

Abstract
Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency is an autosomal recessive disorder and is the most common cause of ambiguous genitalia in the newborn. The genes encoding 21-hydroxylase, CYP21A2, and tenascin-X (TNX), TNXB, are located within the HLA complex, in a region of high gene density termed the RCCX module. The module has multiple pseudogenes as well as tandem repeat sequences that promote misalignment during meiosis leading to complex gene rearrangements, deletions and gene conversion events. CYP21A2 mutations cause CAH, and TNX deficiency has been identified as a cause of hypermobility type Ehlers-Danlos syndrome (EDS). Here we report on a three-generation family with a heterozygous deletion encompassing CYP21A2 and TNXB that initially came to medical attention due to the diagnosis of CAH in the proband. Southern blotting and PCR-based analysis of the RCCX module revealed a CYP21A2 deletion extending into TNXB in one allele and a CYP21A2 point mutation in the other allele. Family history is notable for joint hypermobility. Additional radiological and clinical investigations showed a quadricuspid aortic valve, single kidney, bicornuate uterus and a bifid uvula in the proposita, and mitral valve prolapse in her mother. These findings further delineate the phenotype of the CAH-TNX contiguous gene deletion syndrome and point to an intersection of connective tissue dysplasias with a common gene-mediated endocrine disorder.
Keywords: congenital adrenal hyperplasia, CAH, CYP21A2, tenascin-X, TNXB, Ehlers-Danlos Syndrome, EDS, hypermobility, aortic valve, TNX, contiguous deletion syndrome
Introduction
Congenital adrenal hyperplasia (CAH) is a general term used to describe a group of inherited disorders of cortisol biosynthesis. Approximately 95 percent of patients with CAH have 21-hydroxylase deficiency. The gene encoding 21-hydroxylase, CYP21A2, and the gene encoding tenascin-X (TNX), TNXB, are located within the HLA complex on Chromosome 6 in a region of high gene density termed the RCCX module (Yang and others 1999). The presence of pseudogenes and tandem repeats within the module lead to increased frequency of deletions and gene conversion events. Recessively inherited CYP21A2 point mutations or deletions cause CAH. Carriership for CYP21A2 mutations is estimated to be as high as 10% globally, with varying frequencies in different populations (Merke and Bornstein 2005).
TNX is a large extracellular matrix protein that plays a role in deposition of collagen by dermal fibroblasts and is expressed in the dermis of skin and in the connective tissue of heart and skeletal muscle (Burch and others 1995; Matsumoto and others 1994). TNXB was discovered due to its 3′ overlap with the human CYP21A2 gene (Morel and others 1989). TNX deficiency, in recessive or dominant form, has been proposed as a cause of hypermobility type Ehlers-Danlos Syndrome (EDS) (Schalkwijk and others 2001; Zweers and others 2003). The clinical spectrum of TNX deficiency described to date predominantly includes joint hypermobility, stretchy hyperextensible skin without abnormal (atrophic) scars, and easy bruising (Burch and others 1997; Lindor and Bristow 2005; Schalkwijk and others 2001; Zweers and others 2003). Large joint dislocations are the most frequent clinically significant finding; however, mitral valve prolapse, chronic obstructive pulmonary disease, diverticulosis, rectal and uterine prolapse, and muscle weakness have also been reported (Bristow and others 2005; Lindor and Bristow 2005; Schalkwijk and others 2001; Voermans and others 2007; Zweers and others 2003).
Here, we report the clinical and molecular features of a three-generation family with a heterozygous deletion encompassing CYP21A2 and TNXB (Fig. 1).
Fig. 1.
