

Abstract
Background
It is a challenge to identify patients who, after undergoing potentially curative treatment for hepatocellular carcinoma, are at greatest risk for recurrence. Such high-risk patients could receive novel interventional measures. An obstacle to the development of genome-based predictors of outcome in patients with hepatocellular carcinoma has been the lack of a means to carry out genomewide expression profiling of fixed, as opposed to frozen, tissue.
Methods
We aimed to demonstrate the feasibility of gene-expression profiling of more than 6000 human genes in formalin-fixed, paraffin-embedded tissues. We applied the method to tissues from 307 patients with hepatocellular carcinoma, from four series of patients, to discover and validate a gene-expression signature associated with survival.
Results
The expression-profiling method for formalin-fixed, paraffin-embedded tissue was highly effective: samples from 90% of the patients yielded data of high quality, including samples that had been archived for more than 24 years. Gene-expression profiles of tumor tissue failed to yield a significant association with survival. In contrast, profiles of the surrounding nontumoral liver tissue were highly correlated with survival in a training set of tissue samples from 82 Japanese patients, and the signature was validated in tissues from an independent group of 225 patients from the United States and Europe (P=0.04).
Conclusions
We have demonstrated the feasibility of genomewide expression profiling of formalin-fixed, paraffin-embedded tissues and have shown that a reproducible gene-expression signature correlated with survival is present in liver tissue adjacent to the tumor in patients with hepatocellular carcinoma.
Introduction
In developing countries, hepatocellular carcinoma often comes to medical attention when the tumors are at an advanced stage and curative therapies are of limited benefit. In developed countries, however, at-risk populations of patients (e.g., those who are infected with hepatitis virus and have cirrhosis) are often under close surveillance; as a result, hepatocellular carcinoma is usually detected when the tumors are small and treatment is more likely to be successful.1,2 Nevertheless, recurrences eventually occur in most patients.1,2 Studies suggest that chemopreventive strategies may suppress recurrence and prolong survival,1,3–6 although these findings are still uncertain. It would be ideal to treat only patients at greatest risk for recurrence. Several methods have been used to predict survival among patients with hepatocellular carcinoma, including the enumeration of anatomical and histopathological attributes (e.g., tumor multinodularity and vascular invasion), but these have become less useful as hepatocellular carcinoma is increasingly diagnosed at earlier stages.
A technical challenge facing the use of gene-expression profiling to predict the outcome of hepatocellular carcinoma has been the lack of suitable specimens from patients. Current methods of genomewide expression profiling require frozen tissue for analysis, whereas tissue banks with clinical outcome data generally have formalin-fixed, paraffin-embedded specimens. Even today, the vast majority of specimens are formalin-fixed; the collection of frozen tissues has yet to become routine clinical practice.
We tested a method for genomewide expression profiling of formalin-fixed, paraffin-embedded tissues. We applied the method to the analysis of the clinical outcome of hepatocellular carcinoma.
Methods
Patients and Samples
The training set consisted of tissue samples from 106 patients who were consecutively treated with surgery for primary hepatocellular carcinoma between 1990 and 2001 at Toranomon Hospital in Tokyo and for whom data on clinical outcomes (over a median follow-up period of 7.8 years) and formalin-fixed, paraffin-embedded blocks of tumor and adjacent tissue were available (Figure 1Figure 1Study Design.). The validation set included tissue samples from 234 patients with hepatocellular carcinoma who consecutively underwent surgery between 1994 and 2005: 92 patients at the Mount Sinai School of Medicine in New York, 46 at Hospital Clínic Barcelona, and 96 at the National Cancer Institute of Milan (members of the HCC Genomic Consortium). Archived formalin-fixed, paraffin-embedded tissues obtained as part of routine clinical care were analyzed, with approval by the local institutional review boards granted on the condition that all samples be made anonymous. Formalin-fixed, paraffin-embedded blocks obtained at the time of resection were cut into three or four sections (each 10 µm thick), macrodissected to isolate tumor and adjacent liver tissue, and subjected to RNA extraction as described in the Supplementary Appendix (available with the full text of this article at www.nejm.org).
Figure 1. Study Design.
