

Abstract
In the mammalian auditory system, the synapse between efferent olivocochlear (OC) neurons and sensory cochlear hair cells is cholinergic, fast, and inhibitory. This efferent synapse is mediated by the nicotinic α9α10 receptor coupled to the activation of SK2 Ca2+-activated K+ channels that hyperpolarize the cell. So far, the ion channels that support and/or modulate neurotransmitter release from the OC terminals remain unknown. To identify these channels, we used an isolated mouse cochlear preparation and monitored transmitter release from the efferent synaptic terminals in inner hair cells (IHCs) voltage clamped in the whole-cell recording configuration. Acetylcholine (ACh) release was evoked by electrically stimulating the efferent fibers that make axosomatic contacts with IHCs before the onset of hearing. Using the specific antagonists for P/Q- and N-type voltage-gated calcium channels (VGCCs), ω-agatoxin IVA and ω-conotoxin GVIA, respectively, we show that Ca2+ entering through both types of VGCCs support the release process at this synapse. Interestingly, we found that Ca2+ entering through the dihydropiridine-sensitive L-type VGCCs exerts a negative control on transmitter release. Moreover, using immunostaining techniques combined with electrophysiology and pharmacology, we show that BK Ca2+-activated K+ channels are transiently expressed at the OC efferent terminals contacting IHCs and that their activity modulates the release process at this synapse. The effects of dihydropiridines combined with iberiotoxin, a specific BK channel antagonist, strongly suggest that L-type VGCCs negatively regulate the release of ACh by fueling BK channels that are known to curtail the duration of the terminal action potential in several types of neurons.
Introduction
In the auditory system of rodents, synapses between sensory inner hair cells (IHCs) and the dendrites of primary afferent spiral ganglion neurons in the cochlea are functional before the onset of hearing (Beutner and Moser, 2001; Glowatzki and Fuchs, 2002). Neonatal IHCs fire calcium action potentials during this prehearing period (Kros et al., 1998; Glowatzki and Fuchs, 2000; Marcotti et al., 2003) driven mainly by ATP released from nearby supporting cells (Tritsch et al., 2007). These Ca2+ spikes promote the release of glutamate at the first auditory synapse, which helps establish and refine synaptic connections in the auditory pathway (Beutner and Moser, 2001; Glowatzki and Fuchs, 2002; Erazo-Fischer et al., 2007; Tritsch and Bergles, 2010). During this period, IHCs are transiently innervated by cholinergic fibers from the medial olivocochlear (MOC) system (Liberman et al., 1990; Simmons, 2002). This synapse is inhibitory and modulates the spiking frequency of IHCs (Glowatzki and Fuchs, 2000; Marcotti et al., 2004; Goutman et al., 2005). Therefore, it has been suggested that olivocochlear (OC) efferents are involved in the proper establishment of the auditory pathways (Beutner and Moser, 2001; Goutman et al., 2005).
The postsynaptic events after activation of the nicotinic cholinergic receptor (nAChR) at this synapse are well characterized. Current data indicate that activation of the α9α10 nAChRs (Elgoyhen et al., 2001) leads to an increase in intracellular Ca2+ and the subsequent opening of Ca2+-activated SK2 channels that hyperpolarize the cells (Fuchs, 1996; Glowatzki and Fuchs, 2000; Oliver et al., 2000; Katz et al., 2004; Gomez-Casati et al., 2005). Previous work showed that transmitter release at the neonatal rat efferent–IHC synapse has a low probability of release that significantly increases after high-frequency stimulation (Goutman et al., 2005).
In mammals, fast synaptic transmission is supported by multiple types of voltage-gated calcium channels (VGCCs) (Katz et al., 1997; Iwasaki and Takahashi, 1998; Iwasaki et al., 2000; Reid et al., 2003; Snutch, 2005). VGCCs are transmembrane proteins formed by four different subunits (α1, α2-δ, β, and sometimes γ). The biophysical and pharmacological diversity of VGCCs that are classified as L, P/Q, N, R, and T type based on their biophysical and pharmacological characteristics arises primarily from the existence of multiple pore-forming α1 subunits (Catterall, 1998). In some neurons, the entry of Ca2+ that triggers transmitter release also activates calcium- and voltage-sensitive K+ channels (BK) that accelerate the repolarization of the terminal membrane (Storm, 1987; Berkefeld and Fakler, 2008) and thereby exert negative feedback on the release process (Petersen and Maruyama, 1984; Robitaille et al., 1993; Lingle et al., 1996; Raffaelli et al., 2004). Investigations of BK channels in various types of neurons show that the activation of these channels requires the delivery of Ca2+ through colocalized VGCCs (Robitaille et al., 1993; Marrion and Tavalin, 1998; Sun et al., 2003; Berkefeld and Fakler, 2008). This work is the first description of the Ca2+ and Ca2+-activated K+ channels that support and regulate the release of acetylcholine (ACh) at the synapse between efferent fibers and sensory hair cells of the mammalian cochlea.
Materials and Methods
Animal procedures and isolation of the organ of Corti.
Procedures for preparing and recording from the postnatal mouse organ of Corti were essentially identical to those published previously (Glowatzki and Fuchs, 2000; Katz et al., 2004). Briefly, apical turns of the organ of Corti were excised from BalbC mice of either sex between postnatal day 9 (P9) and P11 (day of birth was considered P0) and used within 3 h. Cochlear preparations were placed in the chamber for electrophysiological recordings, mounted under a Leica LFS microscope, and viewed with differential interference contrast using a 40× water-immersion objective and a camera with contrast enhancement (Hamamatsu C275410). All experimental protocols were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (publication number 80-23, revised in 1978).
Electrophysiological recordings.
IHCs were identified visually and by the size of their capacitance (7–12 pF) and by their characteristic voltage-dependent currents (Kros et al., 1998). The cochlear preparation was superfused continuously by means of a peristaltic pump (Gilson Minipulse 3, with 8 channels; Bioesanco) containing an extracellular saline solution of an ionic composition similar to that of the perilymph (in mm): 155 NaCl, 5.8 KCl, 1.3 CaCl2, 0.7 NaH2PO4, 5.6 d-glucose, and 10 HEPES buffer, pH 7.4. Working solutions containing the different drugs and toxins used were made up in this same saline and delivered through the perfusion system. The pipette solution contained the following (in mm): 150 KCl, 3.5 MgCl2, 0.1 CaCl2, glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (5 mm EGTA), 5 HEPES buffer, 2.5 Na2ATP, pH 7.2. Some cells were removed to access IHCs, but mostly the pipette moved through the tissue using positive fluid flow to clear the tip. Currents in IHCs were recorded in the whole-cell patch-clamp mode using an Axopatch 200B amplifier, low-pass filtered at 2–10 kHz, and digitized at 5–20 kHz with a Digidata 1322A board (Molecular Devices). Recordings were made at room temperature (22–25°C). Glass pipettes (1.2 mm inner diameter) had resistances of 7–10 MΩ. Indicated holding potentials were not corrected for liquid junction potentials (−4 mV).
Electrical stimulation of the MOC efferent axons.
Neurotransmitter release was evoked by bipolar electrical stimulation of the MOC efferent axons as described previously (Goutman et al., 2005). Briefly, the electrical stimulus was delivered via a 20- to 80-μm-diameter theta glass pipette placed at 20–60 μm modiolar to the base of the IHC under study, voltage clamped at −90 mV. The position of the pipette was adjusted until postsynaptic currents in the IHC were consistently activated. An electrically isolated constant current source (model DS3; Digitimer) was triggered via the data-acquisition computer to generate pulses up to 30 mA, 200–2000 μs.
Estimation of the quantal content of transmitter release.
The quantal content of transmitter release (m) was estimated as the ratio between the mean amplitude of evoked synaptic currents (eIPSC) and the mean amplitude of spontaneous synaptic currents (sIPSC) (del Castillo and Katz, 1957). To estimate eIPSC mean amplitude, protocols of 200 stimuli were applied at a frequency of 1 Hz. sIPSC were recorded during and after the stimulation protocols. To study the calcium cooperativity of the MOC–IHC synapse, the relationship between transmitter release and Ca2+ was evaluated by estimating m under different external Ca2+ concentrations in the absence or presence of 0.9 mm Mg2+ (the physiological Mg2+ concentration in the perilymph that bathes the basolateral membrane of IHCs). Mg2+ was used as a control to compare our data with those previously reported for the relationship between transmitter release and extracellular Ca2+ but was not used in the rest of the experiments reported in this work, as it is known to partially block the postsynaptic α9α10 nAChR (Katz et al., 2000; Weisstaub et al., 2002; Gomez-Casati et al., 2005). Data were fitted with a power equation: m = K([Ca2+]o)n, where K is the proportionality constant and n is the coefficient of the power relation (Dodge and Rahamimoff, 1967). Cooperativity of transmitter release (n) was estimated by fitting all the data points (m values) obtained in the different cells after variation of the extracellular calcium concentration.