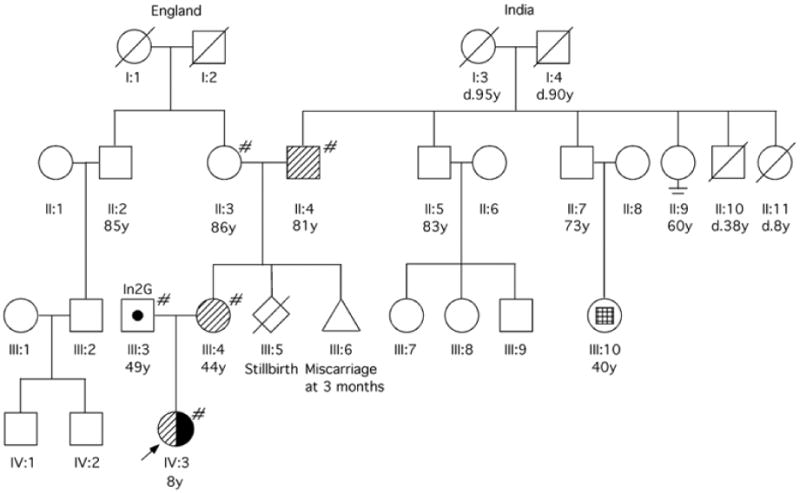
Pedigree of the family. # Genetic analysis performed;  Carrier of TNXB/XA fusion gene (CYP21A2 deletion);
Carrier of TNXB/XA fusion gene (CYP21A2 deletion);  Carrier of CYP21A2 point mutation (In2G);
Carrier of CYP21A2 point mutation (In2G);  Affected by CAH, compound heterozygote (TNXB/XA, In2G).
Affected by CAH, compound heterozygote (TNXB/XA, In2G).  Facial asymmetry with ipsilateral ear dysplasia and unilateral deafness, but normal intelligence.
Facial asymmetry with ipsilateral ear dysplasia and unilateral deafness, but normal intelligence.
Clinical Report
The proposita with ambiguous genitalia, weighing 3.3 kg and 54.6 cm in length, was born at 41 weeks gestation by spontaneous vaginal delivery to a healthy 36 year old woman. An ultrasound of her genitourinary tract showed a single kidney, a bicornuate uterus, and grade III vesicoureteral reflux. Peripheral blood karyotype was 46, XX (20 metaphases analyzed). A serum 17-hydroxyprogesterone level of 7,974 ng/dl (normal < 200 ng/dl) confirmed the diagnosis of CAH due to 21-hydroxylase deficiency. At 7 months of age, the patient underwent urogenital surgery which included ureteral reimplantation.
At 8 years of age, physical examination showed a well-developed, well-nourished child of normal height and weight, bifid uvula, single palmar crease on the left hand, normal skin, mild arachnodactyly and hyperextensible joints with a Beighton score of 8 (Fig. 2).
Fig. 2.
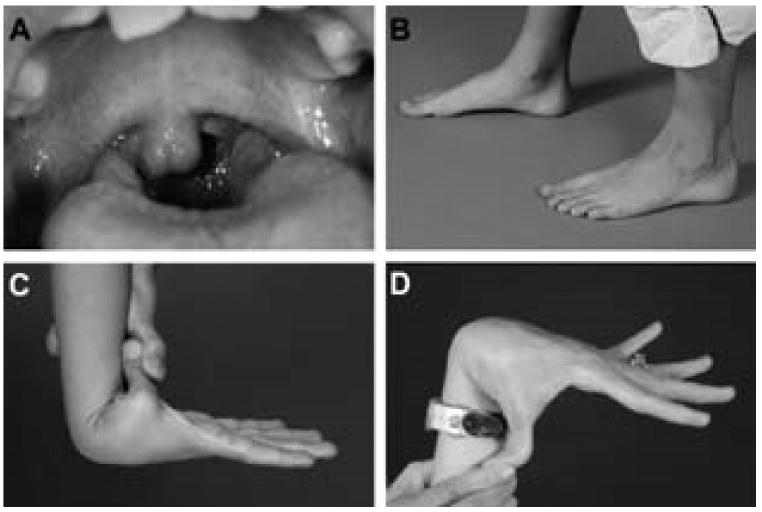
Phenotypic features include: A: bifid uvula (proposita); B: mild arachnodactyly (proposita); C: hyperextensible joints (proposita); D: hyperextensible joints (mother).