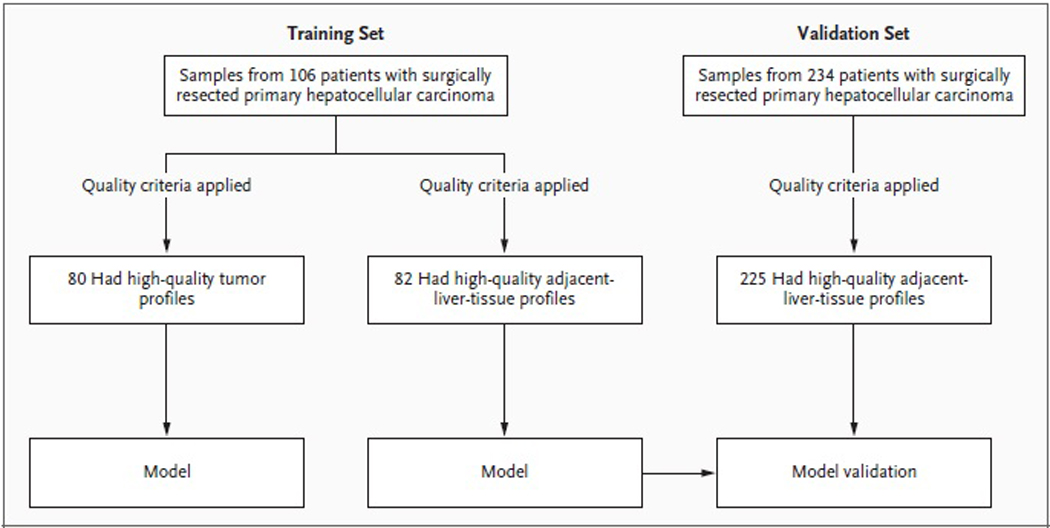
In the training set, tumor tissue and liver tissue adjacent to the tumor were profiled separately, and each was used to generate an outcome model. The model based on adjacent liver tissue was validated with the use of an independent validation set.
Analysis of Gene Expression
Gene-expression profiling was performed according to the complementary DNA–mediated annealing, selection, extension, and ligation (DASL) assay7,8 (Illumina), and we selected 6100 transcriptionally informative genes for analysis (see the Supplementary Appendix). (Microarray data are available at www.ncbi.nlm.nih.gov/geo/, accession numbers GSE10143 and GPL5474.) Genes whose expression was associated with disease-specific survival and time to recurrence were selected with the use of the Cox score (see the Supplementary Appendix). The value of the signature was assessed on the basis of overall survival. Late recurrence was defined as tumor recurrence occurring more than 2 years after surgery.9,10 Outcome association analysis was performed with the use of a nearest-neighbor–based method (see the Supplementary Appendix).
Statistical Analysis
Functional annotation was performed by means of gene set enrichment analysis (GSEA, www.broad.mit.edu/gsea/).11 Survival analyses were performed with the use of the log-rank test and Cox regression modeling. Subgroup analysis was performed on data from patients with a longer duration of follow-up (treated no later than 2004) and those with carcinoma classified as stage 0 or stage A according to the Barcelona Clinic Liver Cancer staging system (BCLC), which ranks hepatocellular carcinoma in five stages, ranging from 0 (very early stage) to D (terminal stage).1,12 The hazard function for tumor recurrence was calculated as previously described.10,13 All analyses were performed with the use of GenePattern14 (www.broad.mit.edu/cancer/software/genepattern/) or the R statistical package (www.r-project.org). (See the Supplementary Appendix for details on the statistical analyses and methods of clonality analysis.)
Results
Validation of the Profiling Method
We first sought a method that was suitable for gene-expression profiling of formalin-fixed, paraffin-embedded material. An approach has been reported for the analysis of several hundred transcripts based on DASL, a multiplex, locus-specific polymerase-chain-reaction (PCR) assay.7,8 However, an unbiased discovery of diagnostic signatures requires a genomewide profiling method. Accordingly, we modified the DASL method for probe selection and analysis and performed a bioinformatic meta-analysis to identify 6000 transcripts that captured the majority of variance in gene expression across the human transcriptome (see the Supplementary Appendix). This 6000-gene DASL assay served as a potential tool for genomewide analysis of formalin-fixed, paraffin-embedded tissues. We found the assay to be highly reproducible (R2>0.96 in replicate experiments), with an overall success rate of 90% among all the specimens, including formalin-fixed, paraffin-embedded tissue blocks collected up to 24 years ago (see the Supplementary Appendix). We found that representing each transcript with one probe only (as opposed to three, as previously reported7,8) resulted in little loss of assay performance (Figure 1 in the Supplementary Appendix).
