

Abstract
ZIPK (zipper-interacting protein kinase) is a Ca2+-independent protein kinase that promotes myosin phosphorylation in both smooth muscle and non-muscle cells. A recent report attempted to clarify a debate over the subcellular localization of ZIPK in non-muscle cells (Shoval et. al. (2007) Plos Genetics. 3: 1884–1883). A species-specific loss of a key phosphorylation site (T299) in murine (mouse and rat) ZIPK seems to direct it to the nucleus, while the presence of the T299 site in human ZIPK correlates with cytoplasmic localization. T299 is immediately adjacent to a putative nuclear localization sequence (NLS) and may mask its function when phosphorylated, therefore explaining the species-specific dichotomy of intracellular localization. However, despite the murine ZIPK (mZIPK) lacking the T299 residue that is critical for controlling human ZIPK (hZIPK) subcellular localization, mutational analysis showed that this NLS control locus is nonfunctional in the murine context. A constitutively active Rho promoted the cytoplasmic retention of a human ZIPK mutant that would otherwise localize to the nucleus. Endogenous hZIPK showed sensitivity to the nuclear export inhibitor leptomysin B, suggesting a continuous shuttling between cytoplasm and nucleus that is dependent upon T299 dephosphorylation. Thus, the C-terminal domain of human and murine ZIPK demonstrated quite divergent nuclear import and export functionality. We conclude that in the case of ZIPK, studies between the species may not be directly comparable to each other given the gross differences in intracellular localization and movement.
Keywords: ZIPK, phosphorylation, subcellular localization, human, murine
1. Introduction
Zipper-interacting protein kinase (ZIPK) is one of several Ca2+-independent protein kinases that regulate myosin function in both smooth muscle and non-muscle cells [1, 2]. In smooth muscle, ZIPK is thought to regulate myosin either directly by phosphorylating its regulatory light chain (LC20) or through inhibition of smooth muscle myosin phosphatase (SMPP1M) [3]. Inhibition of SMPP1M functions to enhance muscle responsiveness to Ca2+ (Ca2+-sensitization) and promote a contractile state [4]. Similar roles have been suggested for ZIPK in non-muscle cells whereby the enzyme is thought to regulate myosin phosphorylation as part of non-apoptotic cell death pathways. In addition to roles in regulating Ca2+-independent myosin phosphorylation in the cytoplasm, human and murine forms of ZIPK have been reported to localize differentiallyto the cytoplasm and nucleus in non-muscle cells, respectively, although its nuclear functions have yet to be defined [5–7].
Human ZIPK (hZIPK) is regulated by an upstream signal transduction cascade mediating Ca2+-sensitization in smooth muscle [1, 2]. Activated G protein-coupled receptors signal to Rho/ROCK which, in turn, phosphorylate hZIPK at several sites to promote activation [8]. While in non-muscle cells, activated DAPK1 can function as an upstream activator of hZIPK, also phosphorylating multiple sites [9]. We have shown that three sites (T180, T225, and T265) in hZIPK are required to be phosphorylated for kinase activity, whereas phosphorylation at T299 regulates cytoplasmic localization [5]. While the T299 site can be the product of autophosphorylation, both ROCK and DAPK1 can also phosphorylate hZIPK at the T299 residue [8, 9]. ZIPK contains four putative NLS sequences, and the T299 site is immediately C-terminal to the second putative NLS motif (NLS2) and may mask its function when phosphorylated. Consistent with this observation, a phosphomimetic mutation (T299D) is exclusively observed in the cytoplasm, and a nonphosphorylatable mutant (TT299/300AA) localizes exclusively to the nucleus [5].