Percentage quantal content (%m) was calculated as , where mc is the estimation of m in the control condition and mt is the quantal content estimated after incubation of the preparation with the drug or toxin under study for the time specified in Results (treated preparations). To quantify the effects of each drug or toxin, the preparation was incubated for the time necessary to reach a plateau in the observed effect. In addition, the same procedure was used with preparations subjected to the same protocols, for the same periods of time, but in which no drug or toxin was added (control preparations) (see Fig. 1b). Failures of release were also computed in the absence or presence of the different drugs or toxins to double check the quantal content of transmitter release under the different conditions by the failures method [mf = ln N/N0, where N0 is the number of failures and N is the total number of successive trials (100 trials at a frequency of 1 Hz)] (Hubbard et al., 1969). Failure analysis to calculate the quantal content of transmitter release was only used in those cases in which the drug or toxin caused an increase in the number of failures. The failures method can only be used when the probability or release is very low and thus follows a Poisson distribution (Hubbard et al., 1969). Therefore, in the cases in which the treatment caused a reduction in the number of failures, we only report the percentage of failures during 100 successive trials at 1 Hz (N0/N)*100 in the absence or presence of the drug or toxin tested.
Figure 1.

Quantal content of evoked release at the mouse transient MOC–IHC synapse. a, Schematic representation of the mouse cochlear preparation used in the present study. MOC efferent axons were stimulated using a bipolar electrode placed modiolar to the base of the IHCs, and the postsynaptic responses were recorded from IHCs using a patch pipette at a holding potential of −90 mV. b, Graph illustrating that the quantal content of transmitter release (m) at this synapse was low (∼1) and stable over recording times of at least 1 h. m was calculated as the ratio between the mean amplitude of eIPSCs (determined from protocols consisting of 200 stimuli applied at a frequency of 1 Hz) and the mean amplitude of sIPSCs. c, ACh release at the efferent–IHC synapse was sensitive to variations in the external Ca2+ concentration. Data were fitted according to Dodge and Rahamimoff (1967) with a power equation m = k(Ca2+)n. Best fit was obtained with n = 2.49 ± 0.68 (n = 8–15 IHCs). Error bars are SEM.
Statistical comparisons (unpaired, one-tailed, Student's t test) were made between control and treated preparations. Values of p < 0.05 were considered significant. All data were expressed as mean ± SEM. Synaptic currents and potentials were analyzed with Minianalysis (Synaptosoft) and Clampfit 9.2 (Molecular Devices).
Immunostaining.
Immunofluorescent staining of organs of Corti from BALB/c mice (age P9–P22; Charles River Laboratories) was performed as described previously (McLean et al., 2009) using the following antibodies: rabbit polyclonal anti-BK channel (APC021; 1:500; Alomone Labs), mouse monoclonal anti-BK channel (L6/23; 1:500; gift from Dr. J. Trimmer, University of California at Davis, Davis, CA), mouse monoclonal anti-calretinin (MAB1568; 1:1000; Millipore), goat polyclonal anti-synapsin (sc-1739; 1:500; Santa Cruz Biotechnology), mouse monoclonal anti-CaV1.2 channel (L57/46; 1:50; NeuroMab), rabbit polyclonal anti-CaV1.2 channel (ACC-003; 1:50; Alomone Labs), and rabbit polyclonal anti-CaV1.3 channel (ACC-005; 1:50; Alomone Labs). The specificity of all primary antibodies was verified by Western blot by the supplier. Secondary antibodies (Alexa Fluor 488 and 594 generated in either goat or donkey) were purchased from Invitrogen and diluted 1:1000 in blocking buffer.
Microscopy and image analysis.
Fluorescent images were acquired using an Olympus Fluoview FV1000 confocal microscope with a 60× Olympus PlanoApo oil-immersion lens (numerical aperture 1.42) under the control of the Olympus Fluoview FV1000 version 1.6a software (Olympus). Z-stacks were collected at 0.2–0.5 μm. Three-dimensional (3D) reconstructions of confocal z-stacks were generated and analyzed using Imaris 6.4 3D image visualization and analysis software (Bitplane). All images are presented as 3D reconstructions through the z-axis.
Drugs and toxins.
Stock solutions of dihydropyridines (DHPs; nitrendipine, nifedipine, and Bay-K) were prepared in dimethylsulfoxide (final concentration, ≤0.1%). Peptidic toxin stock solutions were prepared in distilled water. Drugs and reagents were purchased from Sigma-Aldrich; ω-conotoxin GVIA was purchased from Alomone Labs, and ω-conotoxin IVA was from Peptides International. All drugs and toxins were thawed and diluted in the extracellular solution just before use.
Results
Quantal content of transmitter release and Ca2+ cooperativity at the mouse transient MOC–IHC synapse
To investigate whether the amount of transmitter released at the mouse transient MOC–IHC synapse is similar to that reported in rats at the same postnatal age (Goutman et al., 2005), we monitored electrically evoked and spontaneously occurring cholinergic postsynaptic currents (eIPSCs and sIPSCs) by whole-cell recordings in P9–P11 IHCs voltage clamped at −90 mV (see schematic representation of this procedure in Fig. 1a). The quantal content of transmitter release (m) was computed as the ratio of the average amplitudes between eIPSC and sIPSCs (see Materials and Methods). In the mouse cochlear preparation, m was 1.1 ± 0.1 (n = 14 cells, 11 mice), a value similar to that reported in rats of the same age group (m = 1 ± 0.5) (Goutman et al., 2005). The quantum size (q), as evaluated by the mean amplitude of sIPSCs, −16.5 ± 0.5 pA (n = 2311 events, 44 IHCs, 11 mice), was similar to that reported in rats (q = −18 ± 2 pA) (Goutman et al., 2005).
As one of the aims of the present study was to investigate the types of Ca2+ channels that support release at this synapse, we checked the stability of the quantal content during the times necessary to thoroughly test the effects of toxins specifically targeting these channels. Figure 1b illustrates that m, evaluated by 200 stimuli given at 1 Hz each 5 min during 60 min, remained constant (30 min, m = 1.1 ± 0.2, n = 8; 60 min, m = 1.2 ± 0.4, n = 4). A one-way ANOVA test was used to compare data points taken at each 5 min; p = 0.99). Similar results were obtained when the quantal content was calculated by computing the failures of release (mf = ln N/N0, see Materials and Methods; 30 min, mf = 1.0 ± 0.3; 60 min, mf = 1.4 ± 0.7).
In most fast chemical synapses, as first described at the frog neuromuscular junction by Dodge and Rahamimoff (1967), the relationship between external Ca2+ and the amount of transmitter released by the synaptic terminal is highly nonlinear (Mintz et al., 1995; Borst and Sakmann, 1996; Takahashi et al., 1996; Wu et al., 1999; Rosato-Siri et al., 2002). Therefore, small variations in the amount of Ca2+ in the external milieu or in the activity of terminal membrane VGCCs might exert a great impact on transmitter release. As illustrated in Figure 1c, at this synapse changes in the extracellular Ca2+ concentration change the probability of release in a cooperative way. The best fit to the power equation relating quantal content to the external calcium concentration (see Materials and Methods) is as follows: m = K([Ca2+]o)n (Dodge and Rahamimoff, 1967) was obtained with a power coefficient (n) of 2.49 ± 0.68, n = 8–15 data points for each [Ca2+]o tested, obtained in 15 IHCs from 10 mice. The addition of a physiological concentration of Mg2+, similar to that present in the perilymph bathing the basolateral membranes of IHCs (0.9 mm), did not produce any significant change in the sensitivity of transmitter release to variations in external Ca2+ (the best fit to the power equation was with 2.57 ± 0.61 (n = 8–15 data points for each [Ca2+]o tested, obtained in 15 IHCs from 10 mice). The addition of this physiological concentration of Mg2+ to the external solution caused a slight positive shift in the m-[Ca2+]o curve (data not shown). We did not further investigate the effects of Mg2+ on the release process at this synapse (Dodge and Rahamimoff, 1967) because this divalent cation also blocks the postsynaptic α9α10 nAChR (Weisstaub et al., 2002; Gomez-Casati et al., 2005).