Patients
Table 1Table 1Characteristics of Patients in the Training Set and in the Validation Set, at the Time of Surgery. summarizes the clinical characteristics of the patients in the training and validation sets. All patients were treated with curative surgical resection, which was, in some cases, followed by second-line treatments at the time of recurrence. By design, the training set included tissue samples from a large proportion of patients with very-early-stage hepatocellular carcinoma (BCLC stage 0), because these patients represent the greatest clinical challenge with respect to outcome prediction. Indeed, no clinical variables, either alone or in combination, were associated with survival among these patients (Table 1 in the Supplementary Appendix). Although there were no significant differences between the training set and validation set with respect to the number of patients with advanced-stage carcinoma (BCLC stage B) or the status of liver function, there was heterogeneity between the two sets with respect to certain tumor characteristics, such as diameter and type of viral infection (Table 1). Such heterogeneity may help to ensure that molecular predictors have real-world applicability across heterogeneous populations of patients.
Table 1.
Patient Characteristics of Training and Validation Sets at Time of Surgery
| Characteristic | Training (n=82) | Validation (n=225) | p-value | |
|---|---|---|---|---|
| Age | 59 [52,64] | 66 [57,71] | <0.001 | |
| Sex (male) | 64 (78%) | 173 (77%) | 0.88 | |
| HCV | 60 (73%) | 104 (48%) | <0.001 | |
| HBV | 17 (21%) | 62 (29%) | 0.25 | |
| Alcohol | 3 (4%) | 19 (9%) | 0.22 | |
| Tumor diameter (cm) | 2.2 [1.7,3.2] | 3.5 [2.3,5.5] | <0.001 | |
| Differentiation | Well | 18 (22%) | 34 (26%) | 0.68 |
| Moderate | 49 (60%) | 80 (60%) | ||
| Poor | 15 (18%) | 19 (14%) | ||
| Vascular invasion | 4 (5%) | 74 (34%) | <0.001 | |
| BCLC stage | 0 | 25 (30%) | 21 (9%) | 0.64* |
| A | 50 (61%) | 186 (83%) | <0.001** | |
| ≥ B | 7 (9%) | 16 (7%) | ||
| Child-Pugh A | 72 (88%) | 204 (97%) | 0.52 | |
| AFP (> 100 ng/mL) | 53 (65%) | 53 (24%) | 0.14 | |
| Median follow-up (yr) | 7.8 | 2.2 | - | |
Median [25%,75%]
HBV: hepatitis B virus, HCV: hepatitis C virus,
BCLC: Barcelona Clinic Liver Cancer staging,
AFP: alpha-fetoprotein
"0+A" vs. ≥ B,
0 vs. A vs. ≥ B
Profiles of Hepatocellular Carcinoma Tumors
We first investigated whether gene-expression profiles of hepatocellular carcinoma tumors were associated with the clinical outcome. For each of the 106 patients in the training set, tumor-containing portions of the formalin-fixed paraffin-embedded blocks were macrodissected away from surrounding liver tissue. Eighty tumors (75%) yielded high-quality gene-expression profiles (see the Supplementary Appendix). Using a leave-one-out cross-validation procedure and a nearest-neighbor–based algorithm, we failed to detect a significant gene-expression correlate of either tumor recurrence (P=0.22) or survival (P=0.70) (Figure 2A in the Supplementary Appendix). Furthermore, a previously reported signature associated with survival among patients with hepatocellular carcinoma15 was not associated with survival in our series of patients (P=0.76) (Figure 2B in the Supplementary Appendix). This failure to identify an outcome-associated signature is unlikely to be due to a technical flaw of the formalin-fixed, paraffin-embedded DASL method, because we observed the same molecular-subclass structure in the formalin-fixed, paraffin-embedded samples as that observed in collections of frozen samples of hepatocellular carcinoma (Figure 2B and 3B in the Supplementary Appendix). Although this result does not exclude the possibility of tumor-derived expression profiles as predictors of the outcome of hepatocellular carcinoma, the data suggest that at least in this training set, the outcome was largely related to other factors.
Survival Signature in Adjacent Liver Tissue
The lack of association between tumor-derived gene-expression profiles and survival led us to consider the pattern of recurrence of early-stage hepatocellular carcinoma. In contrast to advanced tumors, which tend to recur rapidly after resection, early-stage tumors, which are increasingly diagnosed in modern clinical practice, recur much later, generally more than 2 years after resection9,10 (Figure 4 in the Supplementary Appendix). This emerging pattern of late recurrence of hepatocellular carcinoma (due at least in part to the diagnosis of hepatocellular carcinoma at an early stage) has led to the notion that a late recurrence may not be an actual recurrence but rather a second primary tumor in an at-risk liver, presumably due to the carcinogenic effects of cirrhosis.1,2,9 We therefore hypothesized that the surrounding liver tissue — not the tumor itself — might harbor a gene-expression signature associated with subsequent recurrence.