Primary sequence alignments of mammalian forms of ZIPK revealed striking non-conserved amino acid divergence within the C-terminal domain of only murine (mouse and rat) when compared with all other species, including close rodent species [6]. Within the 181 amino acids of the mZIPK C-terminus, 54 substitutions are present, of which, 23 are non-conserved and five deletions (compared to human). These variations suggest that ZIPK has evolved additional functions that impact its activity in the murine context but are not found in other species including humans. When overexpressed as recombinant enzymes, the murine specific sequence differences result in exclusive localization of ZIPK to the nucleus. In contrast, the human form (hZIPK) resides exclusively within the cytoplasm, either associated with actin filaments or soluble in non-muscle cells. The difference in localization between species seems attributable to a single T-to-A residue divergence at T299 in murine ZIPK, removing a key phosphorylation site serving as a locus of control [6]. Thus, the subcellular localization of mZIPK behaves like the nonphosphorylatable mutant (TT299/300AA) of hZIPK and exhibits nuclear localization. At first sight one would expect that the loss of a critical phosphorylation site is explained by unmasking of a juxtaposed putative NLS (NLS2); however, another group reported that the NLS4 of mZIPK is the functional NLS, leaving the issue unresolved [10]. Regardless of the mode of nuclear entry for mZIPK, its presence in the nucleus begs the question of its functional conservation with the cytoplasmically localized hZIPK. The additional C-terminal sequence divergence of mZIPK has been suggested as a gain-of-function mechanism to conserve ZIPK cytoplasmic function between these species. When overexpressed, the tumor suppressor protein PAR-4 can bind to nuclear mZIPK to promote a stepwise nuclear export of the complex, then association with actin filaments [11, 12]. Consistent with a gain-of-function divergence in the mZIPK sequence, PAR-4 exclusively binds to mZIPK and not hZIPK [6]. Although why nature would promote nuclear localization in murine only, and then salvage this phenotype by mutating several dozen other C-terminal amino acids is certainly a biological conundrum, especially since PAR-4 is highly conserved across all mammalian species examined.
In the present study, we have exploited the natural sequence differences between murine and human ZIPKs to determine the molecular mechanisms by which phosphorylation of T299 normally regulate ZIPK nuclear localization in human cells. We show that in the human context, the NLS2 functions as the nucleus-directing motif, but only upon dephosphorylation of the adjacent T299 residue. However, in the case of mZIPK, the NLS2 is nonfunctional for transport. In humans we show that the Rho pathway regulates the intracellular localization of hZIPK in vivo. We also demonstrate that hZIPK can constantly shuttle between the cytoplasmic and nuclear compartments, perhaps demonstrating some functional commonality with mZIPK. Our conclusions show that the C-terminal domain of ZIPK differentially regulates the intracellular localization of both human and murine ZIPK and dramatically affects the function of these proteins.
2. Materials and Methods
2.1 Cell culture, transfection, and indirect immunofluorescence microscopy
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (MediaTech) supplemented with 10% fetal bovine serum (Gibco). Cells (1 × 105) were grown on coverslips overnight and transfected for 16 hours then prepared for indirect immunofluorescence microscopy. Transfections were performed using Fugene (Roche) as per manufacturer’s instructions. Leptomycin B (Calbiochem) was used at 50 nM. Briefly, cells were fixed in 4% paraformaldehyde in PBS (pH 7.6) and lysed in 0.2% Triton X-100. Nonspecific sites were blocked with 5% normal goat serum (NGS) in PBS followed by incubation with primary (anti-FLAG (1:1000 dilution) or anti-ZIPK 301–454 (1:200)). Samples were then incubated with the appropriate anti-mouse or rabbit secondary antibodies (Alexa 488 or 568 (1:200)). Actin filaments were visualized by Alexa 568-conjugated phalloidin (Molecular Probes). DAPI (0.1 μg/ml) in PBS was used to counterstain nuclei. Images were obtained on a Zeiss Axio Imager controlled by MetaMorph in the Duke University Light Microscopy Core Facility.
2.2 Antibodies, Western blotting, and Immunoprecipitation
Anti-FLAG (Sigma) and anti-ROCK1 (Santa Cruz) antibodies and immobilized anti-FLAG M2 antibody gel were used for Western blotting and immunoprecipitation. Custom polyclonal anti-phosphoT299 ZIPK antibody [8] and anti-human ZIPK (raised to residues 301–454) were generated by Proteintech (Chicago, IL). Cells were lysed on ice for 10 minutes in 50 mM TRIS-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM dithiothreitol, 2 mM EDTA, and Complete Mini protease inhibitor cocktail (Roche) or sonicated. Lysates were spun 10 minutes at 13,000 × g, and protein concentrations were determined by BioRad assay and normalized for Western blotting and immunoprecipitation.
