expression of SLC16A7 in transformed B cells from healthy donors. In arthritis patients this allele was associated with a poor response to MTX. Although it is difficult to extrapolate directly from control B cell data, to peripheral blood cells from patients with arthritis, this result does strongly suggest an association between SLC genotype at the rs3763980 SNP, and gene expression level. Further functional studies will be required to elucidate how altered protein levels of SLC16A7 influence response to MTX.
The SLC16A7 (MCT2) protein is a transporter of lactate and pyruvate, whose expression is enhanced by activation of the PI3K/AKT pathway, the most significant pathway identified in our pathway analysis (Table 2). There is evidence that SLC16A7 is co-regulated with PPARalpha, (also upregulated by MTX), which inhibits production of many inflammatory cytokines[54],[55]. At present the functional relationship of SLC16A7 expression and MTX mechanisms, or the coding change (T455S) caused by the SNP associated with non-response remain unclear, but warrant further investigation.
In conclusion, we believe this to be the first study in which gene expression profiling, in cells from a readily available source (peripheral blood) have been used to select novel gene candidates for analysis of genetic components to response to MTX, in childhood arthritis. We have identified pathways which are altered by MTX, many of which are central regulating pathways for the immune system. In addition for a small number of genes within these pathways we have gone one step further to determine whether genetic variation plays a role in determining differences in response. In one of these genes, SLC16A7, we have identified a SNP that is associated with risk of non-response to MTX, with validation in an independent cohort. Our findings will require validation in larger studies and other cohorts of patients treated with MTX for arthritis, or other inflammatory conditions. Further investigation and functional studies of pathways and genes identified here should contribute valuable insights into efficacy of MTX in inflammatory arthritis. This approach to identifying pathways and genes that may be involved in the efficacy and response to a commonly used drug in childhood arthritis may ultimately allow us to develop methods to predict response and thereby select individual treatment for patients with greater accuracy.
Supplementary Material
Acknowledgments
We thank all the patients and their families for participation in this study, the ward and clinic staff, for help collecting samples, and members of the CHARMS and laboratory teams for sample handling and processing. We thank N. Jina for technical assistance with Gene expression profiling.
Sources of support: HM, SU LW, and the SPARKS-CHARMS study, were funded by SPARKS UK and the Big Lottery Fund UK. The study was supported by the UK Medicines for Children Research Network. WT, PM and AH were funded by the Arthritis Research UK (grant reference 17522). DNG, SDT, TW and the US cohort were supported by NIH/NIAMS N01-AI042272, P30-AR046373, P01-AR048929, P60-AR047784 and the Children's Hospital Research Foundation.
Abbreviations
- JIA
Juvenile idiopathic arthritis
- MTX
methotrexate
- CHARMS
childhood arthritis response to medication study
- SLC
solute carrier
- FDR
false discovery rate
- SNP
single nucleotide polymorphism
- DOI
definition of improvement
- CHAQ
childhood health assessment questionnaire
- PBMC
peripheral blood mononuclear cells
- NR
non-responder
Footnotes
Conflicts of Interest: NONE
Contributor Information
Halima Moncrieffe, Rheumatology Unit, UCL Institute of Child Health, 30 Guilford Street London WC1N 1EH.
Anne Hinks, Arthritis Research UK Epidemiology Unit, Manchester Academic Health Science Centre, 3333 Burnet Ave, Cincinnati, OH 45241 USA.
Simona Ursu, Rheumatology Unit, UCL Institute of Child Health, 30 Guilford Street London WC1N 1EH.
Laura Kassoumeri, Rheumatology Unit, UCL Institute of Child Health, 30 Guilford Street London WC1N 1EH.
Angela Etheridge, Rheumatology Unit, UCL Institute of Child Health, 30 Guilford Street London WC1N 1EH.
Mike Hubank, Molecular Haematology and Cancer Biology Units, UCL Institute of Child Health, 30 Guilford Street London WC1N 1EH.
Paul Martin, Arthritis Research UK Epidemiology Unit, Manchester Academic Health Science Centre, 3333 Burnet Ave, Cincinnati, OH 45241 USA.
Tracey Weiler, Division of Rheumatology, Cincinnati Children's Hospital Medical Center, 3333 Burnet Ave, Cincinnati, OH 45241 USA.
David N Glass, Division of Rheumatology, Cincinnati Children's Hospital Medical Center, 3333 Burnet Ave, Cincinnati, OH 45241 USA.
Susan D. Thompson, Division of Rheumatology, Cincinnati Children's Hospital Medical Center, 3333 Burnet Ave, Cincinnati, OH 45241 USA
Wendy Thomson, Arthritis Research UK Epidemiology Unit, Manchester Academic Health Science Centre, 3333 Burnet Ave, Cincinnati, OH 45241 USA.