The proposita's father, a healthy man with Ashkenazi Jewish ancestry had a Beighton score of 0. The proband's mother, of British and Indian ancestry, had hyperextensible joints with a Beighton score of 7. She reported a medical history of urethral prolapse at age10 years that was repaired, a hiatal hernia resulting in gastroesophageal reflux, and a diagnosis of sarcoidosis at 41 years. The parents denied consanguinity. The maternal grandmother, of British ancestry, has coronary artery disease, Paget disease and osteoporosis. The maternal grandfather, of Indian ancestry, has a history of myocardial infarction at age 79 years, and has had a liver transplant for cryptogenic cirrhosis. He has hyperextensible thumbs. The mother's paternal cousin was reported to have hemifacial microsomia with a shortened jaw, abnormal ear formation and deafness (Fig. 1).
Methods
Mutation Detection
DNA samples were isolated from peripheral blood. Functional CYP21A2 gene was amplified by PCR using gene-specific primers and conditions (Krone and others 2002).
Southern Blot
Restriction enzymes TaqI and PshAI (New England Biolabs, Ipswich, MA) were used in Southern Blots conducted per standard protocol. Probes E and F, used for hybridization to the CYP21 and TNX genes, respectively, are previously described (Chung and others 2005). The RP gene probe, a 743 bp fragment corresponding to the end of the DNA sequence in RP1 and RP2, was an amplicon made by primers 5′ CACCTTTCCCCTTTCCTGT 3′ and 5′ ATCCCACCTTCCTATTTCAA 3′ from commercial human genomic DNA (BD, Franklin Lakes, NJ).
PCR Detection
The PCR method to detect TNXB/XA fusion genes used primers to amplify genomic DNA as described previously (Burch and others 1997). Primer design is depicted in Figure 3A. The PCR reaction was performed with ExTaq DNA polymerase (Hot-Start Version; Takara Bio USA, Madison, WI) in the presence of 1 μl DMSO (Sigma, St Louis, MO). PCR conditions were as follows: initial cycle of 94°C for 4 min, followed by 30 cycles of 94°C for 30s, 62°C (for TNXB or 60°C for TNXA) for 45s and 72°C for 1 min, and a final cycle of 72°C for 10 min. PCR products were resolved on 1% agarose gels.
Fig. 3.
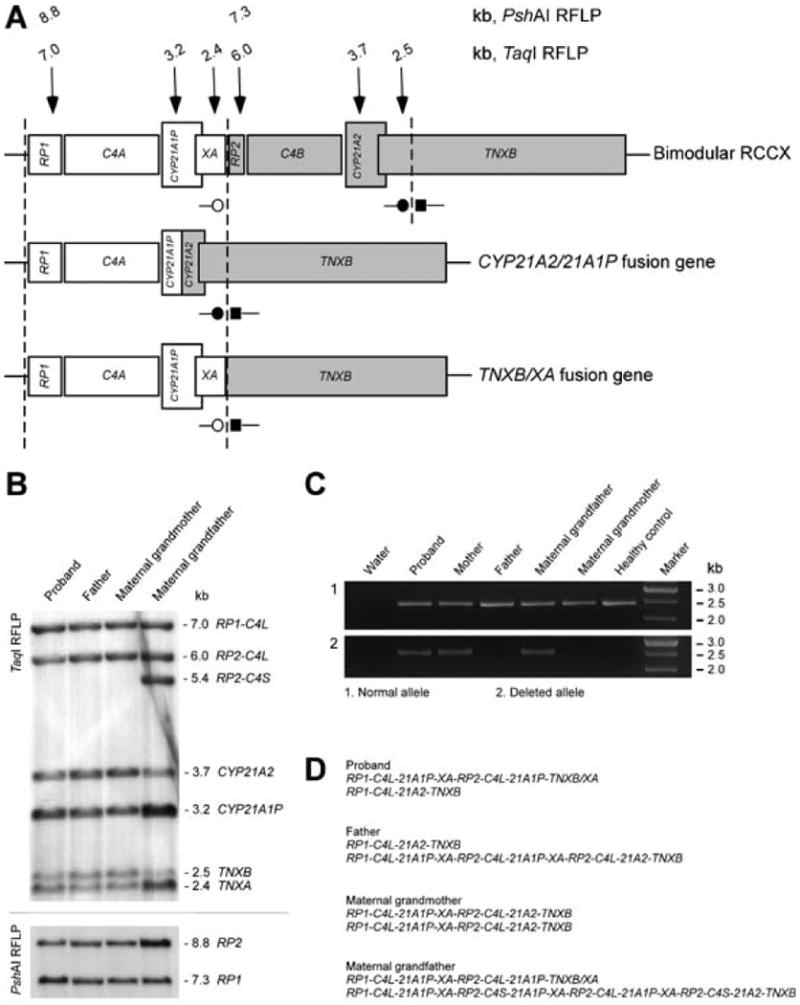
A. Genomic structure of a bimodular haplotype for RCCX and deletions leading to two different fusion genes. Dashed lines (¦) indicate duplication boundaries. Vertical arrows (↓) indicate the locations of DNA probes and sizes of digested fragments from Southern blots. ■– TNXB-specific forward primer; ●– TNXB-specific reverse primer; ○– TNXA-specific reverse primer. The common 30 kb deletion in CAH patients, covering the TNXA-specific reverse primer, results in a non-functional CYP21A1P/21A2 fusion gene (middle panel). Recombination between TNXB and TNXA results in a deletion, TNXB/XA fusion gene and 2.4 kb product (lower panel). B. Southern blots detecting the TNXB/XA fusion gene. TaqI RFLP demonstrated the