To test this hypothesis, we assessed the gene-expression profiles of the liver tissue surrounding the resected tumor in the 106 formalin-fixed, paraffin-embedded blocks that constituted the training set. Eighty-two samples (77%) yielded high-quality gene-expression profiles (see the Supplementary Appendix). Using a standard leave-one-out cross-validation procedure, we found the liver signature to be significantly correlated with survival (P=0.02) (Figure 2AFigure 2Survival Signatures and Survival Curves in the Training Set.). The aggregate survival-correlated signature contained 186 genes (Figure 2B and 2C, and Table 2 in the Supplementary Appendix) and was tested in the validation set. Using GSEA, which shows whether a defined set of genes has a significant association with a phenotype of interest, we found the good-prognosis signature to contain genes associated with normal liver function (Table 2 and Table 3 in the Supplementary Appendix), including the plasma proteins C4, C5, C8, C9, and F9 and several drug-metabolizing enzymes: the alcohol dehydrogenases ADH5 and ADH6, the aldo–keto-reductases AKR1A1 and AKR1D1, the aldehyde dehydrogenase ALDH9A1, the cytochrome P450 CYP2B6, and hepatic lipase (LIPC). These findings are consistent with the association between impaired liver function and a poor outcome.1 In addition, the poor-prognosis signature contained gene sets associated with inflammation, including those related to interferon signaling, activation of nuclear factor-κB, and signaling by tumor necrosis factor α. Histologic features of liver inflammation were not found to be associated with the outcome (Figure 2D, and Table 4 and Figure 5 in the Supplementary Appendix). Of particular interest, GSEA showed that the downstream targets of interleukin-6 were strongly associated with the poor-prognosis signature, which is consistent with the finding that disruption of interleukin-6 signaling protects mice from chemically induced hepatocellular carcinoma.16
Figure 2. Survival Signatures and Survival Curves in the Training Set.
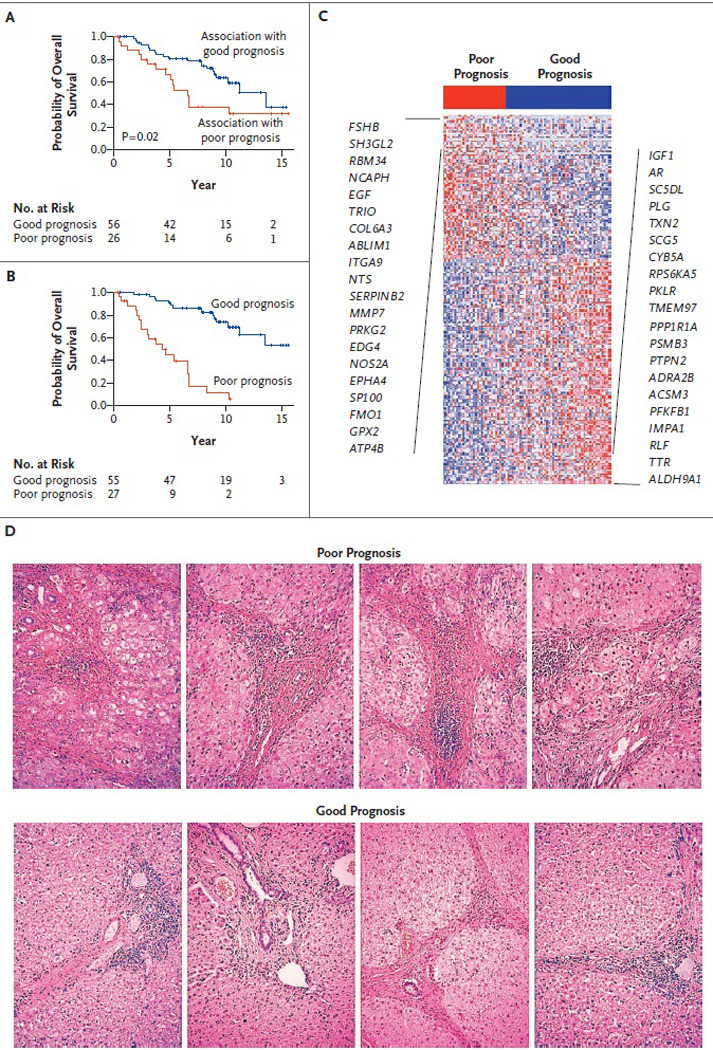
Curves are shown for survival according to the association of the gene signature with survival, based on leave-one-out cross-validation testing (Panel A), and for overall survival according to the level of expression of the 186 signature genes (Panel B); of these, 113 were associated with a good prognosis and 73 with a poor prognosis. Panel C shows the expression pattern of the survival signature (comprising 186 genes). The 20 genes most closely associated with a poor prognosis are listed on the left, and the 20 most closely associated with a good prognosis on the right. Red indicates high expression; blue indicates low expression. Panel D shows representative photomicrographs of sections of liver tissue adjacent to tumor that were profiled in this study; there were no histologic correlates with survival. Staining was with hematoxylin and eosin.