Types of calcium channels that support transmitter release at the transient efferent-IHC synapse
The two high-voltage VGCCs that more commonly support fast release at mammalian synapses, namely P/Q and N (Reid et al., 2003; Snutch, 2005), can be readily distinguished by their sensitivity to the spider toxin ω-agatoxin IVA (ω-AgaIVA) and to the marine snail toxin ω-conotoxin GVIA (ω-CgTx), which specifically block P/Q- and N-type VGCCs, respectively (Mintz et al., 1992; Olivera et al., 1994; Randall and Tsien, 1995; Doering and Zamponi, 2003). To study which types of Ca2+ channels support transmitter release at the transient efferent synapse to IHCs, we tested the effects of the above-mentioned toxins on the quantal content of electrically evoked transmitter release. The amplitude of eIPSCs was greatly reduced after 15 min incubation with 200 nm ω-AgaIVA (Fig. 2a); whereas sIPSCs amplitude was not significantly affected by this toxin (control, −11.8 ± 0.6 pA; 200 nm ω-AgaIVA, −11.1 ± 0.8 pA; p = 0.1279; 753 events, 4 cells, 4 mice) (Fig. 2d). Under this condition, m was reduced to 44.6 ± 6.8% of the initial value (m = 0.9 ± 0.1; p < 0.001, 6 cells from 6 mice) (Fig. 2c). Consistently, failures of release ([N0/N]*100) significantly increased from 40.0 ± 6.8% to 70.5 ± 5.1% (p < 0.001, 6 cells, 6 mice). The quantal content calculated by the failures method was reduced from mf = 1.1 ± 0.3 (control) to mf = 0.4 ± 0.1 (ω-AgaIVA). We did not test higher concentrations of ω-AgaIVA because application of 200 nm ω-AgaIVA for 15 min has been reported to completely block both P- and Q-type channels in other preparations (Mintz et al., 1992; Randall and Tsien, 1995; Katz et al., 1997; Bourinet et al., 1999). Moreover, higher concentrations of this toxin have nonspecific effects on N-type VGCCs (Sidach and Mintz, 2000). Thus, this result indicates that P/Q-type VGCCs contribute to ACh release at this synapse only partially. Therefore, we also evaluated the effects of the N-type VGCC antagonist ω-CgTx (300 nm). This toxin also greatly diminished eIPSC amplitude (Figs. 2a,b) without affecting the amplitude of sIPSCs (control, −13.4 ± 0.9 pA; 300 nm ω-CgTx, −13 ± 0.9 pA; p = 0.4095; 1002 events, 7 cells from 7 mice) (Fig. 2d). In this case, transmitter release was also partially blocked after 20 min of incubation with 300 nm ω-CgTx, m was reduced to 55.8 ± 15.2 of the initial value (Fig. 2b) (control, m = 1.1 ± 0.5; p < 0.01, n = 6 cells from 6 mice). In agreement with this result, failures of release ([N0/N]*100) significantly increased from 37.8 ± 9.8 to 49.7 ± 13.2% (p < 0.05; 6 cells from 6 mice), and mf was reduced from 0.9 ± 0.2 (control) to 0.6 ± 0.2 (ω-CgTx). As ω-CgTx is sometimes used at higher concentrations to block N-type VGCCs in other preparations (Boland et al., 1994; Katz et al., 1997), we also tested the effects of a 1 μm concentration. This higher concentration, however, did not further reduce m (p = 0.5644 with respect to the reduction observed with 300 nm ω-CgTx; n = 3 cells from 3 mice; data not illustrated).
Figure 2.
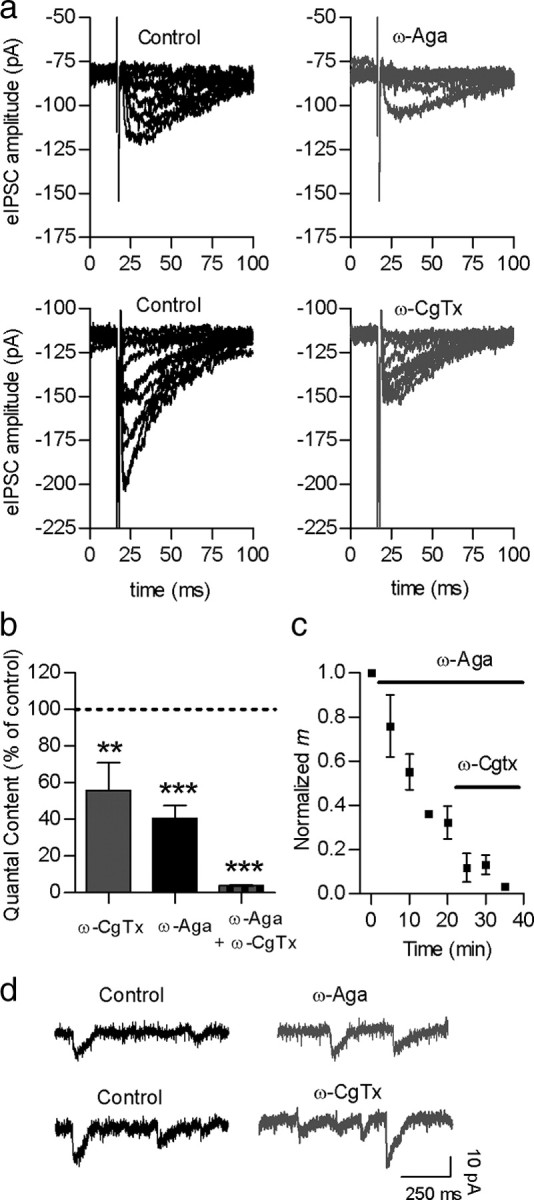
Both P/Q- and N-type VGCCs support transmitter release at the MOC–IHC synapse. a, Representative traces of eIPSCs recorded at a membrane potential of −90 mV before and after incubation with either 200 nm ω-Aga IVA or 300 nm ω-CgTx, specific antagonists of P/Q- and N-type VGCCs, respectively. b, Bar graph illustrating the effects on m of each of the toxins applied separately or at the same time. c, Graph showing that the sequential application of ω-Aga IVA and ω-CgTx almost completely abolished the release of ACh. d, Representative records of sIPSC before and after incubation with ω-Aga IVA (top) and ω-CgTx (bottom) show no change in sIPSC amplitude. Error bars are SEM. **p < 0.01, ***p < 0.001.
We tested the effects of both toxins applied together to evaluate whether transmission was completely blocked by blocking both P/Q- and N-type VGCCs. Applying 200 nm ω-AgaIVA and 300 nm ω-CgTx, either sequentially or simultaneously, almost completely blocked the release of ACh. As illustrated in Figure 2, b and c, in the presence of both toxins m was reduced to 3.9 ± 0.4% of the control value (control, m = 0.8 ± 0.2 p < 0.0001, n = 6 cells from 6 mice). Consistently, the quantal content evaluated by the failures method was reduced from mf = 0.8 ± 0.3 (control) to mf = 0.07 ± 0.01(ω-AgaIVA plus ω-CgTx). These results indicate that calcium entering through both P/Q and N-type VGCC support the release of ACh with little, if any, participation of R- or L-type VGCCs in triggering this process. Notwithstanding, as L-type VGCCs were shown to be present in the cochlea (Platzer et al., 2000; Waka et al., 2003; Brandt et al., 2005; Layton et al., 2005; Knirsch et al., 2007) and to participate in the release process in many fast synapses under certain particular experimental or developmental conditions (Catterall, 2000; Flink and Atchison, 2003; Perissinotti et al., 2008), we investigated whether they had any role in the release process at the OC–IHC synapse.
L-type calcium VGCCs negatively regulate the amount of evoked transmitter release at this transient efferent synapse
L-type VGCCs support the graded release of neurotransmitters and hormones and, although they are present at many synaptic terminals, they do not support transmitter release at fast synapses under normal conditions (Stanley and Atrakchi, 1990; Catterall, 2000). They have, however, been shown to participate in this process at reinnervating and developing neuromuscular synapses (Katz et al., 1996; Sugiura and Ko, 1997), and also under other particular conditions (Urbano et al., 2001; Flink and Atchison, 2003; Perissinotti et al., 2008). In addition, they have been shown to be involved in the regulation of transmitter release at several synapses either by the activation of calcium-dependent conductances (Robitaille et al., 1993; Sugiura and Ko, 1997; Marrion and Tavalin, 1998; Prakriya and Lingle, 1999; Sun et al., 2003; Berkefeld et al., 2006; Loane et al., 2007; Marcantoni et al., 2007; Muller et al., 2007; Berkefeld and Fakler, 2008; Fakler and Adelman, 2008; Grimes et al., 2009) or by the activation of second-messenger cascades that eventually also regulate the influx of Ca2+ entering synaptic terminals (Sugiura and Ko, 1997; Jensen et al., 1999). L-type VGCCs are highly sensitive to micromolar concentrations of DHPs that, either negatively (i.e., nifedipine, nitrendipine) or positively (i.e., Bay K), modulate their activity (Brown et al., 1984; Hess et al., 1984; Doering and Zamponi, 2003; Catterall and Few, 2008). Therefore, to study the participation of L-type VGCC in ACh release at the transient efferent synapse, we tested the effects of nifedipine, nitrendipine, and Bay-K on m.