RCCX haplotype and gene contents at the RCCX locus (upper panel). PshAI RFLP confirmed RP genotyping (lower panel). The father had equal intensities from each gene pair (RP1:RP2, CYP21A2:CYP21A1P, TNXB:TNXA). The proposita had reduced intensities from CYP21A2 and TNXB, indicating a large deletion encompassing both genes. C4L, longer copy of C4; C4S, shorter copy of C4. C. PCR amplification of genomic DNA from five family members and a healthy control. A reaction with only water (no DNA) as template is also shown. All family members and the control demonstrated a 2.4 kb fragment produced by the TNXB-specific forward primer and TNXB-specific reverse primer (upper panel). The proposita, mother and maternal grandfather exhibited a 2.4 kb fragment produced by the TNXB-specific forward primer and TNX-specific reverse primer (lower panel), indicating heterozygosity for the TNXB/XA fusion gene. D. The haplotype organizations of RCCX in each subject with Southern blot.
Echocardiography and Cardiac MRI
Cardiovascular magnetic resonance imaging (MRI) of the heart, including the aortic valve, was performed on a 1.5 Tesla scanner (Avanto 1.5T; Siemens, Malvern, PA) using steady-state free precession cine, black-blood turbo spin echo, and velocity-encoding MRI techniques. Transthoracic 2-dimensional echocardiography (Vivid i; GE Healthcare, Chalfont St. Giles, UK) was performed in standard parasternal, apical, and subcostal views, with the subject in the left lateral recumbent position.
Results
Mutation Detection
For the proposita, the multiplex mini-sequencing assay detected a heterozygous In2G mutation in CYP21A2. No heterozygosity was detected for the single nucleotide polymorphic markers (SNPs) screened suggesting that the patient has the In2G mutation on one allele and a deletion of the other allele. Multiplex mini-sequencing of the father's DNA sample detected the In2G mutation, whereas the mother's DNA revealed lack of heterozygosity at 12 known SNPs, consistent with a deletion of one copy of CYP21A2.
Southern blot and PCR-based analysis of the RCCX module
The proposita had reduced ratios of CYP21A2:CYP21A1P and TNXB:TNXA, confirming the large deletion (Fig. 3B). PCR-based detection showed that the deletion was present in the proposita's mother and maternal grandfather (Fig. 3C).
Echocardiography and Cardiac MRI
The proposita's quadricuspid aortic valve was best visualized in diastole and systole using steady-state free precession cine MRI (Fig. 4). Black-blood MRI also clearly depicted four leaflets during diastole. Velocity-encoded MRI showed systolic blood flow through a 4-sided polygonal-shaped aortic orifice with no stenosis. There was trivial aortic regurgitation. A transthoracic echocardiogram documented the quadricuspid configuration in diastole, however, it was difficult to resolve the four leaflets from the walls of the aortic root during systole. Systolic function was normal.