Validation of the Liver-Derived Survival Signature
We next tested the 186-gene survival signature in an independent set of tissue samples from eligible patients at three treatment centers in the United States and Europe. Of the 234 samples in this validation set, 225 (96%) yielded gene-expression profiles of high quality (see the Supplementary Appendix). The survival signature (Figure 3AFigure 3Survival Signatures and Survival Curves in the Validation Set.) was associated with significant differences in survival among patients (P=0.04) (Figure 3B), despite the modest duration of follow-up (median, 2.2 years). The separation of the survival curves was even more pronounced when, in a prespecified subgroup analysis, we limited our attention to the 168 patients with a longer duration of follow-up (median, 2.8 years; P=0.01) (Figure 3C). These results support the validity of the survival signature and highlight the potential role of nontumoral liver tissue in predicting the outcome for patients with early hepatocellular carcinoma.
Figure 3. Survival Signatures and Survival Curves in the Validation Set.
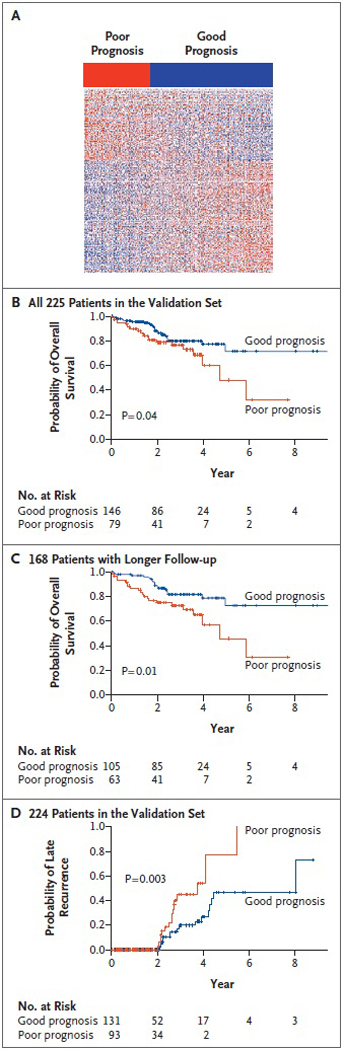
Panel A shows the expression pattern of the 186-gene survival signature. Red indicates a poor prognosis; blue indicates a good prognosis. Survival curves are shown for overall survival according to the level of expression of the 186 signature genes among all 225 patients whose tissue samples constituted the validation set (Panel B) and among the 168 patients with a longer duration of follow-up (treated no later than 2004) (Panel C). Panel D shows the probability of late recurrence according to the level of expression of the late-recurrence gene signature. Data are missing for one patient in Panel D.
Recurrence-Associated Signature
We performed a similar analysis using tumor recurrence as the clinical end point. A 132-gene late-recurrence signature defined in the training set was tested in the validation set. Whereas the recurrence signature did not show an association with recurrence within the first 2 years after surgery (a finding that was consistent with its development in association with late recurrence) (Figure 6A and 6B in the Supplementary Appendix), it was significantly associated with late recurrence (P=0.003) (Figure 3D). Not surprisingly, a nonparametric enrichment test indicated that the survival and late-recurrence signatures were closely associated (P<0.001) (Figure 6C in the Supplementary Appendix).