2.3 Kinase Assays
Purified ROCK1 (Invitrogen) was added at 0, 50, or 100 ng amounts to kinase buffer (50 mM TRIS, pH 7.5, 10 mM MgCl2, 100 μM ATP (2500 cpm/nmol), and 1 mM dithiothreitol) with 20 ng ZIPK and incubated for 10 minutes at room temperature. The reaction was stopped with sample buffer, separated on a denaturing gel, and transferred to PVDF membranes for Western blotting and autoradiography. Bands of phospho-T299 blots were quantitated by ImageJ version 1.41 and graphed and statistically analyzed in Microsoft Excel.
3. Results
A recent report provided clarification to many previously conflicting ZIPK subcellular localization accounts by highlighting sequence differences in a key regulatory phosphorylation site, T299, between human and murine ZIPK proteins ([6] and Figure 1A). We had previously demonstrated that an alanine substitution at T299 results in dramatic relocalization of human Flag-ZIPK from the cytoplasm to the nucleus in human cells ([5] and Figure 1B (panel c compared to panel a)). Given the proximity of T299 to a potential nuclear localization sequence (NLS), we hypothesized that phosphorylation at this residue functions to block access of the NLS to nuclear import machinery. Introducing a phosphomimetic mutation at this site (T299D) resulted in cytoplasmic localization of hZIPK (Figure 1B (panel e)). In contrast, mZIPK, which lacks an analogous phosphorylation site but contains a well-conserved NLS, demonstrated punctuate nuclear localization ([6, 7] Figure 1B (panel g)). Therefore, herein, we aimed to elucidate if this control locus operated similarly between species.
Figure 1. Sequence and localization differences between human and murine ZIPK.

(A) Amino acid alignment between human and murine ZIPK at the putative NLS2 region showed that NLS2 (underlined) between species had very good conservation, but the T299 phosphorylation site (shown in bold) was absent in the murine context, highlighting differences that may control differential subcellular localization between species. (B) Immunoflourescence microscopy in HeLa cells showed subcellular localization of wild-type Flag-human ZIPK (a), Flag-human ZIPK TT299/300AA (c), Flag-human ZIPK T299D (e), and GFP-murine ZIPK (g). Corresponding nuclei were stained in panels b, d, f, and h.
To test a model where T299 phosphorylation blocks an immediately adjacent putative NLS (NLS2) function, we truncated hZIPK to evaluate possible alternative function of the other three putative NLS (Figure 2A schematic). Wild-type, full-length hZIPK was phosphorylated on T299 and localized to the cytoplasm. C-terminal truncations at either residues 400 or 342 remained phosphorylated at T299 and cytoplasmic (Figure 2A panels a, c, and e and 2B). The kinase domain construct, 1–273, remained cytoplasmic, indicating the functional NLS of ZIPK is contained in the C-terminal residues 274–454 (Figure 2A panel g and 2B). Deleting the kinase domain leaves the 274–454 construct which localized to the nucleus and did not show T299 phosphorylation (Figure 2A panel i and 2B). The final construct is 343–454, which lacked the NLS2 region, and demonstrated cytoplasmic localization (Figure 2A, panel k, and 2B). Additionally, introduction of a T299D mutation within the context of the 274–454 protein resulted in cytoplasmic localization (Figure 2C panel c). In aggregate, these data suggest that the putative NLS2 region and the phosphorylation status of T299 control the subcellular localization of human ZIPK.
Figure 2. Phosphorylation and localization of human ZIPK truncation mutants.

(A) Various FLAG-ZIPK constructs were tested for cellular localization following transfection into HeLa cells. Full-length (1–454) wild-type ZIPK (panel a), 1–400 (c), 1–342 (e), 1–273 (g), 274–454 (i), and 343–454 (k) were visualized by FLAG immunofluoresence microscopy. Corresponding nuclei were stained in panels below the FLAG staining (b, d, f, h, j, and l). The schematic of Flag-ZIPK truncations to individually isolate the four putative NLS motifs (black rectangles) for localization control analysis is shown to the right of panel b. The kinase domain is shown in gray. The localization of each construct in HeLa cells is given next to the schematic of each deletion construct. (B) The Flag-tagged proteins and their T299 phosphorylation status were immunoblotted. (C) The Flag-274–454 (a) and Flag-274–454 T299D constructs (c) were imaged in HeLa cells with an anti-Flag antibody, and corresponding nuclei were stained (b and d).