FIG. 4.
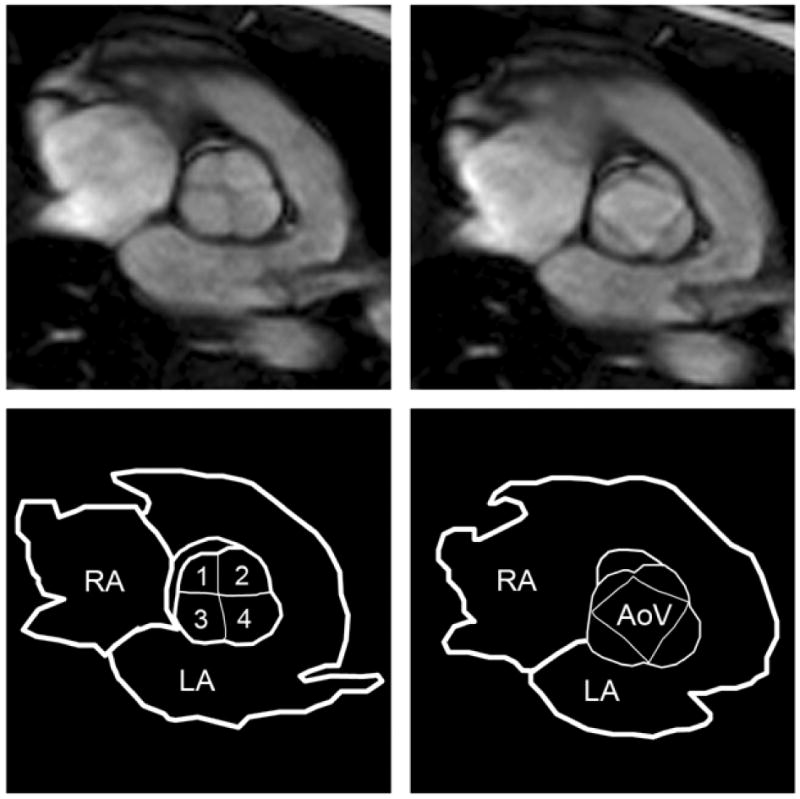
Cardiac magnetic resonance steady-state free precession images. The quadricuspid aortic valve is depicted in diastole (left) and in systole (right) with corresponding line drawings to label the anatomy. RA, right atrium; LA, left atrium; AoV, aortic valve; 1, 2, 3, 4 indicate cusps of the aortic valve.
Transthoracic echocardiogram of the mother revealed a redundant anterior mitral valve leaflet, with mild mitral prolapse and trace regurgitation. She had a normal tricuspid aortic valve.
Discussion
Here, we describe a novel phenotype that includes CAH, joint hypermobility, midline defects and major cardiac valvular abnormalities in a proposita who is a compound heterozygote of the TNXB/XA fusion gene, with CYP21A2 deletion of one allele and a CYP21A2 point mutation in the other allele, further delineating the phenotype of the CAH-TNX contiguous gene deletion syndrome.
The first patient with CAH due to 21-hydroxylase deficiency associated with an EDS phenotype was a 26 year old male with skin hyperextensibility and joint hypermobility, but without the atrophic scars and poor wound healing characteristic of classic EDS (Burch and others 1997). Absent TNX mRNA and protein, a maternal CYP21A2 mutation, and a paternal TNXB/XA fusion gene with a deletion of CYP21A2 were found. An autosomal recessive pattern of inheritance was postulated, as the father and sister were heterozygous for the TNXB/XA fusion gene, and both relatives were phenotypically normal. However, a second TNXB mutation on the maternal allele was not identified. Subsequently, in a cohort of 151 patients with classic EDS, five patients were found to have autosomal recessive TNX deficiency based on undetectable serum TNX (Schalkwijk and others 2001). Four of these five patients were found to have a large deletion and/or truncating mutation of TNXB on both alleles, one patient was heterozygous for the TNXB/XA fusion gene, and one patient who was homozygous for the TNXB/XA fusion gene also had CAH. Autosomal recessive TNX deficiency without CAH was described in 2 additional patients, with an EDS phenotype of marked skin hyperextensibility, easy bruising and joint laxity (Lindor and Bristow 2005).