Lucy R Wedderburn, Rheumatology Unit, UCL Institute of Child Health, 30 Guilford Street London WC1N 1EH.
References
- 1.Ramanan AV, Whitworth P, Baildam EM. Use of methotrexate in juvenile idiopathic arthritis. Arch Dis Child. 2003;88:197–200. doi: 10.1136/adc.88.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wessels JA, Huizinga TW, Guchelaar HJ. Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology. 2008;47:249–55. doi: 10.1093/rheumatology/kem279. [DOI] [PubMed] [Google Scholar]
- 3.Woo P, Southwood TR, Prieur AM, Dore CJ, Grainger J, David J, et al. Randomized, placebo-controlled, crossover trial of low-dose oral methotrexate in children with extended oligoarticular or systemic arthritis. Arthritis Rheum. 2000;43:1849–57. doi: 10.1002/1529-0131(200008)43:8<1849::AID-ANR22>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 4.Ruperto N, Murray KJ, Gerloni V, Wulffraat N, Feitosa De Oliveira SK, Falcini F, et al. A randomized trial of parenteral methotrexate comparing an intermediate dose with a higher dose in children with juvenile idiopathic arthritis who failed to respond to standard doses of methotrexate. Arthritis Rheum. 2004;50:2191–201. doi: 10.1002/art.20288. [DOI] [PubMed] [Google Scholar]
- 5.Nistala K, Woo P, Wedderburn LR. Juvenile Idiopathic Arthritis. In: Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley's textbook of Rheumatology. 2008. [Google Scholar]
- 6.Cespedes-Cruz A, Gutierrez-Suarez R, Pistorio A, Ravelli A, Loy A, Murray KJ, et al. Methotrexate improves the health-related quality of life of children with juvenile idiopathic arthritis. Ann Rheum Dis. 2008;67:309–14. doi: 10.1136/ard.2007.075895. [DOI] [PubMed] [Google Scholar]
- 7.Foster HE, Marshall N, Myers A, Dunkley P, Griffiths ID. Outcome in adults with juvenile idiopathic arthritis: a quality of life study. Arthritis Rheum. 2003;48:767–75. doi: 10.1002/art.10863. [DOI] [PubMed] [Google Scholar]
- 8.Duffy CM. Measurement of health status, functional status, and quality of life in children with juvenile idiopathic arthritis: clinical science for the pediatrician. Rheum Dis Clin North Am. 2007;33:389–402. doi: 10.1016/j.rdc.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 9.van Ede AE, Laan RF, Blom HJ, De Abreu RA, van de Putte LB. Methotrexate in rheumatoid arthritis: an update with focus on mechanisms involved in toxicity. Semin Arthritis Rheum. 1998;27:277–92. doi: 10.1016/s0049-0172(98)80049-8. [DOI] [PubMed] [Google Scholar]
- 10.Ranganathan P, McLeod HL. Methotrexate pharmacogenetics: the first step toward individualized therapy in rheumatoid arthritis. Arthritis Rheum. 2006;54:1366–77. doi: 10.1002/art.21762. [DOI] [PubMed] [Google Scholar]
- 11.Cronstein BN, Merrill JT. Mechanisms of the effects of methotrexate. Bull Rheum Dis. 1996;45:6–8. [PubMed] [Google Scholar]
- 12.van Ede AE, Laan RF, Rood MJ, Huizinga TW, van de Laar MA, van Denderen CJ, et al. Effect of folic or folinic acid supplementation on the toxicity and efficacy of methotrexate in rheumatoid arthritis: a forty-eight week, multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2001;44:1515–24. doi: 10.1002/1529-0131(200107)44:7<1515::AID-ART273>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Wessels JA, de Vries-Bouwstra JK, Heijmans BT, Slagboom PE, Goekoop-Ruiterman YP, Allaart CF, et al. Efficacy and toxicity of methotrexate in early rheumatoid arthritis are associated with single-nucleotide polymorphisms in genes coding for folate pathway enzymes. Arthritis Rheum. 2006;54:1087–95. doi: 10.1002/art.21726. [DOI] [PubMed] [Google Scholar]
- 14.Ranganathan P, Eisen S, Yokoyama WM, McLeod HL. Will pharmacogenetics allow better prediction of methotrexate toxicity and efficacy in patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62:4–9. doi: 10.1136/ard.62.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hider SL, Thomson W, Mack LF, Armstrong DJ, Shadforth M, Bruce IN. Polymorphisms within the adenosine receptor 2a gene are associated with adverse events in RA patients treated with MTX. Rheumatology. 2008;47:1156–9. doi: 10.1093/rheumatology/ken182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wessels JA, van der Kooij SM, le Cessie S, Kievit W, Barerra P, Allaart CF, et al. A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum. 2007;56:1765–75. doi: 10.1002/art.22640. [DOI] [PubMed] [Google Scholar]