As illustrated by the representative records in Figure 3a, after 5 min of incubating the cochlear preparation with 3 μm nifedipine, the amplitude of eIPSCs increased, and the failures of release diminished ([N0/N]*100 = 28.7 ± 7.1% and 8.1 ± 3.7% failures in the absence or presence of nifedipine, respectively; p < 0.01; 7 cells from 7 mice). No effect was observed on the amplitude of sIPSCs (p = 0.3654; 7 cells, 1670 events) (Fig. 3c,d). In the presence of this drug, m increased to 195.2 ± 22.6% of the initial value (m = 1.5 ± 0.2; n = 7 cells from 7 mice; p < 0.0005) (Fig. 3b).
Figure 3.

L-type VGCC activity negatively regulates transmitter release at the MOC–IHC synapse. a, Representative traces of eIPSCs recorded at a holding potential of −90 mV before and after incubation with different DHPs: nifedipine (Nif), nitrendipine (Nitren), and Bay-K. b, Bar graph showing that both L-type VGCC antagonists Nif (3 μm) and Nitren (10 μm) caused a significant increase in m, whereas the agonist Bay-K (10 μm) caused a significant reduction in this parameter. c, Bar graph illustrating the effects of DHPs on the amplitude of sIPSC. Nifedipine at a concentration of 3 μm had no effects on this parameter. Both Nitrendipine and Bay-K, which were used at higher concentrations (10 μm), slightly but significantly reduced sIPSC amplitude. d, Representative traces showing the average of 10–15 sIPSCs before and after incubation with DHPs. Error bars are SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
In addition, in the presence of 10 μm nitrendipine, there was also an increase in the amplitude of eIPSCs with a reduction in the number of failures ([N0/N]*100 = 41.0 ± 9.6% and 24.8 ± 8.9 failures in the absence or presence of nitrendipine, respectively; p < 0.005; 5 cells from 4 mice). This drug, however, slightly but significantly, reduced the amplitude of sIPSCs (control, −14.6 ± 1.2 pA; nitrendipine, −11.5 ± 0.3; p < 0.05; n = 5 cells, 642 events from 4 mice), suggesting that apart from targeting the presynaptic L-type VGCC, it could be affecting the activity of the postsynaptic α9α10 nAChR receptor (Elgoyhen et al., 2001; Gomez-Casati et al., 2005). Notwithstanding, when normalizing the mean amplitude of eIPSCs to the mean amplitude of sIPSCs to calculate m, we found that the amount of transmitter release was increased to 206.1 ± 31.6% of the initial value (m = 1.0 ± 0.2; p < 0.05; n = 5 from 4 mice) by this DHP.
When the cochlear preparation was incubated with Bay-K (10 μm), a DHP that increases the current through L-type VDCC by stabilizing their open state (Brown et al., 1984; Hess et al., 1984; Doering and Zamponi, 2003; Catterall and Few, 2008), the amplitude of eIPSCs was reduced, the number of failures of release increased ([N0/N]*100 = 20.2 ± 3.5% and 35.3 ± 6.6% failures in the absence or presence of Bay-K, respectively; p < 0.05; 4 cells from 4 mice) (Fig. 3a,b), and mf was reduced from 0.9 ± 0.2 (control) to 0.6 ± 0.2 (Bay-K). This drug had no effect on the amplitude of sIPSCs (p > 0.05, n = 4 cells, 831 events) (Fig. 3c,d). Consistent with the positive effect that the antagonist DHPs (nifedipine and nitrendipine) exerted on m, Bay-K had a negative effect on this parameter and therefore significantly reduced the amount of transmitter release to 50.2 ± 5.6% of the initial value (m = 1.4 ± 0.1; p < 0.01; n = 4 cells from 4 mice) (Fig. 3b).
BK channels also negatively regulate the amount of evoked release at the transient efferent–IHC synapse
Our findings that L-type VGCC antagonists (nifedipine and nitrendipine) increase transmitter release while the L-type VGCC-positive modulator (Bay K) decreases transmitter release suggest that Ca2+ entering through L-type VGCCs somehow negatively modulate release at the efferent–IHC synapse. This reduction could be accomplished by either activating a Ca2+-dependent second-messenger cascade (Sugiura and Ko, 1997; Jensen et al., 1999) or by activating a Ca2+-dependent K+ conductance that would serve to accelerate the repolarization of the terminal membrane and thereby reduce the width of the action potential (Marcantoni et al.; Storm, 1987). Because BK Ca2+-activated K channels have been shown to contribute to the repolarization of the action potential (Storm, 1987) and influence the release of neurotransmitters in a variety of other synapses (Petersen and Maruyama, 1984; Robitaille et al., 1993; Lingle et al., 1996; Raffaelli et al., 2004), we investigated whether BK Ca2+-activated K channels were also involved in regulating release at the OC–IHC synapse.
To test the involvement of BK channels, we incubated the cochlear preparation with iberiotoxin (IbTx), a specific BK channel antagonist (Galvez et al., 1990). As illustrated by the representative records in Figure 4, after 5 min of incubating the cochlear preparation with 100 nm IbTx, there was a dramatic increase in the amplitude of eIPSCs and a reduction or complete disappearance in the number of failures of release ([N0/N]*100 = 14.2 ± 5.5% and 4.1 ± 3.2% failures in the absence or presence of IbTx, respectively; p < 0.01; 7 cells from 7 mice). This toxin did not exert any effect on the amplitude of sIPSCs (p = 0.1812; n = 7 cells from 7 mice; 2559 events) (Fig. 4, inset). In the presence of IbTx, the quantal content of transmitter release increased to 243.8 ± 45% of the initial value without toxin (control, m = 2.2 ± 0.7; p < 0.0001, n = 7 cells) (Fig. 4). This result indicates that the activity of BK channels reduces the amount of transmitter released per nerve impulse at the efferent–IHC synapse.
Figure 4.

BK channels also negatively regulate transmitter release at this synapse. a, Representative traces of eIPSCs recorded at a holding potential of −90 mV before (left) and after incubation (right) with 100 nm IbTx, a specific BK channel antagonist. The inset shows that IbTx did not affect the amplitude of sIPSCs, also recorded at a holding potential of −90 mV.
L-type VDCC and BK channels are functionally coupled at the efferent–IHC synapse
The observation that both BK and L-type VDCC antagonists cause a significant increase in the amount of transmitter released by these synaptic terminals suggests that as described in other preparations (Prakriya and Lingle, 1999; Sun et al., 2003; Grunnet and Kaufmann, 2004; Berkefeld et al., 2006; Marcantoni et al., 2007; Berkefeld and Fakler, 2008), there could be a functional coupling between these two types of ionic channels. To test this hypothesis, we first studied the effects of nifedipine in cochlear preparations preincubated with 100 nm IbTx to suppress the activity of BK channels. Under this condition, 3 μm nifedipine did not further increase the amount of transmitter being released (Fig. 5a) (control, m = 1.7 ± 0.5; IbTx, m = 3.3 ± 0. 7; IbTx plus nifedipine, m = 3.0 ± 0.6; n = 6 cells from 6 mice; p = 0.1550, comparing the latter two values). Consistently, if we first blocked L-type VGCCs with 3 μm nifedipine, 100 nm IbTx failed to further increase the amount of transmitter released (Fig. 5b) (control, m = 1.3 ± 0.1; nifedipine, m = 2.5 ± 0. 4; nifedipine plus IbTx, m = 2.5 ± 0.5; n = 6 cells from 6 mice; p = 0.6868, comparing the latter two values).
Figure 5.