Although EDS due to TNXB mutations was initially described as autosomal recessive, haploinsufficient subjects have approximately half of the normal level of TNX in their serum, and clinically may have joint hypermobility, joint subluxations and chronic musculoskeletal pain (Zweers and others 2003). Interestingly, a female preponderance has been found in heterozygote individuals who have clinical findings of hypermobility type EDS. Whether or not having CAH in addition to haploinsufficiency for a TNXB mutation changes the risk of having an EDS phenotype is unknown. A study of 39 Dutch families with CAH due to 21-hydroxylase deficiency found a TNXB/XA fusion gene on 4 out of 77 chromosomes (Koppens and others 2002), suggesting that carriership of a TNXB mutation may be common in CAH.
Our patient had a quadricuspid aortic valve found on cardiac MRI and transthoracic echocardiogram at 8 years of age. Quadricuspid aortic valves are rare and occur with a prevalence of approximately 1-5 per 10,000 people (Feldman and others 1990). Aortic regurgitation is the predominant functional abnormality associated with quadricuspid aortic valve, and is postulated to be mostly due to a small accessory cusp (Armen and others 2008; Feldman and others 1990). It is typically asymptomatic in childhood but can progress over time (Armen and others 2008; Feldman and others 1990; Holt and others 2007). Valve replacement is often required by the fifth or sixth decade. Early detection of quadricuspid aortic valve is essential for the prevention of valvular and ventricular dysfunction (Armen and others 2008).
Cardiac defects associated with EDS vary depending on the subtype. In hypermobility type EDS, mitral valve prolapse was reported to be the most common cardiac defect (Peeters and others 2004), although mildly dilated aortic root, mitral, tricuspid and aortic regurgitation, and impaired left ventricular relaxation have also been noted (McDonnell and others 2006). Quadricuspid aortic valve has been reported in a 16 year old boy with a clinical diagnosis of EDS Type II (Dotti and others 1999) which would now be classified as the classical form of EDS (Beighton and others 1998), and may be caused by a homozygous TNXB defect (Burch and others 1997; Schalkwijk and others 2001). A case of recessive form of EDS in Japanese female sibs without accompanying molecular etiology was reported to involve congenital heart anomalies including atrial septal defect, ventricular septal defect, bicuspid right atrioventricular valve and persistent atrioventricular canal (Maeda and others 1996). Recently, a recessive cardiac valvular form of EDS due to COL1A2 mutations was reported (Schwarze and others 2004). Mitral valve prolapse has been reported in patients with TNX deficiency and EDS (Lindor and Bristow 2005; Schalkwijk and others 2001).
To our knowledge, there are no prior reports of quadricuspid aortic valves or congenital heart malformations associated with CAH. Similarly, midline defects are not characteristic of CAH. However, an increased incidence of upper-tract genitourinary malformations has been reported in CAH (Nabhan and Eugster 2007). Our patient had several additional midline defects including a bifid uvula, a single kidney and bicornuate uterus. There are two earlier clinical reports of cleft palate in association with presumed diagnosis of EDS: a 6-year old female with a cleft lip and palate and classic EDS (Okamura and Matsumoto 1984), and an infant with cleft palate, micrognathia, mitral valve prolapse and EDS Type II associated with dermal collagen abnormalities (Rizzo and others 1987). However, cleft palate or bifid uvula is not frequently encountered in patients with EDS in current clinical practice.
The propostia's mother, who carries the TNXB/XA fusion gene and is heterozygous for CYP21A1 deletion, has clinical features suggestive of TNX deficiency including hyperextensible joints, a history of urethral prolapse, a hiatal hernia with gastroesophageal reflux, and mitral valve prolapse.
The family presented here represents the genetic and phenotypic complexity of the RCCX module. The optimal medical management of patients with TNX deficiency is unclear. However, the findings in our patient suggest that patients with CAH due to 21-hydroxylase deficiency who have clinical features suggestive of TNX deficiency or develop a cardiac murmur should be screened with a baseline echocardiogram to detect possible valvular abnormalities. This clinical report underscores the need for future clinical studies of CAH patients who may have an associated TNX deficiency.