- 17.Stephenson T. How children's responses to drugs differ from adults. Br J Clin Pharmacol. 2005;59:670–3. doi: 10.1111/j.1365-2125.2005.02445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmeling H, Biber D, Heins S, Horneff G. Influence of methylenetetrahydrofolate reductase polymorphisms on efficacy and toxicity of methotrexate in patients with juvenile idiopathic arthritis. J Rheumatol. 2005;32:1832–6. [PubMed] [Google Scholar]
- 19.Albers HM, Wessels JA, van der Straaten RJ, Brinkman DM, Suijlekom-Smit LW, Kamphuis SS, et al. Time to treatment as an important factor for the response to methotrexate in juvenile idiopathic arthritis. Arthritis Rheum. 2009;61:46–51. doi: 10.1002/art.24087. [DOI] [PubMed] [Google Scholar]
- 20.Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–2. [PubMed] [Google Scholar]
- 21.Giannini EH, Ruperto N, Ravelli A, Lovell DJ, Felson DT, Martini A. Preliminary definition of improvement in juvenile arthritis. Arthritis Rheum. 1997;40:1202–1209. doi: 10.1002/1529-0131(199707)40:7<1202::AID-ART3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 22.Nugent J, Ruperto N, Grainger J, Machado C, Sawhney S, Baildam E, et al. The British version of the Childhood Health Assessment Questionnaire (CHAQ) and the Child Health Questionnaire (CHQ) Clin Exp Rheumatol. 2001;19:S163–7. [PubMed] [Google Scholar]
- 23.Griffin TA, Barnes MG, Ilowite NT, Olson JC, Sherry DD, Gottlieb BS, et al. Gene expression signatures in polyarticular juvenile idiopathic arthritis demonstrate disease heterogeneity and offer a molecular classification of disease subsets. Arthritis Rheum. 2009;60:2113–23. doi: 10.1002/art.24534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnes MG, Grom AA, Thompson SD, Griffin TA, Pavlidis P, Itert L, et al. Subtype-specific peripheral blood gene expression profiles in recent-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:2102–12. doi: 10.1002/art.24601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 26.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartoli M, Taro M, Magni-Manzoni S, Pistorio A, Traverso F, Viola S, et al. The magnitude of early response to methotrexate therapy predicts long-term outcome of patients with juvenile idiopathic arthritis. Ann Rheum Dis. 2008;67:370–4. doi: 10.1136/ard.2007.073445. [DOI] [PubMed] [Google Scholar]
- 28.Dervieux T, Furst D, Lein DO, Capps R, Smith K, Walsh M, et al. Polyglutamation of methotrexate with common polymorphisms in reduced folate carrier, aminoimidazole carboxamide ribonucleotide transformylase, and thymidylate synthase are associated with methotrexate effects in rheumatoid arthritis. Arthritis Rheum. 2004;50:2766–74. doi: 10.1002/art.20460. [DOI] [PubMed] [Google Scholar]
- 29.Weisman MH, Furst DE, Park GS, Kremer JM, Smith KM, Wallace DJ, et al. Risk genotypes in folate-dependent enzymes and their association with methotrexate-related side effects in rheumatoid arthritis. Arthritis Rheum. 2006;54:607–12. doi: 10.1002/art.21573. [DOI] [PubMed] [Google Scholar]
- 30.Buckler JL, Liu X, Turka LA. Regulation of T-cell responses by PTEN. Immunol Rev. 2008;224:239–48. doi: 10.1111/j.1600-065X.2008.00650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu MY, Tsai TF, Beaudet AL. Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the PWS/AS domain. Genes Dev. 2006;20:2859–70. doi: 10.1101/gad.1452206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajaram V, Knezevich S, Bove KE, Perry A, Pfeifer JD. DNA sequence of the translocation breakpoints in undifferentiated embryonal sarcoma arising in mesenchymal hamartoma of the liver harboring the t(11;19)(q11;q13.4) translocation. Genes Chromosomes Cancer. 2007;46:508–13. doi: 10.1002/gcc.20437. [DOI] [PubMed] [Google Scholar]
- 34.Guffanti A, Iacono M, Pelucchi P, Kim N, Solda G, Croft LJ, et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009;10:163. doi: 10.1186/1471-2164-10-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fellenberg J, Dechant MJ, Ewerbeck V, Mau H. Identification of drug-regulated genes in osteosarcoma cells. Int J Cancer. 2003;105:636–43. doi: 10.1002/ijc.11135. [DOI] [PubMed] [Google Scholar]
- 36.Johnson AD, O'Donnell CJ. An open access database of genome-wide association results. BMC Med Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatzikyriakidou A, Georgiou I, Voulgari PV, Papadopoulos CG, Tzavaras T, Drosos AA. Transcription regulatory polymorphism -43T>C in the 5′-flanking region of SLC19A1 gene could affect rheumatoid arthritis patient response to methotrexate therapy. Rheumatol Int. 2007;27:1057–61. doi: 10.1007/s00296-007-0339-0. [DOI] [PubMed] [Google Scholar]