L-type VGCC and BK channels are functionally coupled. a, Bar graph showing that the effects of 3 μm nifedipine on transmitter release were occluded by prior incubation of the cochlear preparation with 100 nm IbTx. b, Bar graph illustrating that the effects of 100 nm IbTx on transmitter release were occluded by prior incubation of the cochlear preparation with 3 μm nifedipine. c, Bar graph shows that elevating external Ca2+ from 1.3 mm to 1.5 mm increases transmitter release even in the presence of 100 nm IbTx, suggesting that the release “machinery” of this synapse is not completely saturated when BK channels are blocked. d, Bar graph showing that Bay-K was not able to reduce m if BK channels had been previously blocked by IbTx. Error bars are SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
To test whether our inability to further increase the amount of evoked neurotransmitter released in response to serial application of BK channel and VGCC antagonists was possibly caused by the functional coupling of these two channels and not simply by saturation of evoked release, we performed two additional experiments. First, we preincubated the preparation with 100 nm IbTx in 1.3 mm Ca2+, and we then increased the Ca2+ concentration to 1.5 mm. As shown in Figure 5c, the quantal content of transmitter release significantly increased (control, m = 1.6 ± 0.3; IbTx, 1.3 mm Ca2+, m = 3.9 ± 0. 5; IbTx, 1.5 mm Ca2+, m = 6.4 ± 0.2; n = 3 cells from 3 mice; p < 0.05), indicating that the system was not saturated and that the amount of ACh being released significantly increased after increasing external Ca2+. The next control was to use the positive L-type VGCC modulator, Bay K, instead of nifedipine. As illustrated by the bar diagram in Figure 5d, 10 μm BayK did not exert any effect on m when BK channels were blocked (control, m = 1.2 ± 0.3; IbTx, m = 2.6 ± 0. 8; IbTx + BayK, m = 2.5 ± 0.6; n = 7 cells from 7 mice; p = 0.7344). Together, these results strongly suggest that Ca2+ entering through L-type VGCCs activate BK channels, thus reducing the amount of transmitter released by each nerve impulse. In addition, these results show that Ca2+ entering through P/Q- or N- type VGCC fails to activate BK channels. If Ca2+ entering through P/Q- and N-type VGCCs were activating BK channels, the amount of transmitter released by each nerve impulse should have increased after incubation with the BK channel antagonist, as was shown at the frog neuromuscular junction, where Ca2+ entering through N-type VGCCs support release and activate BK channels that are closely associated with the release sites (Robitaille et al., 1993).
BK channels are expressed at the efferent–IHC synapse
To verify the expression of BK channels in the efferent terminals that transiently contact IHCs, we immunostained identical cochlear preparations from P9–P11 mice with a rabbit polyclonal antibody against the BK channel and a mouse monoclonal antibody against calretinin, a cytoplasmic marker of IHCs as well as of the afferent fibers contacting them (Dechesne et al., 1991, 1993; Zheng and Gao, 1997). BK-positive puncta are clearly visible near the base of IHCs (Fig. 6a–c). Although these BK-positive puncta do not directly contact the base of IHCs, the pattern of immunoreactivity is, nevertheless, as would be expected for a cytoplasmic label of the postsynaptic cell (calretinin) and a membrane-bound protein of the presynaptic terminal (BK channel).
Figure 6.

Immunostaining reveals transient expression of BK channels in the region of the MOC efferent–IHC synapses. a, b, Cochlear preparations from P10 mice were immunostained with a rabbit polyclonal antibody against the BK channel (a) and a mouse monoclonal antibody against calretinin (b), a cytoplasmic marker of IHCs as well as the afferent fibers contacting IHCs. c, BK channel immunoreactivity is visible as small puncta in the region below IHCs. d, e, To verify the specificity of BK channel immunolabeling, cochlear preparations from similarly aged mice were immunostained with both a rabbit polyclonal (APC-021; d) and a mouse monoclonal (L6/23; e) antibody against the BK channel. f, The colocalized immunoreactivity suggests that both antibodies are specifically detecting the BK channel. g, h, To determine whether BK-positive puncta below IHCs are associated with efferent terminals, cochlear preparations were immunostained with a rabbit polyclonal antibody against the BK channel (g) and a goat polyclonal antibody against synapsin (h), a cytoplasmic marker of efferent presynapses. i, BK-positive puncta indeed correspond with synapsin-positive efferent terminals. j, k, Finally, cochlear preparations from P20 mice were immunostained with a rabbit polyclonal antibody against the BK channel (j) and a mouse monoclonal antibody against calretinin (k). l, At this age, BK channel immunoreactivity is no longer visible as small puncta below IHCs but is instead restricted to larger puncta surrounding the neck of IHCs. Scale bars, 10 μm.
Observation of BK-positive puncta at this age was initially surprising since previous experiments found no BK channel immunoreactivity in mice of this age (Pyott et al., 2004). However, longer fixation times in those previous experiments (2 h compared with ≤50 min in these experiments) most likely abolished BK channel immunoreactivity. Nonetheless, to confirm the specificity of BK channel immunoreactivity, we immunostained cochlear preparations from similarly aged mice with both a rabbit polyclonal and a mouse monoclonal antibody against the BK channel. These antibodies recognize discrete locations of the BK channel. (APC-021 recognizes an epitope between amino acid residues 1089 and 1196, and L6/23 recognizes an epitope between amino acid residues 682 and 859.) The colocalized immunoreactivity (Fig. 6d–f) suggests that both antibodies are specifically detecting the BK channel.
To determine whether the BK-positive puncta at the bases of IHCs indeed colocalize with efferent terminals, we immunostained cochlear preparations with a rabbit polyclonal antibody against the BK channel and a goat polyclonal antibody against synapsin, a cytoplasmic marker of efferent presynapses (Pyott et al., 2007; McLean et al., 2009). BK-positive puncta are clearly associated with synapsin-positive efferent terminals (Fig. 6g–i). We also observed BK-positive puncta further below the base of the IHCs (Fig. 6c,i) that associate with synapsin and, because of their location, may correspond to efferents of the lateral OC system contacting the afferent dendrites (Liberman et al., 1990; Simmons, 2002) or to medial efferents that might also transiently contact the afferent dendrites (Simmons et al., 1996).
Finally, we examined expression of the BK channel in cochlear preparations from hearing mice (P20–P22). At this age, BK channel immunoreactivity is no longer visible as small puncta contacting the bases of IHCs but is instead restricted to larger puncta surrounding the necks of these cells (Fig. 6j–l), consistent with previous reports (Pyott et al., 2004; Ruttiger et al., 2004; Hafidi et al., 2005). Although one group has reported BK channel immunoreactivity at the base of IHCs in similarly aged mice (Hafidi et al., 2005), we found no such immunoreactivity even in conditions of reduced fixation. Thus, the expression of the BK channel in efferent fibers contacting IHCs or IHC afferent fibers appears to be transient and likely restricted to the transient efferent innervation that occurs after birth and disappears after the onset of hearing (Liberman et al., 1990; Simmons, 2002; Katz et al., 2004).
In addition to characterizing the expression of BK channels by immunostaining, we also used immunofluorescence to identify the particular L-type VGCCs responsible for the activation of BK channels observed by electrophysiology in the efferent terminals. We immunostained organs of Corti from P9 and P10 mice with various antibodies against CaV1.2 and CaV1.3 using fixation conditions established previously (Platzer et al., 2000; Waka et al., 2003; Brandt et al., 2005; Layton et al., 2005; Knirsch et al., 2007). We observed no immunoreactivity with either of the two CaV1.2 antibodies examined (data not shown), supporting previous work reporting the absence of CaV1.2 immunoreactivity in the mouse organ of Corti before P14 (Waka et al., 2003). CaV1.3 immunoreactivity did not colocalize with BK channel immunoreactivity (data not shown) and, instead, appeared restricted to the inner and outer hair cell ribbon synapses as described previously (Knirsch et al., 2007; Zampini et al., 2010).
Discussion
Properties of the mouse efferent–IHC synapse
To identify the ion channels regulating synaptic transmission at the efferent–IHC synapse, we monitored electrically evoked and spontaneously occurring cholinergic postsynaptic currents in P9–P11 IHCs. The quantal content, m, of evoked release is low, as reported in the rat MOC–IHC synapse (Goutman et al., 2005). In addition, we determined that the relationship between m and the external Ca2+ concentration is nonlinear with a power coefficient of 2.5. This suggests the cooperative involvement of at least two Ca2+ in triggering the release of each vesicle of ACh (Dodge and Rahamimoff, 1967). This value falls in the lower range of those reported for other mammalian synapses, where it varies between 2 and 5 (Mintz et al., 1995; Borst and Sakmann, 1996; Takahashi et al., 1996; Wu et al., 1999; Rosato-Siri et al., 2002) and may reflect differences in the release machinery and/or the types of VGCCs supporting release at this synapse.
P/Q- and N-type calcium channels support evoked release of ACh at the efferent–IHC synapse
We show that transmitter release at the efferent–IHC synapse is supported by both N- and P/Q-type VGCCs. At many mammalian synapses, P/Q- and N-type VGCCs mediate synaptic transmission (Reid et al., 2003; Fedchyshyn and Wang, 2005; Snutch, 2005) with their relative contribution varying between synapses and postnatal age (Iwasaki et al., 2000; Ishikawa et al., 2005). In general, N-type channels support transmitter release at immature synapses, but their contribution decreases with development, being replaced by P/Q-type VGCCs (Rosato Siri and Uchitel, 1999; Iwasaki et al., 2000; Rosato-Siri et al., 2002). There is evidence suggesting that release is more steeply dependent on the intraterminal Ca2+ concentration for P/Q- than for N-type VGCCs. Cooperativity in cerebellar synapses was estimated to be 4 and 2.5 for P/Q- and N-type VGCCs, respectively (Mintz et al., 1995). Although this does not hold for many synapses and is still controversial (Evans and Zamponi, 2006), it would be interesting to study whether N- or P/Q-type channels are equally efficient in triggering release at the MOC–IHC synapse or even whether their contribution varies during the short period (P1 to P13–P14) during which this synapse is functional (Katz et al., 2004).