Acknowledgments
We thank the family who participated in this research. We thank the NIA Core Lab Staff for DNA extraction and purification, and LCI staff Ronnie Black and Tina Roberson for help with sample transmittals. We are indebted to Dr. CY Yu (Center for Molecular and Human Genetics, Columbus Children's Research Institute, Columbus, OH) for a kind gift of probes for the RCCX module. We thank Dr. Michel Bernier for assistance with the preparation of the figures. Patient samples and clinical data were collected under NICHD IRB# 06CH001 (Clinical Trials number NCT00250159). All work was supported by funds originating at the intramural programs of NIA/NICHD/NIH and (in part) by the Congenital Adrenal Hyperplasia Research, Education and Support (CARES) Foundation.
References
- Armen TA, Vandse R, Bickle K, Nathan N. Three-dimensional echocardiographic evaluation of an incidental quadricuspid aortic valve. Eur J Echocardiogr. 2008;9(2):318–20. doi: 10.1016/j.euje.2007.03.041. [DOI] [PubMed] [Google Scholar]
- Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Am J Med Genet. 1998;77(1):31–7. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Bristow J, Carey W, Egging D, Schalkwijk J. Tenascin-X, collagen, elastin, and the Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2005;139C(1):24–30. doi: 10.1002/ajmg.c.30071. [DOI] [PubMed] [Google Scholar]
- Burch GH, Bedolli MA, McDonough S, Rosenthal SM, Bristow J. Embryonic expression of tenascin-X suggests a role in limb, muscle, and heart development. Dev Dyn. 1995;203(4):491–504. doi: 10.1002/aja.1002030411. [DOI] [PubMed] [Google Scholar]
- Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, Miller WL, Bristow J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet. 1997;17(1):104–8. doi: 10.1038/ng0997-104. [DOI] [PubMed] [Google Scholar]
- Chung EK, Wu YL, Yang Y, Zhou B, Yu CY. Human complement components C4A and C4B genetic diversities: complex genotypes and phenotypes. Curr Protoc Immunol. 2005;Chapter 13 doi: 10.1002/0471142735.im1308s68. Unit 13 8. [DOI] [PubMed] [Google Scholar]
- Dotti MT, De Stefano N, Mondillo S, Agricola E, Federico A. Neurological involvement and quadricuspid aortic valve in a patient with Ehlers-Danlos syndrome. J Neurol. 1999;246(7):612–3. doi: 10.1007/s004150050414. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Khandheria BK, Warnes CA, Seward JB, Taylor CL, Tajik AJ. Incidence, description and functional assessment of isolated quadricuspid aortic valves. Am J Cardiol. 1990;65(13):937–8. doi: 10.1016/0002-9149(90)91446-d. [DOI] [PubMed] [Google Scholar]
- Holt NF, Sivarajan M, Mandapati D, Printsev Y, Elefteriades JA. Quadricuspid aortic valve with aortic insufficiency: case report and review of the literature. J Card Surg. 2007;22(3):235–7. doi: 10.1111/j.1540-8191.2007.00399.x. [DOI] [PubMed] [Google Scholar]
- Koppens PF, Hoogenboezem T, Degenhart HJ. Carriership of a defective tenascin-X gene in steroid 21-hydroxylase deficiency patients: TNXB -TNXA hybrids in apparent large-scale gene conversions. Hum Mol Genet. 2002;11(21):2581–90. doi: 10.1093/hmg/11.21.2581. [DOI] [PubMed] [Google Scholar]
- Krone N, Braun A, Weinert S, Peter M, Roscher AA, Partsch CJ, Sippell WG. Multiplex minisequencing of the 21-hydroxylase gene as a rapid strategy to confirm congenital adrenal hyperplasia. Clin Chem. 2002;48(6 Pt 1):818–25. [PubMed] [Google Scholar]
- Lindor NM, Bristow J. Tenascin-X deficiency in autosomal recessive Ehlers-Danlos syndrome. Am J Med Genet A. 2005;135(1):75–80. doi: 10.1002/ajmg.a.30671. [DOI] [PubMed] [Google Scholar]