- 38.James HM, Gillis D, Hissaria P, Lester S, Somogyi AA, Cleland LG, et al. Common polymorphisms in the folate pathway predict efficacy of combination regimens containing methotrexate and sulfasalazine in early rheumatoid arthritis. J Rheumatol. 2008;35:562–71. [PubMed] [Google Scholar]
- 39.Postigo AA, Depp JL, Taylor JJ, Kroll KL. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. Embo J. 2003;22:2453–62. doi: 10.1093/emboj/cdg226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams TM, Moolten D, Burlein J, Romano J, Bhaerman R, Godillot A, et al. Identification of a zinc finger protein that inhibits IL-2 gene expression. Science. 1991;254:1791–4. doi: 10.1126/science.1840704. [DOI] [PubMed] [Google Scholar]
- 41.Kilding R, Iles MM, Timms JM, Worthington J, Wilson AG. Additional genetic susceptibility for rheumatoid arthritis telomeric of the DRB1 locus. Arthritis Rheum. 2004;50:763–9. doi: 10.1002/art.20043. [DOI] [PubMed] [Google Scholar]
- 42.Allcock RJ, Williams JH, Price P. The central MHC gene, BAT1, may encode a protein that down-regulates cytokine production. Genes Cells. 2001;6:487–94. doi: 10.1046/j.1365-2443.2001.00435.x. [DOI] [PubMed] [Google Scholar]
- 43.Quinones-Lombrana A, Lopez-Soto A, Ballina-Garcia FJ, Alperi-Lopez M, Queiro-Silva R, Lopez-Vazquez A, et al. BAT1 promoter polymorphism is associated with rheumatoid arthritis susceptibility. J Rheumatol. 2008;35:741–4. [PubMed] [Google Scholar]
- 44.Lieberson R, Mowen KA, McBride KD, Leautaud V, Zhang X, Suh WK, et al. Tumor necrosis factor receptor-associated factor (TRAF)2 represses the T helper cell type 2 response through interaction with NFAT-interacting protein (NIP45) J Exp Med. 2001;194:89–98. doi: 10.1084/jem.194.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mowen KA, Schurter BT, Fathman JW, David M, Glimcher LH. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes. Mol Cell. 2004;15:559–71. doi: 10.1016/j.molcel.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 46.Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–24. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen R, Morgan AA, Dudley J, Deshpande T, Li L, Kodama K, et al. FitSNPs: highly differentially expressed genes are more likely to have variants associated with disease. Genome Biol. 2008;9:R170. doi: 10.1186/gb-2008-9-12-r170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gorlov IP, Gallick GE, Gorlova OY, Amos C, Logothetis CJ. GWAS meets microarray: are the results of genome-wide association studies and gene-expression profiling consistent? Prostate cancer as an example. PLoS One. 2009;4:e6511. doi: 10.1371/journal.pone.0006511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang RS, Duan S, Kistner EO, Zhang W, Bleibel WK, Cox NJ, et al. Identification of genetic variants and gene expression relationships associated with pharmacogenes in humans. Pharmacogenet Genomics. 2008;18:545–9. doi: 10.1097/FPC.0b013e3282fe1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sorich MJ, Pottier N, Pei D, Yang W, Kager L, Stocco G, et al. In vivo response to methotrexate forecasts outcome of acute lymphoblastic leukemia and has a distinct gene expression profile. PLoS Med. 2008;5:e83. doi: 10.1371/journal.pmed.0050083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh PT, Buckler JL, Zhang J, Gelman AE, Dalton NM, Taylor DK, et al. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J Clin Invest. 2006;116:2521–31. doi: 10.1172/JCI28057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buckler JL, Walsh PT, Porrett PM, Choi Y, Turka LA. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J Immunol. 2006;177:4262–6. doi: 10.4049/jimmunol.177.7.4262. [DOI] [PubMed] [Google Scholar]
- 53.Parker A, Izmailova ES, Narang J, Badola S, Le T, Roubenoff R, et al. Peripheral blood expression of nuclear factor-kappab-regulated genes is associated with rheumatoid arthritis disease activity and responds differentially to anti-tumor necrosis factor-alpha versus methotrexate. J Rheumatol. 2007;34:1817–22. [PubMed] [Google Scholar]
- 54.Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169:453–9. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 55.Jansen S, Cashman K, Thompson JG, Pantaleon M, Kaye PL. Glucose deprivation, oxidative stress and peroxisome proliferator-activated receptor-alpha (PPARA) cause peroxisome proliferation in preimplantation mouse embryos. Reproduction. 2009;138:493–505. doi: 10.1530/REP-09-0038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.