Transmitter release is almost completely abolished by the combined use of the P/Q- and N-type VGCCs antagonists. The percentage block by each selective antagonist sums 100%. At some synapses controlled by both P/Q- and N-type VGCCs, the percentage block of synaptic currents by each antagonist sums to >100%. Supra-additivity was taken as evidence of a mixed population of P/Q-and N-type VGCCs coexisting at a single release site and contributing jointly to the local Ca2+ transient that triggers transmitter release (Reid et al., 2003). Although other experimental approaches should be used to address this issue, our results suggest that at this synapse N- and P/Q-type VGCCs could be segregated at different release sites.
L-type VGCCs functionally coupled to the activation of BK channels negatively regulate ACh release
We find that transmitter release at the efferent–IHC synapse is enhanced by antagonists of both L-type VGCCs and BK channels. Fast release of transmitter occurs when the action potential invades and depolarizes the synaptic terminal, thus promoting the opening of Ca2+ channels with the consequent increase in cytosolic Ca2+ (Katz and Miledi, 1969). BK channels are synergistically activated by membrane depolarization and intracellular Ca2+ (Fakler and Adelman, 2008). Therefore, the two conditions necessary to activate BK channels are achieved when neurotransmitter is being released. In various types of neurons, activation of BK channels requires the delivery of Ca2+ through closely associated L-type VGCCs (Storm, 1987; Lingle et al., 1996; Prakriya and Lingle, 1999). This prompted us to test whether L-type VGCCs and BK channels were functionally coupled at the efferent–IHC synapse. Indeed, L-type VGCC antagonists or agonists failed to further enhance or reduce, respectively, release in cochlear preparations that had been previously treated with the BK channel antagonist. Moreover, pretreatment with DHPs completely occluded the effect of the BK channel blocker supporting the idea of a functional coupling between L-type VGCCs and BK channels at this synapse. This result also indicates that Ca2+ entering through N- and P/Q- type VGCCs does not activate BK channels possibly located farther away from the release sites.
We verified the expression of BK channels by immunofluorescence and found, in the region below IHCs, BK-positive puncta that correspond to synapsin-positive efferent terminals in prehearing mice (P9–P10). Paralleling the retraction of the transient efferent innervation to IHCs (Liberman et al., 1990; Simmons, 2002; Katz et al., 2004), BK-positive efferent–IHC terminals are no longer observed in hearing mice (P20–P22). Together with electrophysiological and pharmacological findings, these results suggest that these BK-positive efferent terminals correspond to the transient MOC terminals making axosomatic contacts with IHCs up to the onset of hearing (Liberman et al., 1990; Simmons et al., 1996; Katz et al., 2004). However, we cannot exclude the possibility that some BK-positive efferent terminals correspond to lateral olivocochlear (LOC) terminals, which contact the afferent dendrites that innervate IHCs and persist throughout life (Liberman et al., 1990; Simmons, 2002). If this were also the case, BK channels would be downregulated in LOC terminals after the onset of hearing.
Activation of BK channels by Ca2+ influx through VGCCs imposes spatial and temporal constraints and suggests the formation of macromolecular complexes between BK channels and their VGCC partners. Such a complex would provide a simple mechanism for reliably activating BK channels without affecting other Ca2+-dependent intracellular processes (Fakler and Adelman, 2008). We suggest that depolarization from an invading action potential activates P/Q-, N-, and L-type VGCCs. Influx of Ca2+ via P/Q- and N-type VGCCs closely associated with the release machinery would support release. In contrast, influx of Ca2+ via L-type VGCCs functionally coupled with BK channels, and possibly farther away from the release machinery (Flink and Atchison, 2003; Urbano et al., 2001), together with membrane depolarization would activate BK channels (this model is schematized in Fig. 7). Activation of BK channels would then accelerate repolarization and curtail release (Storm, 1987; Marcantoni et al., 2007).
Figure 7.

Schematic representation of the ion channels that support and regulate transmitter release from the MOC efferent presynaptic terminals innervating IHCs. Our results indicate that after invasion of the terminal action potential, Ca2+ entering through both P/Q- and N-type VGCCs support the release of ACh. In addition, they strongly suggest that Ca2+ entering though L-type Ca2+ channels exert a negative control on transmitter release by activating BK channels which accelerate the repolarization of the terminal action potential and thereby reduce the amount of ACh released per nerve impulse.
Functional significance of negative feedback to the transient efferent–IHC synapse
We show that functionally coupled L-type VGCCs and BK channels provide a negative feedback loop that reduces ACh release from these synaptic terminals. From previous work, we know that this synapse is functional (Glowatzki and Fuchs, 2000; Katz et al., 2004) and can prevent spontaneous Ca2+ action potentials in neonatal IHCs (Glowatzki and Fuchs, 2000; Goutman et al., 2005). These Ca2+ action potentials (Glowatzki and Fuchs, 2000; Tritsch et al., 2007) trigger glutamate release at the first synapse of the auditory system before the onset of hearing (Beutner and Moser, 2001; Glowatzki and Fuchs, 2002) and are thought to be critical for the establishment and refinement of synaptic connections in the auditory system (Erazo-Fischer et al., 2007; Tritsch and Bergles, 2010). If the quantal output of the efferent–IHC synapse were too strong, the resulting hyperpolarization of the IHC would inhibit the generation of action potentials and likely silence the entire afferent auditory pathway before the onset of hearing, probably disrupting its proper development.
The MOC efferent–IHC synapses might subtly contribute to the prenatal activity of IHCs. In the prehearing cat, firing of auditory nerve fibers in response to prolonged sound stimuli is rhythmic (Pujol, 1972), and this rhythmicity is lost if the MOC pathway is transected (Walsh et al., 1998). Coupled L-type VGCCs and BK channels curtail ACh release from the efferent terminals and may play an important role in achieving the patterned activity at the first auditory synapse that likely contributes to the correct establishment of synapses throughout the auditory pathway.
Interestingly, the transient MOC efferent–IHC synapse facilitates after high-frequency stimulation (Goutman et al., 2005). The role of BK channels in modulating release after high-frequency stimulation is not known for this synapse. However, in hippocampal neurons, BK channels inactivate when the frequency of the action potential-triggered activation of BK channels exceeds their rate of recovery from inactivation. BK channels become progressively unavailable to regulate firing frequency as the action potential train progresses (Shao et al., 1999). At the efferent–IHC synapse, high-frequency stimulation could cause inactivation of BK channels and facilitate release from the efferent terminal. In addition, Ca2+ accumulation in the terminal caused by high-frequency stimulation could also promote Ca2+-induced inactivation of L-type VGCCs (Morad and Soldatov, 2005), thus reducing BK channel activation. In either case, the reduction in the number of BK channels available would cause the terminal spike to broaden (Marcantoni et al. 2007; Storm, 1987), increasing thereby the quantal output. Studies performed in IHCs from adult mice with a targeted deletion in the α-subunit of the BK channel demonstrate the essential role of these channels for the precise timing of cochlear signaling (Oliver et al., 2006). Future experiments will investigate whether BK channels also contribute to the stimulus-induced facilitation of this synapse (Goutman et al., 2005) and should help clarify the role of BK channels in both the intrinsic excitability of the transient MOC efferent–IHC synapse as well as in the developing auditory network.
Footnotes
This work was supported by research grants from the National Organization for Hearing Research (NOHR 2006 and 2007) and by grants from Consejo Nacional de Investigaciones Científicas y Técnicas and the University of Buenos Aires to E.K. Immunostaining was supported by a research grant from the Deafness Research Foundation to S.P. The monoclonal antibody against the BK channel (L6/23) was generously provided by Dr. James Trimmer (University of California at Davis, Davis, CA). We especially thank Drs. Paul Fuchs and Ana Belén Elgoyhen, who also contributed to the support of this work with their grants from the National Institutes of Health (R01 DC001508) and the Howard Hughes Medical Institute, respectively. E.K. thanks Drs. Juan Goutman and Elisabeth Glowatzki, for generously teaching her how to evoke transmitter release at the OC-IHC synapse by electrical stimulation of the efferent axons in the acute preparation of the rodent organ of Corti; and Drs. Ana Belén Elgoyhen, Juan Goutman, and Paul Fuchs for critically reading this manuscript.