- Maeda T, Suzuki Y, Haeno S, Asada M, Hiramatsu R, Tanaka F, Okada M, Suzuki T. Ehlers-Danlos syndrome and congenital heart anomalies. Intern Med. 1996;35(3):200–2. doi: 10.2169/internalmedicine.35.200. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Saga Y, Ikemura T, Sakakura T, Chiquet-Ehrismann R. The distribution of tenascin-X is distinct and often reciprocal to that of tenascin-C. J Cell Biol. 1994;125(2):483–93. doi: 10.1083/jcb.125.2.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell NB, Gorman BL, Mandel KW, Schurman SH, Assanah-Carroll A, Mayer SA, Najjar SS, Francomano CA. Echocardiographic findings in classical and hypermobile Ehlers-Danlos syndromes. Am J Med Genet A. 2006;140(2):129–36. doi: 10.1002/ajmg.a.31035. [DOI] [PubMed] [Google Scholar]
- Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365(9477):2125–36. doi: 10.1016/S0140-6736(05)66736-0. [DOI] [PubMed] [Google Scholar]
- Morel Y, Bristow J, Gitelman SE, Miller WL. Transcript encoded on the opposite strand of the human steroid 21-hydroxylase/complement component C4 gene locus. Proc Natl Acad Sci U S A. 1989;86(17):6582–6. doi: 10.1073/pnas.86.17.6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabhan ZM, Eugster EA. Upper-tract genitourinary malformations in girls with congenital adrenal hyperplasia. Pediatrics. 2007;120(2):e304–7. doi: 10.1542/peds.2006-2993. [DOI] [PubMed] [Google Scholar]
- Okamura H, Matsumoto Y. A case of Ehlers-Danlos syndrome associated with cleft lip and palate. J Laryngol Otol. 1984;98(3):311–5. doi: 10.1017/s0022215100146626. [DOI] [PubMed] [Google Scholar]
- Peeters AC, Kucharekova M, Timmermans J, van den Berkmortel FW, Boers GH, Novakova IR, Egging D, den Heijer M, Schalkwijk J. A clinical and cardiovascular survey of Ehlers-Danlos syndrome patients with complete deficiency of tenascin-X. Neth J Med. 2004;62(5):160–2. [PubMed] [Google Scholar]
- Rizzo R, Contri MB, Micali G, Quaglino D, Pavone L, Ronchetti IP. Familial Ehlers-Danlos syndrome type II: abnormal fibrillogenesis of dermal collagen. Pediatr Dermatol. 1987;4(3):197–204. doi: 10.1111/j.1525-1470.1987.tb00778.x. [DOI] [PubMed] [Google Scholar]
- Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345(16):1167–75. doi: 10.1056/NEJMoa002939. [DOI] [PubMed] [Google Scholar]
- Schwarze U, Hata R, McKusick VA, Shinkai H, Hoyme HE, Pyeritz RE, Byers PH. Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Hum Genet. 2004;74(5):917–30. doi: 10.1086/420794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voermans NC, Jenniskens GJ, Hamel BC, Schalkwijk J, Guicheney P, van Engelen BG. Ehlers-Danlos syndrome due to tenascin-X deficiency: muscle weakness and contractures support overlap with collagen VI myopathies. Am J Med Genet A. 2007;143A(18):2215–9. doi: 10.1002/ajmg.a.31899. [DOI] [PubMed] [Google Scholar]
- Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular variations of the human major histocompatibility complex class III genes for serine/threonine kinase RP, complement component C4, steroid 21-hydroxylase CYP21, and tenascin TNX (the RCCX module). A mechanism for gene deletions and disease associations. J Biol Chem. 1999;274(17):12147–56. doi: 10.1074/jbc.274.17.12147. [DOI] [PubMed] [Google Scholar]
- Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, Kucharekova M, Boezeman JB, Schalkwijk J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73(1):214–7. doi: 10.1086/376564. [DOI] [PMC free article] [PubMed] [Google Scholar]