We have previously shown that phosphorylation of T299 in hZIPK is the result of both autophosphorylation and ROCK phosphorylation [8]. To test for in vivo rho/ROCK signaling in the regulation of hZIPK localization, we exploited a mutant form of hZIPK that demonstrates nuclear localization. The mutation of three valines to alanine in the leucine zipper motif (ΔLZ) resulted in hZIPK protein both impaired in oligomerization and localized mainly to the nucleus [5, 13]. We illustrate here that the ΔLZ has reduced T299 phosphorylation which allowed us to observe a potential change in intracellular localization (Figure 3A). Overexpression of a constitutively active GFP-rhoA63L activated rho/ROCK signaling and resulted in enhanced cytoplasmic localization of the ΔLZ mutant (Figure 3B (panel j compared to panel d)). Quantitation of the localization change of the ΔLZ mutant with or without rho coexpression showed a dramatic relocalization of ΔLZ to the cytoplasm only in the presence of rho (Figure 3C). Using an in vitro kinase assay, we demonstrate that purified ROCK protein can increase ZIPK phosphorylation at T299 above levels generated by ZIPK autophosphorylation (Figure 3D). These data suggest that active rho/ROCK signaling directs hZIPK, via T299 phosphorylation, to remain in the cytoplasm upon rho/ROCK activation and perform distal functions of the pathway.
Figure 3. Human Flag-ZIPKΔLZ mutant showed decreased T299 phosphorylation and nuclear localization that could be overcome by coexpression with constitutively active RhoA.

(A) HeLa cells were transfected with FLAG-ZIPK or FLAG-ZIPK ΔLZ and blotted for phospho-T299 and FLAG antibodies. (B) HeLa cells were transfected with GFP and either Flag- ZIPK (a–c) or Flag-ZIPK ΔLZ (d–f). Cells were transfected with GFP-RhoA 63L and either FLAG-ZIPK (g–i) or FLAG-ZIPK ΔLZ(j –l). Cells were counterstained with DAPI (panels b, e, h, and k) and showed GFP fluorescence (panels c, f, i, and l) (C) HeLa cells transfected with Flag-ZIPK ΔLZ in the presence or absence of Rho were counted for any change in localization of the Flag-ZIPK ΔLZ. Light bars showed percent cells with exclusively nuclear Flag staining, while dark bars demonstrated the percent of cells with Flag staining in both cytoplasmic and nuclear compartments. Twenty fields of cells were counted, and averages were graphed with standard deviation. Results of Student’s t-tests demonstrated significant differences between pairs tested, giving p values<0.005. * indicates a comparison of exclusively nuclear Flag staining (light bars) between FLAG-ZIPK ΔLZ and FLAG-ZIPK ΔLZ plus Rho. **indicates a comparison of both cytoplasmic and nuclear Flag staining (dark bars) between FLAG-ZIPK ΔLZ and FLAG-ZIPK ΔLZ plus Rho. (D) In vitro phosphorylation of purified ZIPK by ROCK1. Increasing amounts of ROCK1 was added to the assay: (1) 0 ng ROCK1, (2) 50 ng ROCK1, and (3) 100 ng ROCK1. Antibodies used for Western blotting are noted next to their corresponding blots. Autoradiograph indicates [γ-32 ATP] incorporation into ZIPK protein. Quantitation of phospho-T299 Western blot is shown in the accompanying graph.
To confirm the role of NLS2 in nuclear import of hZIPK, we mutated two basic residues within the putative NLS2 to alanines at positions 294 and 295. This RR294–295AA construct (mNLS2) demonstrated cytoplasmic localization but, interestingly, retained phosphorylation at T299, therefore, providing incomplete information on the effect of this mutation (Figure 4C). Coupling a non-phosphorylatable TT299–300AA mutation with mutNLS2 eliminated the possibility of T299 phosphorylation and also resulted in cytoplasmic localization of this construct (Figure 4D). Thus, NLS2 functions as the bona fide nuclear import signal in the human ZIPK protein.
Figure 4. Mutation of NLS2 impairs nuclear import in hZIPK.