References
- Berkefeld H, Fakler B. Repolarizing responses of BKCa-Cav complexes are distinctly shaped by their Cav subunits. J Neurosci. 2008;28:8238–8245. doi: 10.1523/JNEUROSCI.2274-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U, Fakler B. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–620. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- Beutner D, Moser T. The presynaptic function of mouse cochlear inner hair cells during development of hearing. J Neurosci. 2001;21:4593–4599. doi: 10.1523/JNEUROSCI.21-13-04593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland LM, Morrill JA, Bean BP. omega-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J Neurosci. 1994;14:5011–5027. doi: 10.1523/JNEUROSCI.14-08-05011.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Brandt A, Khimich D, Moser T. Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J Neurosci. 2005;25:11577–11585. doi: 10.1523/JNEUROSCI.3411-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Kunze DL, Yatani A. The agonist effect of dihydropyridines on Ca channels. Nature. 1984;311:570–572. doi: 10.1038/311570a0. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium. 1998;24:307–323. doi: 10.1016/s0143-4160(98)90055-0. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Dechesne CJ, Winsky L, Kim HN, Goping G, Vu TD, Wenthold RJ, Jacobowitz DM. Identification and ultrastructural localization of a calretinin-like calcium-binding protein (protein 10) in the guinea pig and rat inner ear. Brain Res. 1991;560:139–148. doi: 10.1016/0006-8993(91)91224-o. [DOI] [PubMed] [Google Scholar]
- Dechesne CJ, Winsky L, Moniot B, Raymond J. Localization of calretinin mRNA in rat and guinea pig inner ear by in situ hybridization using radioactive and non-radioactive probes. Hear Res. 1993;69:91–97. doi: 10.1016/0378-5955(93)90096-j. [DOI] [PubMed] [Google Scholar]
- del Castillo J, Katz B. Interaction at endplate receptors between different choline derivatives. Proc R Soc Lond B Biol Sci. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering CJ, Zamponi GW. Molecular pharmacology of high voltage-activated calcium channels. J Bioenerg Biomembr. 2003;35:491–505. doi: 10.1023/b:jobb.0000008022.50702.1a. [DOI] [PubMed] [Google Scholar]
- Elgoyhen A, Vetter D, Katz E, Rothlin C, Heinemann S, Boulter J. Alpha 10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A. 2001;98:3501–3506. doi: 10.1073/pnas.051622798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo-Fischer E, Striessnig J, Taschenberger H. The role of physiological afferent nerve activity during in vivo maturation of the calyx of Held synapse. J Neurosci. 2007;27:1725–1737. doi: 10.1523/JNEUROSCI.4116-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM, Zamponi GW. Presynaptic Ca2+ channels–integration centers for neuronal signaling pathways. Trends Neurosci. 2006;29:617–624. doi: 10.1016/j.tins.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Fakler B, Adelman JP. Control of K(Ca) channels by calcium nano/microdomains. Neuron. 2008;59:873–881. doi: 10.1016/j.neuron.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Developmental transformation of the release modality at the calyx of held synapse. J Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flink MT, Atchison WD. Iberiotoxin-induced block of Ca2+-activated K+ channels induces dihydropyridine sensitivity of ACh release from mammalian motor nerve terminals. J Pharmacol Exp Ther. 2003;305:646–652. doi: 10.1124/jpet.102.046102. [DOI] [PubMed] [Google Scholar]
- Fuchs PA. Synaptic transmission at vertebrate hair cells. Curr Opin Neurobiol. 1996;6:514–519. doi: 10.1016/s0959-4388(96)80058-4. [DOI] [PubMed] [Google Scholar]
- Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem. 1990;265:11083–11090. [PubMed] [Google Scholar]
- Glowatzki E, Fuchs P. Transmitter release at the hair cell ribbon synapse. Nat Neurosci. 2002;5:147–154. doi: 10.1038/nn796. [DOI] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA. Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science. 2000;288:2366–2368. doi: 10.1126/science.288.5475.2366. [DOI] [PubMed] [Google Scholar]
- Gomez-Casati ME, Fuchs PA, Elgoyhen AB, Katz E. Biophysical and pharmacological characterization of nicotinic cholinergic receptors in rat cochlear inner hair cells. J Physiol. 2005;566:103–118. doi: 10.1113/jphysiol.2005.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutman JD, Fuchs PA, Glowatzki E. Facilitating efferent inhibition of inner hair cells in the cochlea of the neonatal rat. J Physiol. 2005;566:49–59. doi: 10.1113/jphysiol.2005.087460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes WN, Li W, Chavez AE, Diamond JS. BK channels modulate pre- and postsynaptic signaling at reciprocal synapses in retina. Nat Neurosci. 2009;12:585–592. doi: 10.1038/nn.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet M, Kaufmann WA. Coassembly of big conductance Ca2+-activated K+ channels and L-type voltage-gated Ca2+ channels in rat brain. J Biol Chem. 2004;279:36445–36453. doi: 10.1074/jbc.M402254200. [DOI] [PubMed] [Google Scholar]
- Hafidi A, Beurg M, Dulon D. Localization and developmental expression of BK channels in mammalian cochlear hair cells. Neuroscience. 2005;130:475–484. doi: 10.1016/j.neuroscience.2004.09.038. [DOI] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- Hubbard JI, Llinás R, Quastel DMJ. Electrophysiological analysis of synaptic transmission. London: Edward Arnold; 1969. [Google Scholar]
- Ishikawa T, Kaneko M, Shin HS, Takahashi T. Presynaptic N-type and P/Q-type Ca2+ channels mediating synaptic transmission at the calyx of Held of mice. J Physiol. 2005;568:199–209. doi: 10.1113/jphysiol.2005.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. J Physiol. 1998;509:419–423. doi: 10.1111/j.1469-7793.1998.419bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD, Takahashi T. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20:59–65. doi: 10.1523/JNEUROSCI.20-01-00059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Jensen MS, Lambert JD. Role of presynaptic L-type Ca2+ channels in GABAergic synaptic transmission in cultured hippocampal neurons. J Neurophysiol. 1999;81:1225–1230. doi: 10.1152/jn.1999.81.3.1225. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. Spontaneous and evoked activity of motor nerve endings in calcium Ringer. J Physiol. 1969;203:689–706. doi: 10.1113/jphysiol.1969.sp008887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Ferro PA, Weisz G, Uchitel OD. Calcium channels involved in synaptic transmission at the mature and regenerating mouse neuromuscular junction. J Physiol. 1996;497:687–697. doi: 10.1113/jphysiol.1996.sp021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Protti DA, Ferro PA, Rosato Siri MD, Uchitel OD. Effects of Ca2+ channel blocker neurotoxins on transmitter release and presynaptic currents at the mouse neuromuscular junction. Br J Pharmacol. 1997;121:1531–1540. doi: 10.1038/sj.bjp.0701290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Verbitsky M, Rothlin CV, Vetter DE, Heinemann SF, Belen Elgoyhen A. High calcium permeability and calcium block of the alpha9 nicotinic acetylcholine receptor. Hear Res. 2000;141:117–128. doi: 10.1016/s0378-5955(99)00214-2. [DOI] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB, Gomez-Casati ME, Knipper M, Vetter DE, Fuchs PA, Glowatzki E. Developmental regulation of nicotinic synapses on cochlear inner hair cells. J Neurosci. 2004;24:7814–7820. doi: 10.1523/JNEUROSCI.2102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knirsch M, Brandt N, Braig C, Kuhn S, Hirt B, Munkner S, Knipper M, Engel J. Persistence of Ca(v)1.3 Ca2+ channels in mature outer hair cells supports outer hair cell afferent signaling. J Neurosci. 2007;27:6442–6451. doi: 10.1523/JNEUROSCI.5364-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP, Rusch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature. 1998;394:281–284. doi: 10.1038/28401. [DOI] [PubMed] [Google Scholar]
- Layton MG, Robertson D, Everett AW, Mulders WH, Yates GK. Cellular localization of voltage-gated calcium channels and synaptic vesicle-associated proteins in the guinea pig cochlea. J Mol Neurosci. 2005;27:225–244. doi: 10.1385/JMN:27:2:225. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Dodds LW, Pierce S. Afferent and efferent innervation of the cat cochlea: quantitative analysis with light and electron microscopy. J Comp Neurol. 1990;301:443–460. doi: 10.1002/cne.903010309. [DOI] [PubMed] [Google Scholar]