Multivariate Analysis
We next examined the signature in the context of the factors that are generally accepted as indicating a poor prognosis for patients with hepatocellular carcinoma (tumor multinodularity, the presence of microvascular invasion, and a high serum alpha-fetoprotein level1,9) in the validation set. These factors were associated with early recurrence (within 2 years after treatment) (Table 5 in the Supplementary Appendix). In contrast, multivariate analysis showed that the late-recurrence signature was the only independent prognostic variable for late recurrence (Table 2Table 2Associations of Gene-Expression Signatures and Clinical Variables with Late Recurrence or Overall Survival, from Multivariate Analysis of the Validation Set.). Prespecified subgroup analyses showed that this association remained significant in both the subgroup of 168 patients with a longer period of follow-up and the subgroup of 207 patients with early-stage hepatocellular carcinomas (BCLC stage 0 or A) (Figure 4Figure 4Hazard Ratios for Poor Survival and Late Recurrence in Selected Subgroups of Patients in the Validation Set., and Table 6 in the Supplementary Appendix). Similarly, the survival signature was independently associated with survival in multivariate analysis (Table 2), and this association persisted in the subgroup of patients with longer follow-up (Table 2). These results indicate that clinical and histopathological factors are associated with early recurrence of hepatocellular carcinoma and that late recurrence is associated with the gene-expression signature of nontumoral liver tissue adjacent to the primary tumor. The latter finding is consistent with the notion that late recurrences are not actually recurrences but rather new primary tumors. In support of this view, we detected highly discordant patterns of gains and losses in gene-copy number (including in regions exhibiting loss of heterozygosity) between the primary and recurrent hepatocellular carcinoma tumors but did not detect such patterns in endometrial, ovarian, renal, or lymphoma tumors (Table 7 and Figure 7 in the Supplementary Appendix). These results strongly suggest that the primary and recurrent hepatocellular carcinoma tumors arise from distinct clones.
Table 2.
Association of Gene Expression Signatures and Clinical Variables with Clinical Outcome in Multivariate Analysis
| A: Late recurrence | ||||
|---|---|---|---|---|
| Variable | Hazard | 95% CI | p-value | |
| ratio | low | high | ||
| Late recurrence signature | 2.94 | 1.39 | 6.20 | 0.005 |
| B: Survival | ||||
|---|---|---|---|---|
| Variable | Hazard | 95% CI | p-value | |
| ratio | low | high | ||
| Survival signature | 2.08 | 1.03 | 4.18 | 0.04 |
| AFP > 100 ng/mL | 2.29 | 1.14 | 4.61 | 0.02 |
| Vascular invasion | 2.01 | 1.01 | 3.99 | 0.05 |
| C: Survival (longer follow-up patients, n=168) | ||||
|---|---|---|---|---|
| Variable | Hazard | 95% CI | p-value | |
| ratio | low | high | ||
| Survival signature | 2.56 | 1.22 | 5.38 | 0.01 |
| AFP > 100 ng/mL | 2.01 | 0.94 | 4.26 | 0.07 |
| Vascular invasion | 2.20 | 1.06 | 4.53 | 0.03 |
AFP: alpha-fetoprotein
Figure 4. Hazard Ratios for Poor Survival and Late Recurrence in Selected Subgroups of Patients in the Validation Set.
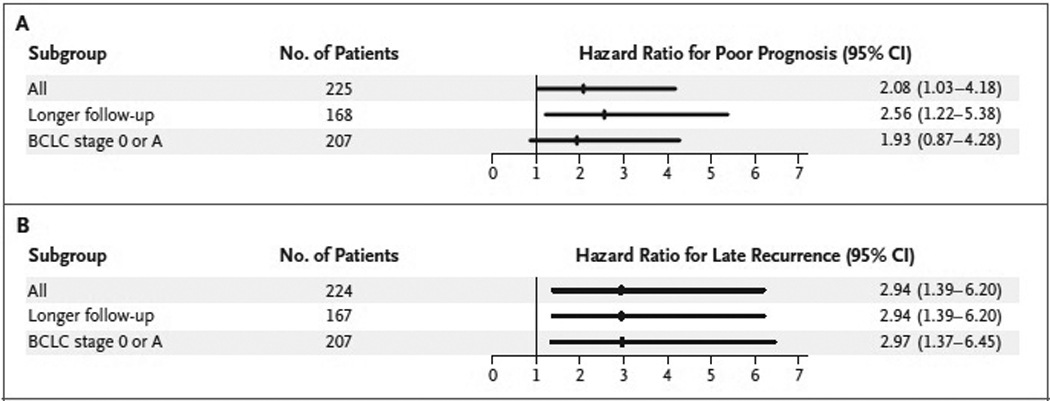
The hazard ratio was for poor survival among patients with the poor-prognosis gene signature (Panel A) or for late recurrence among patients with the late-recurrence gene signature (Panel B), as compared with those without the signature. BCLC denotes Barcelona Clinic Liver Cancer staging system, which ranks hepatocellular carcinoma in five stages, ranging from 0 (very early stage) to D (terminal stage). Data are missing for one patient in Panel B.