HeLa cells were transfected with Flag-tagged wild-type (A), TT299/300AA (B), mNLS2 (C), and TT299/300AA and mNLS2 (D) ZIPK constructs and visualized by Flag immunofluorescence microscopy. Cell lysates were blotted with anti-phospho-T299 and Flag antibodies. Quantitation of localization differences among the four constructs (A, B, C, and D, as above) was graphed. Light bars indicate the percentage of cells with cytoplasmic staining, while dark bars indicate the percentage of cells with nuclear staining. * Student’s t-test demonstrated significant differences between the cytoplasmic staining of WT (A) and TT299/300AA (B), giving a p value<0.001.
Given our results defining the human NLS2 locus, we expected this model of regulation to apply to mZIPK as well despite a report of NLS4 as the functional NLS of mZIPK. Shoval et al generated an AA299–300TT mutation in mZIPK in an attempt to reestablish regulation in this region of the protein. They saw exclusive nuclear staining with this mutant, but did not test for possible phosphorylation at this mutant site [6]. We repeated their results with the AA299–300TT mutant and extended the investigation of this NLS2 region by introducing analogous A299D and NLS2 mutations into mZIPK. In agreement with previous results, AA299–300TT did remain in the nucleus despite the introduction of phosphorylatable residues (Figure 5C). Quite unexpectedly, both A299D and RR294–295AA (mutant NLS) rodent ZIPK constructs remained localized to the nucleus as well (Figure 5E and G, respectively), highlighting that the sequence differences in the C-termini of these two ZIPK species markedly alter their nuclear localization and functionality. Thus, NLS4, not NLS2, likely appears to be the only functional NLS of mZIPK as reported previously [10].
Figure 5. NLS2 and 299 mutations have no localization effects in mZIPK.

HeLa cells were transfected with GFP-mZIPK (A), GFP-mZIPK AA299/300TT (C), GFP-mZIPK A299D (E), and GFP-mZIPK mNLS2 (G) constructs. Corresponding nuclei were stained with DAPI (B, D, F, and H)
The nuclear localized mZIPK has demonstrated the ability to transit to the cytoplasm but only upon overexpression of an interacting protein, Par-4 [11, 12]. Other than overexpression of mutant constructs, hZIPK has not been observed in the nucleus. Our findings that the T299A mutation promotes nuclear localization strongly imply that dephosphorylation of this residue alone would promote nuclear localization, perhaps conserving some function in the murine context. In searching for conditions where hZIPK is nuclear, we tested various kinase inhibitors to block ZIPK autophosphorylation/activation, serum-starvation, and stages of the cell cycle; however, no change in localization was observed in HeLa cells. However, following treatment with leptomycin B (LMB), a nuclear export inhibitor, significant amounts of both recombinant (Figure 6B) and endogenous hZIPK (Figure 6F) did accumulate in the nuclear compartment, suggesting that the human enzyme constantly shuttles into and out of the nucleus via the Crm1 exporter. Not all of the ZIPK demonstrated shuttling activity as a portion remained colocalized with actin upon LMB treatment (Figure 6H). The LMB sensitivity of hZIPK suggested that a nuclear export sequence (NES) is present. NES prediction software indicated a potential leucine-rich NES near residues 378–386; however, alanine substitution of one or all of these leucine residues did not show nuclear accumulation as was consequent with LMB addition. We cannot rule out the possibility of hZIPK interacting with other NES-containing proteins for export. Our data suggest that under some cellular circumstances, hZIPK has the capacity to shuttle in and out of the nucleus and that this requires regulation by dephosphorylation of T299. However, in addition to the physiological stimulus that results in T299 dephosphorylation, the identity of the phosphatase regulating T299 dephosphorylation also remains to be defined.
Figure 6. Inhibiting nuclear export results in nuclear localization of hZIPK.