- Lingle C, Solaro C, Prakriya M, Ding J. Calcium-activated potassium channels in adrenal chromaffin cells. Ion Channels. 1996;4:261–301. doi: 10.1007/978-1-4899-1775-1_7. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Lima PA, Marrion NV. Co-assembly of N-type Ca2+ and BK channels underlies functional coupling in rat brain. J Cell Sci. 2007;120:985–995. doi: 10.1242/jcs.03399. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger-Brauns MJ, Striessnig J, Carbone E. Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci. 2007;30:491–504. doi: 10.1523/JNEUROSCI.4961-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Holley MC, Kros CJ. Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J Physiol. 2003;548:383–400. doi: 10.1113/jphysiol.2002.034801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Kros CJ. A transiently expressed SK current sustains and modulates action potential activity in immature mouse inner hair cells. J Physiol. 2004;560:691–708. doi: 10.1113/jphysiol.2004.072868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- McLean WJ, Smith KA, Glowatzki E, Pyott SJ. Distribution of the Na,K-ATPase alpha subunit in the rat spiral ganglion and organ of corti. J Assoc Res Otolaryngol. 2009;10:37–49. doi: 10.1007/s10162-008-0152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Morad M, Soldatov N. Calcium channel inactivation: possible role in signal transduction and Ca2+ signaling. Cell Calcium. 2005;38:223–231. doi: 10.1016/j.ceca.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Muller A, Kukley M, Uebachs M, Beck H, Dietrich D. Nanodomains of single Ca2+ channels contribute to action potential repolarization in cortical neurons. J Neurosci. 2007;27:483–495. doi: 10.1523/JNEUROSCI.3816-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Klocker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000;26:595–601. doi: 10.1016/s0896-6273(00)81197-6. [DOI] [PubMed] [Google Scholar]
- Oliver D, Taberner AM, Thurm H, Sausbier M, Arntz C, Ruth P, Fakler B, Liberman MC. The role of BKCa channels in electrical signal encoding in the mammalian auditory periphery. J Neurosci. 2006;26:6181–6189. doi: 10.1523/JNEUROSCI.1047-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera BM, Miljanich GP, Ramachandran J, Adams ME. Calcium channel diversity and neurotransmitter release: the omega-conotoxins and omega-agatoxins. Annu Rev Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- Perissinotti PP, Giugovaz Tropper B, Uchitel OD. L-type calcium channels are involved in fast endocytosis at the mouse neuromuscular junction. Eur J Neurosci. 2008;27:1333–1344. doi: 10.1111/j.1460-9568.2008.06113.x. [DOI] [PubMed] [Google Scholar]
- Petersen OH, Maruyama Y. Calcium-activated potassium channels and their role in secretion. Nature. 1984;307:693–696. doi: 10.1038/307693a0. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lingle CJ. BK channel activation by brief depolarizations requires Ca2+ influx through L- and Q-type Ca2+ channels in rat chromaffin cells. J Neurophysiol. 1999;81:2267–2278. doi: 10.1152/jn.1999.81.5.2267. [DOI] [PubMed] [Google Scholar]
- Pujol R. Development of tone-burst responses along the auditory pathway in the cat. Acta Otolaryngol. 1972;74:383–391. doi: 10.3109/00016487209128467. [DOI] [PubMed] [Google Scholar]
- Pyott SJ, Glowatzki E, Trimmer JS, Aldrich RW. Extrasynaptic localization of inactivating calcium-activated potassium channels in mouse inner hair cells. J Neurosci. 2004;24:9469–9474. doi: 10.1523/JNEUROSCI.3162-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyott SJ, Meredith AL, Fodor AA, Vazquez AE, Yamoah EN, Aldrich RW. Cochlear function in mice lacking the BK channel alpha, beta1, or beta4 subunits. J Biol Chem. 2007;282:3312–3324. doi: 10.1074/jbc.M608726200. [DOI] [PubMed] [Google Scholar]
- Raffaelli G, Saviane C, Mohajerani MH, Pedarzani P, Cherubini E. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J Physiol. 2004;557:147–157. doi: 10.1113/jphysiol.2004.062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Bekkers JM, Clements JD. Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci. 2003;26:683–687. doi: 10.1016/j.tins.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Rosato Siri MD, Uchitel OD. Calcium channels coupled to neurotransmitter release at neonatal rat neuromuscular junctions. J Physiol. 1999;514:533–540. doi: 10.1111/j.1469-7793.1999.533ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato-Siri MD, Piriz J, Tropper BA, Uchitel OD. Differential Ca2+-dependence of transmitter release mediated by P/Q- and N-type calcium channels at neonatal rat neuromuscular junctions. Eur J Neurosci. 2002;15:1874–1880. doi: 10.1046/j.1460-9568.2002.02015.x. [DOI] [PubMed] [Google Scholar]
- Ruttiger L, Sausbier M, Zimmermann U, Winter H, Braig C, Engel J, Knirsch M, Arntz C, Langer P, Hirt B, Muller M, Kopschall I, Pfister M, Munkner S, Rohbock K, Pfaff I, Rusch A, Ruth P, Knipper M. Deletion of the Ca2+-activated potassium (BK) alpha-subunit but not the BKbeta1-subunit leads to progressive hearing loss. Proc Natl Acad Sci U S A. 2004;101:12922–12927. doi: 10.1073/pnas.0402660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao LR, Halvorsrud R, Borg-Graham L, Storm JF. The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol. 1999;521:135–146. doi: 10.1111/j.1469-7793.1999.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidach SS, Mintz IM. Low-affinity blockade of neuronal N-type Ca channels by the spider toxin omega-agatoxin-IVA. J Neurosci. 2000;20:7174–7182. doi: 10.1523/JNEUROSCI.20-19-07174.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DD. Development of the inner ear efferent system across vertebrate species. J Neurobiol. 2002;53:228–250. doi: 10.1002/neu.10130. [DOI] [PubMed] [Google Scholar]
- Simmons DD, Mansdorf NB, Kim JH. Olivocochlear innervation of inner and outer hair cells during postnatal maturation: evidence for a waiting period. J Comp Neurol. 1996;370:551–562. doi: 10.1002/(SICI)1096-9861(19960708)370:4<551::AID-CNE10>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Snutch TP. Targeting chronic and neuropathic pain: the N-type calcium channel comes of age. NeuroRx. 2005;2:662–670. doi: 10.1602/neurorx.2.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EF, Atrakchi AH. Calcium currents recorded from a vertebrate presynaptic nerve terminal are resistant to the dihydropyridine nifedipine. Proc Natl Acad Sci U S A. 1990;87:9683–9687. doi: 10.1073/pnas.87.24.9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Ko CP. Novel modulatory effect of L-type calcium channels at newly formed neuromuscular junctions. J Neurosci. 1997;17:1101–1111. doi: 10.1523/JNEUROSCI.17-03-01101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Gu XQ, Haddad GG. Calcium influx via L- and N-type calcium channels activates a transient large-conductance Ca2+-activated K+ current in mouse neocortical pyramidal neurons. J Neurosci. 2003;23:3639–3648. doi: 10.1523/JNEUROSCI.23-09-03639.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science. 1996;274:594–597. doi: 10.1126/science.274.5287.594. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Bergles DE. Developmental regulation of spontaneous activity in the mammalian cochlea. J Neurosci. 2010;30:1539–1550. doi: 10.1523/JNEUROSCI.3875-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–55. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- Urbano FJ, Depetris RS, Uchitel OD. Coupling of L-type calcium channels to neurotransmitter release at mouse motor nerve terminals. Pflugers Arch. 2001;441:824–831. doi: 10.1007/s004240000489. [DOI] [PubMed] [Google Scholar]
- Waka N, Knipper M, Engel J. Localization of the calcium channel subunits Cav1.2 (alpha1C) and Cav2.3 (alpha1E) in the mouse organ of Corti. Histol Histopathol. 2003;18:1115–1123. doi: 10.14670/HH-18.1115. [DOI] [PubMed] [Google Scholar]
- Walsh E, McGee J, McFadden S, Liberman M. Long-term effects of sectioning the olivocochlear bundle in neonatal cats. J Neurosci. 1998;18:3859–3869. doi: 10.1523/JNEUROSCI.18-10-03859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisstaub N, Vetter DE, Elgoyhen AB, Katz E. The alpha9alpha10 nicotinic acetylcholine receptor is permeable to and is modulated by divalent cations. Hear Res. 2002;167:122–135. doi: 10.1016/s0378-5955(02)00380-5. [DOI] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampini V, Johnson SL, Franz C, Lawrence ND, Munkner S, Engel J, Knipper M, Magistretti J, Masetto S, Marcotti W. Elementary properties of CaV1.3 Ca(2+) channels expressed in mouse cochlear inner hair cells. J Physiol. 2010;588:187–199. doi: 10.1113/jphysiol.2009.181917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JL, Gao WQ. Analysis of rat vestibular hair cell development and regeneration using calretinin as an early marker. J Neurosci. 1997;17:8270–8282. doi: 10.1523/JNEUROSCI.17-21-08270.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]