Discussion
The full potential of gene-expression profiling of cancer has been hindered in part by technical limitations — in particular, the requirement of frozen material for analysis. Although frozen tissues are increasingly being banked at tertiary care centers, the duration of clinical follow-up of these collections is usually short, and the vast majority of tumor-biopsy specimens and resections are performed outside of major research hospitals. There is therefore a need for methods that allow for the genomewide expression profiling of formalin-fixed tissue samples, which are routinely collected in the clinical setting. Such approaches have been described,17 but their extensive validation has yet to be reported. We describe here a DASL-based method capable of profiling approximately 6000 human transcripts, and we have tested the method on more than 2000 formalin-fixed, paraffin-embedded blocks collected as long as 24 years ago. Through the assay of 6000 genes across the genome that show maximal variation in expression, this approach is expected to capture the bulk of transcriptional differences in any collection of samples. However, recent increases in array density support the analysis of all human genes on a single array (whole-genome DASL assay, Illumina).
The DASL-based discovery method that we describe here should be distinguished from candidate-gene profiling methods based on the reverse transcriptase (RT)-PCR assay, such as those used in the commercially available OncotypeDx test for determining the prognosis in patients with breast cancer.18 Whereas standard RT-PCR methods can measure a small number of transcripts in formalin-fixed, paraffin-embedded samples, genomewide discovery studies are not feasible with the use of RT-PCR–based methods. In addition, we speculate that the use of formalin-fixed, paraffin-embedded tissue specimens will aid the transition from exploratory research to clinical implementation. We applied the DASL profiling method to an increasingly important challenge in the care of patients with hepatocellular carcinoma. Tumors are often small at the time of diagnosis (owing to increased surveillance and advanced imaging in patients at risk), and existing prognostic factors are less informative for patients with small tumors than for those with larger tumors.
We did not observe a significant association between the expression profiles of the tumors themselves and the outcome for patients with surgically resected early hepatocellular carcinoma. In contrast, others have described tumor-derived prognostic signatures for hepatocellular carcinoma.15,19 The populations of patients in those studies, however, tended to have more advanced disease. Our training set primarily exhibited a pattern of late recurrence that is typical of small tumors.1,9 Accordingly, it is likely that early recurrence (reflecting locally invasive and incompletely resected tumor) is associated with molecular features of the primary tumor, but such features are not associated with late recurrences, which seem to result from new primary tumors arising in a damaged organ (the “field effect”) rather than the proliferation of residual tumor cells derived from the original tumor.
Also supporting the concept that late recurrence of hepatocellular carcinoma represents new primary tumors in patients at risk, we found little correlation between the molecular characteristics of tumors resected at initial diagnosis and those from the same patients at the time of recurrence. In particular, the results of clonality analysis indicated that the late recurrences of hepatocellular carcinoma tended to derive from a different clone than the preceding primary tumors. In addition, the obvious measures of liver damage (e.g., the extent of cirrhosis and the Child–Pugh stage20) were not associated with survival in our study, given that we restricted our analysis to patients with preserved liver function. Our findings indicate a field effect, in which environmental exposure (e.g., viral infection) leads to an increased potential for future malignant transformation. This has in general been overlooked by genomic approaches to studying cancer that have focused only on tumor cells. Our results suggest that a gene-expression signature can serve as a sensitive “readout” of the biologic state of the liver in at-risk patients. It is likely that the survival signature reflects the extent of liver damage and the presence or absence of a proinflammatory milieu, which is mediated in part by gene products involved in an inflammatory response. A heritable basis for the signature, although improbable, cannot be ruled out. Additional work is needed to fully understand the biologic basis of the signature.
Further clinical validation of the survival signature will be needed before it is introduced into clinical practice; our observation that the signature is associated with the outcome across heterogeneous populations of patients is encouraging. We envision the use of this test to identify the patients at highest risk for recurrence of hepatocellular carcinoma and to target intensive clinical follow-up or chemopreventive strategies in such patients.21 This article (10.1056/NEJMoa0804525) was published at www.nejm.org on October 15, 2008.
Supplementary Material
Acknowledgments
Supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK076986-01, to Dr. Llovet), the National Cancer Institute (5U54 CA112962-03, to Dr. Golub), the Samuel Waxman Cancer Research Foundation (to Dr. Llovet), the Spanish National Health Institute (SAF-2007-61898, to Dr. Llovet), Institució Catalana de Recerca i Estudis Avançats (to Dr. Llovet), Centro de Investigaciónes en Red de Enfermedades Hepáticas y Digestivas (to Drs. Llovet and Bruix), the Fund for Health of Spain of the Institute of Health Carlos III (PI05-0150, to Dr. Bruix), the National Institutes of Health (DK37340, to Dr. Friedman), the Italian Association for Cancer Research (to Dr. Mazzaferro), Helse Vest and Norwegian Cancer Society, Harald Andersens grant (to Dr. Salvesen), the Charles A. King Trust fellowship (to Dr. Hoshida), and Fundación Pedro Barrié de la Maza, the Sheila Sherlock Fellowship, and the National Cancer Center Fellowship (all to Dr. Villanueva).