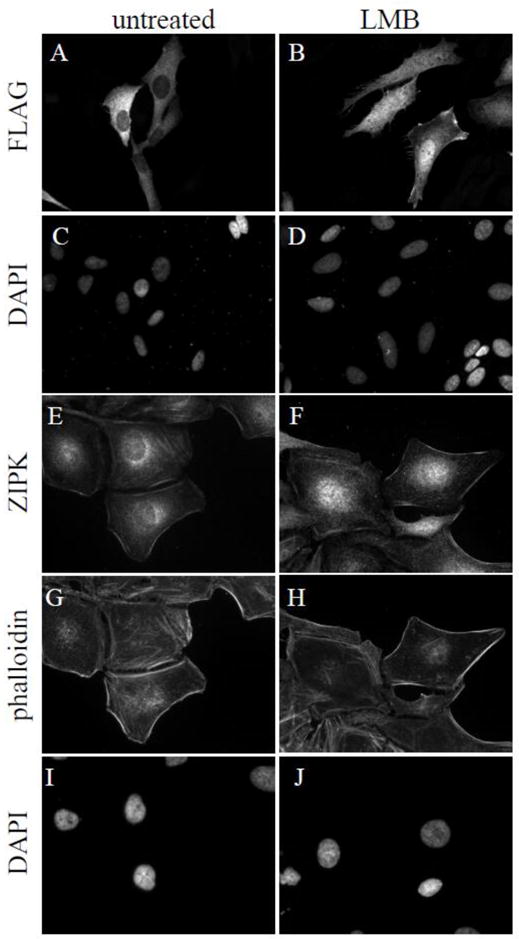
HeLa cells were either transfected with Flag-hZIPK or untransfected then treated with 50 nM LMB for 4 or 16 hours, respectively (panels B, D, F, H, and J) or untreated (A, C, E, G, and I). Flag-hZIPK was visualized with Flag immunostaining (A and B), while endogenous hZIPK was stained with anti-hZIPK antibodies (E and F). Actin was visualized with Alexa-568-phalloidin (G and H), and nuclei were stained with DAPI (C, D, I and J).
4. Discussion
A previous account reported that species-specific differences in ZIPK protein at a key regulatory phosphorylation site resulted in dramatic differences in subcellular localization between human and murine [6]. By comparing human ZIPK with the murine protein, we have detailed the functionality of the NLS juxtaposed to the phosphorylation site at T299. When phosphorylated by the rho/ROCK pathway, phospho-T299 appears to block access of the NLS to the nuclear import machinery and promote cytoplasmic localization of ZIPK. Conversely, a non-phosphorylatable TT299/300AA mutation both mimicked dephosphorylation and resulted in nuclear accumulation. In the context of the TT299/300AA mutation, substitution of two NLS2 residues (RR294/295) to alanine blocked nuclear entry, demonstrating NLS2 as a bona fide NLS in hZIPK. Importantly, nuclear localization of another member of the DAPK family, DRAK2, has also been shown to be regulated by phosphorylation of an immediately adjacent residue, suggesting a conserved mode of regulation [14].
Our finding that mutations that promoted cytoplasmic localization of human ZIPK did not act similarly in the murine context highlight important interspecies differences within the ZIPKs and may have implications for understanding the physiological relevance of the protein in vivo. Using the murine form, we introduced either a phosphomimetic A299D or analogous RR294/295AA (mutant NLS2) mutation but, unexpectedly, observed continued nuclear localization. Such a lack of conservation of function at the NLS2 locus begs the question of why human and murine orthologs of ZIPK differ so greatly not only in their subcellular localization but presumable their function as well. The bulk of the protein sequence differences between the two species occur in the C-terminus of the proteins, exhibiting 66% identity and 79% similarity among residues 273–454. Since the NLS2 region is mostly conserved in sequence, such functional divergence for the mode of ZIPK nuclear entry must be the result of gross differences in their C-termini primary sequence or secondary structure. The hZIPK C-terminus acts as an autoinhibitory domain, whereby the kinase domain alone (1–273) demonstrated a six-fold increase in activity towards its known substrates, myosin and MYPT [5]. Perhaps the folding of the C-terminus of mZIPK prevents the access of the NLS2, thereby rendering the residue at the 299 position unimportant as a localization determinant. Alternatively, interacting proteins may mask NLS2 access. Regardless, we have shown that the straightforward conclusion that a residue change in mZIPK at a key phosphorylation site altered its localization properties does not apply and that the C-terminus of mZIPK has altered functionality compared to hZIPK. While NLS4 has been reported to be the functional NLS of mZIPK, C-terminal truncation mutants lacking NLS4 exhibited both cytoplasmic and nuclear staining suggesting a secondary mode of nuclear entry [10]. Under these circumstances, C-terminal truncation may merely act in a gain-of-function manner to expose the remaining NLS2 region of mZIPK and essentially allow nuclear import to operate by the hZIPK mechanism.
While the human T299 site can be an autophosphorylation site, upstream signaling by the Rho/ROCK pathway can also regulate the site. We have previously shown that ROCK can not only phosphorylate hZIPK at T265 for activation, but T299 as well [8]. We exploited the ΔLZ mutant form of hZIPK, which had previously demonstrated both impaired oligomerization and nuclear localization, to show that activating the Rho/ROCK pathway can affect the localization of hZIPK. These data suggest that Rho/ROCK signaling ensures that hZIPK is properly localized to the cytoplasm to complete the distal operations on substrates such as MYPT and mLC20. DAPK1 likely regulates T299 of hZIPK in a similar manner to manage its downstream cell death processes.
The divergent C-termini of ZIPKs differentially direct nuclear export. Murine ZIPK may lack cis elements that export the protein from the nucleus. This is overcome experimentally by overexpression of Par-4, which can associate with mZIPK and promote nuclear export of the complex [11, 12]. However, hZIPK cannot interact with Par-4 and must exit the nucleus differently [6]. We have shown that both exogenous and endogenous hZIPK export is mediated via the Crm1 export pathway because of its sensitivity to the nuclear export inhibitor LMB. To our knowledge, this is the first report of endogenous hZIPK localizing to the nucleus of tissue culture cells. While our microscopy shows snapshots of ZIPK localization, these data indicate that hZIPK can continuously shuttle between the nucleus and cytoplasm to constantly sample both environments of the cell and that nuclear entry is likely dependent upon T299 dephosphorylation. Perhaps a signal exists to promote an acute ZIPK dephosphorylation and nuclear entry, but we have not yet observed an example. We have mutated several residues in a potential NES (residues 378–386) to alanine, which would hypothetically cause a partial nuclear localization as LMB treatment did. However, no nuclear accumulation of these FLAG-tagged putative NES mutants was observed. These data indicate that either another NES is present or an interacting protein modulates the export of hZIPK.
Our findings herein therefore question the validity of studying ZIPK function in mouse and rat to understand the role of the enzyme in human physiology. We and others have demonstrated important roles for ZIPK in the regulation of myosin phosphorylation to control Ca+2-independent smooth muscle contraction as well as non-apoptotic cell death in non-muscle cells [15–18]. Sequence alignment of the C-termini of ZIPK across a broad range of species single out rat and mouse as oddities [6]. For example, human shares closer sequence identity in the C-terminus of ZIPK to evolutionary distinct species such as platypus than it does to mouse or rat. Indeed, mouse, rat, and guinea pigs are the only species defined to date which exhibit T299A mutations. Interestingly, the kangaroo rat protein has a C-terminal truncation that deletes the NLS4 but does retain a T299, suggesting that either an intact NLS2 or 4 must be present for a functional protein [6]. Taken together, the data shows that it is not possible to simply rescue the mZIPK nuclear localization by A299T mutations alone and suggest that mZIPK has evolved additional functions that are relevant to these species and no other. mZIPK has been shown to interact with the ATF4, Stat3, and AATF transcription factors, indicating a transcriptional control function; however, its nuclear function remains unknown [13, 19, 20]. As such, knockout studies of ZIPK in mouse may lack comprehensive relevance to human physiology. Additionally, hZIPK serves as a possible drug target for smooth muscle related disorders such as asthma or hypertension, but an animal model system other than mouse or rat must be chosen for this analysis.
Acknowledgments
We acknowledge NIH grant RO1 DK065954-05, and thank KH Scheidtmann for his generous gift of GFP-mZIPK. We thank Elizabeth Snyder for help with the preparation of figures and this manuscript.