We thank David Peck, Jun Lu, Aravind Subramanian, and Oleg Iartchouk for technical advice; Joshua Gould, Heidi Kuehn, and Barbara Hill for technical help; David Harrington for critical reading of a draft of the manuscript, and Mariko Kobayashi and Jadwiga Grabarek for general support. Prostate samples and lymphoma cell lines for pilot DASL experiments were kindly provided by Sunita Setlur, Mark Rubin, Kunihiko Takeyama, and Jeffery L. Kutok. Single-nucleotide-polymorphism profiling data for endometrial, ovarian, and renal cancers and lymphoma were provided by Rameen Beroukhim, Matthew Meyerson, Mark Rubin, Stefano Monti, and Margaret Shipp.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Bruix J. Novel advancements in the management of hepatocellular carcinoma in 2008. J Hepatol. 2008;48 Suppl 1:S20–S37. doi: 10.1016/j.jhep.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda K, Arase Y, Saitoh S, et al. Interferon beta prevents recurrence of hepatocellular carcinoma after complete resection or ablation of the primary tumor: a prospective randomized study of hepatitis C virus-related liver cancer. Hepatology. 2000;32:228–232. doi: 10.1053/jhep.2000.9409. [DOI] [PubMed] [Google Scholar]
- 4.Muto Y, Moriwaki H, Ninomiya M, et al. Prevention of second primary tumors by an acyclic retinoid, polyprenoic acid, in patients with hepatocellular carcinoma. N Engl J Med. 1996;334:1561–1567. doi: 10.1056/NEJM199606133342402. [DOI] [PubMed] [Google Scholar]
- 5.Takayama T, Sekine T, Makuuchi M, et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet. 2000;356:802–807. doi: 10.1016/S0140-6736(00)02654-4. [Erratum, Lancet 2000;356:1690.] [DOI] [PubMed] [Google Scholar]
- 6.Lau WY, Leung TW, Ho SK, et al. Adjuvant intra-arterial iodine-131-labelled lipiodol for resectable hepatocellular carcinoma: a prospective randomised trial. Lancet. 1999;353:797–801. doi: 10.1016/s0140-6736(98)06475-7. [DOI] [PubMed] [Google Scholar]
- 7.Fan JB, Yeakley JM, Bibikova M, et al. A versatile assay for high-throughput gene expression profiling on universal array matrices. Genome Res. 2004;14:878–885. doi: 10.1101/gr.2167504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bibikova M, Talantov D, Chudin E, et al. Quantitative gene expression profiling in formalin-fixed, paraffin-embedded tissues using universal bead arrays. Am J Pathol. 2004;165:1799–1807. doi: 10.1016/S0002-9440(10)63435-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llovet JM, Schwartz M, Mazzaferro V. Resection and liver transplantation for hepatocellular carcinoma. Semin Liver Dis. 2005;2:181–200. doi: 10.1055/s-2005-871198. [DOI] [PubMed] [Google Scholar]
- 10.Imamura H, Matsuyama Y, Tanaka E, et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J Hepatol. 2003;38:200–207. doi: 10.1016/s0168-8278(02)00360-4. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 13.Mazzaferro V, Romito R, Schiavo M, et al. Prevention of hepatocellular carcinoma recurrence with alpha-interferon after liver resection in HCV cirrhosis. Hepatology. 2006;44:1543–1554. doi: 10.1002/hep.21415. [DOI] [PubMed] [Google Scholar]
- 14.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 15.Lee JS, Chu IS, Heo J, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–676. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- 16.Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 17.Coudry RA, Meireles SI, Stoyanova R, et al. Successful application of microarray technology to microdissected formalin-fixed, paraffin-embedded tissue. J Mol Diagn. 2007;9:70–79. doi: 10.2353/jmoldx.2007.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Habel LA, Shak S, Jacobs MK, et al. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res. 2006;8:R25–R25. doi: 10.1186/bcr1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye QH, Qin LX, Forgues M, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–423. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 20.Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. doi: 10.1002/bjs.1800600817. [DOI] [PubMed] [Google Scholar]
- 21.Llovet JM, Di Bisceglie AM, Bruix J, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:698–711. doi: 10.1093/jnci/djn134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.