Footnotes
Abbreviations. ZIPK, zipper-interacting protein kinase; mZIPK, murine ZIPK; hZIPK, human ZIPK; LC20, 20-kDa regulatory light chain; SMPP1M, myosin light chain phosphatase; MYPT, myosin phosphatase targeting subunit; mNLS2, mutant NLS2; ROCK, rho kinase; DAPK1, death-associated protein kinase 1; DRAK2, DAPK-related apoptosis-inducing kinase 2; Par-4, prostate apoptosis response gene 4
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haystead TA. Cell Signal. 2005;17(11):1313–1322. doi: 10.1016/j.cellsig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Ihara E, MacDonald JA. Can J Physiol Pharmacol. 2007;85(1):79–87. doi: 10.1139/y06-103. [DOI] [PubMed] [Google Scholar]
- 3.Murthy KS. Annu Rev Physiol. 2006;68:345–74. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
- 4.Somlyo AP, Somlyo AV. Physiol Rev. 2003;83(4):1325–358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 5.Graves PR, Winkfield KM, Haystead TA. J Biol Chem. 2005;280(10):9363–374. doi: 10.1074/jbc.M412538200. [DOI] [PubMed] [Google Scholar]
- 6.Shoval Y, Pietrokovski S, Kimchi A. PLoS Genet. 2007;3(10):1884–893. doi: 10.1371/journal.pgen.0030180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kogel D, Plottner O, Landsberg G, Christian S, Scheidtmann KH. Oncogene. 1998;17(20):2645–654. doi: 10.1038/sj.onc.1202204. [DOI] [PubMed] [Google Scholar]
- 8.Hagerty L, Weitzel DH, Chambers J, Fortner CN, Brush MH, Loiselle D, Hosoya H, Haystead TA. J Biol Chem. 2007;282(7):4884–893. doi: 10.1074/jbc.M609990200. [DOI] [PubMed] [Google Scholar]
- 9.Shani G, Marash L, Gozuacik D, Bialik S, Teitelbaum L, Shohat G, Kimchi A. Mol Cell Biol. 2004;24(19):8611–626. doi: 10.1128/MCB.24.19.8611-8626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kogel D, Bierbaum H, Preuss U, Scheidtmann KH. Oncogene. 1999;18(51):7212–218. doi: 10.1038/sj.onc.1203169. [DOI] [PubMed] [Google Scholar]
- 11.Page G, Kogel D, Rangnekar V, Scheidtmann KH. Oncogene. 1999;18(51):7265–273. doi: 10.1038/sj.onc.1203170. [DOI] [PubMed] [Google Scholar]
- 12.Vetterkind S, Illenberger S, Kubicek J, Boosen M, Appel S, Naim HY, Scheidtmann KH, Preuss U. Exp Cell Res. 2005;305(2):392–08. doi: 10.1016/j.yexcr.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Matsumoto M, Takeda K, Sanjo H, Akira S. Mol Cell Biol. 1998;18(3):1642–651. doi: 10.1128/mcb.18.3.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuwahara H, Nishizaki M, Kanazawa H. J Biochem. 2008;143(3):349–58. doi: 10.1093/jb/mvm236. [DOI] [PubMed] [Google Scholar]
- 15.Endo A, Surks HK, Mochizuki S, Mochizuki N, Mendelsohn ME. J Biol Chem. 2004;279(40):42055–2061. doi: 10.1074/jbc.M403676200. [DOI] [PubMed] [Google Scholar]
- 16.MacDonald JA, Borman MA, Muranyi A, Somlyo AV, Hartshorne DJ, Haystead TA. Proc Natl Acad Sci U S A. 2001;98(5):2419–2424. doi: 10.1073/pnas.041331498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niiro N, Ikebe M. J Biol Chem. 2001;276(31):29567–29574. doi: 10.1074/jbc.M102753200. [DOI] [PubMed] [Google Scholar]
- 18.Murata-Hori M, Fukuta Y, Ueda K, Iwasaki T, Hosoya H. Oncogene. 2001;20(57):8175–8183. doi: 10.1038/sj.onc.1205055. [DOI] [PubMed] [Google Scholar]
- 19.Sato N, Kawai T, Sugiyama K, Muromoto R, Imoto S, Sekine Y, Ishida M, Akira S, Matsuda T. Int Immunol. 2005;17(12):1543–1552. doi: 10.1093/intimm/dxh331. [DOI] [PubMed] [Google Scholar]
- 20.Page G, Lodige I, Kogel D, Scheidtmann KH. FEBS Lett. 1999;462(1–2):187–191. doi: 10.1016/s0014-5793(99)01529-x. [DOI] [PubMed] [Google Scholar]