

Abstract
Insulin resistance is characteristic of obesity, type 2 diabetes, and components of the cardiometabolic syndrome, including hypertension and dyslipidemia, that collectively contribute to a substantial risk for cardiovascular disease. Metabolic actions of insulin in classic insulin target tissues (eg, skeletal muscle, fat, and liver), as well as actions in nonclassic targets (eg, cardiovascular tissue), help to explain why insulin resistance and metabolic dysregulation are central in the pathogenesis of the cardiometabolic syndrome and cardiovascular disease. Glucose and lipid metabolism are largely dependent on mitochondria to generate energy in cells. Thereby, when nutrient oxidation is inefficient, the ratio of ATP production/oxygen consumption is low, leading to an increased production of superoxide anions. Reactive oxygen species formation may have maladaptive consequences that increase the rate of mutagenesis and stimulate proinflammatory processes. In addition to reactive oxygen species formation, genetic factors, aging, and reduced mitochondrial biogenesis all contribute to mitochondrial dysfunction. These factors also contribute to insulin resistance in classic and nonclassic insulin target tissues. Insulin resistance emanating from mitochondrial dysfunction may contribute to metabolic and cardiovascular abnormalities and subsequent increases in cardiovascular disease. Furthermore, interventions that improve mitochondrial function also improve insulin resistance. Collectively, these observations suggest that mitochondrial dysfunction may be a central cause of insulin resistance and associated complications. In this review, we discuss mechanisms of mitochondrial dysfunction related to the pathophysiology of insulin resistance in classic insulin-responsive tissue, as well as cardiovascular tissue.
Keywords: mitochondrial dysfunction, insulin resistance, cardiovascular disease
There are at least 47 million people in the United States who have the cardiometabolic syndrome, a precursor to diabetes and subsequent cardiovascular complications.1 Furthermore, the development of insulin resistance, the cardinal feature of the cardiometabolic syndrome, is associated with increased tissue renin–angiotensin system activity and increasingly appears to be a nexus between components of the syndrome.2,3 The metabolic actions of insulin maintain glucose homeostasis by promoting glucose uptake in skeletal muscle and suppressing glucose production in the liver. Insulin resistance is typically defined as decreased sensitivity to these metabolic actions of insulin. Insulin-resistant individuals are at higher risk of developing type 2 diabetes mellitus (T2DM) and cardiovascular disease compared with subjects with normal insulin sensitivity.2,4 Studies from our group have shown that increased tissue expression of angiotensin II via stimulation of the angiotensin II type 1 receptor (AT1R) can cause mitochondrial morphological and functional abnormalities in skeletal muscle, and liver (Figure 1) as well as cardiovascular tissue (Figure 2).5–7 Furthermore, we and others have shown that blockade of the AT1R reduces oxidative stress and mitochondrial structure and functional abnormalities in rodent models of excessive tissue renin–angiotensin system activity.7,8
Figure 1.
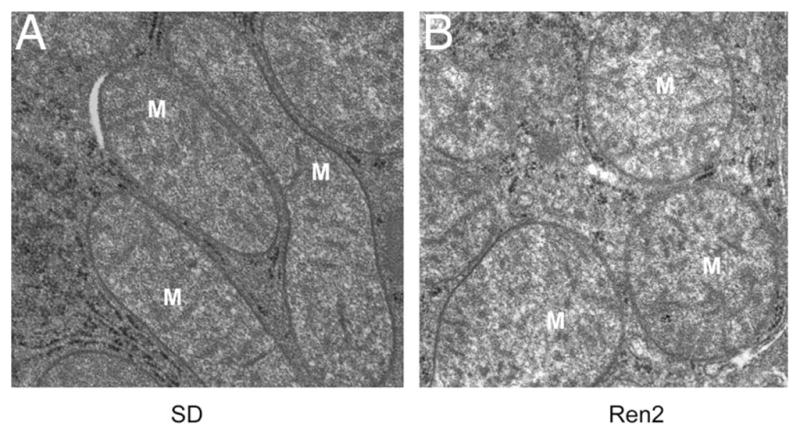
Mitochondrial abnormality in the liver of transgenic Ren2 male rat at 12 weeks of age. A, The normal hepatic mitochondrial morphology in the Sprague–Dawley control male rat model at 12 weeks of age. B, Mitochondrial abnormality in morphology in the 12-week-old transgenic Ren2 male rat model of hypertension and insulin resistance. Note the swollen and decreased matrix density by transmission electronic microscopy. Magnification, ×25 000.
Figure 2.

Mitochondrial biogenesis in the transgenic Ren2 male rat at 10 weeks of age. A, The longitudinal normal myocardial mitochondrial morphology in the Sprague–Dawley control male rat model at 10 weeks of age. Note the orderly and linearly arranged sarcomeres (closed arrows) and subsarcolemmal (sarcoplasmic reticulum) mitochondria (open arrows). Normal intercalated disc (arrowheads). Magnification, ×10 000. Bar=500 nm. B, Mitochondrial biogenesis in the 10-week-old transgenic Ren2 untreated control (Ren2C) rats, which display insulin resistance and abnormalities in both systolic and diastolic cardiac functions. Note the biogenesis of increased myocardial interdigitating mitochondria. Also note the loss of the orderly and linearly arranged sarcomeres. Magnification, ×10 000. Bar=500 nm.
Metabolic regulation is largely dependent on mitochondria, which play an important role in energy homeostasis by metabolizing nutrients and producing ATP and heat. Imbalance between energy intake and expenditure leads to mitochondrial dysfunction, characterized by a reduced ratio of energy production (ATP production) to respiration.9 Genetic and environmental factors including exercise, diet, aging, and stress affect both mitochondrial function and insulin sensitivity.10,11 Importantly, it has been shown that mitochondrial dysfunction is associated with insulin resistance in skeletal muscle,12–14 as well as in other tissues, including liver, fat, heart, vessels, and pancreas.15–18 Thus, insulin resistance caused in part by mitochondrial dysfunction may contribute to a common pathophysiologic etiology for many chronic diseases.
Mitochondrial Function
Aerobic organisms consume oxygen to produce energy from nutrients. In eukaryotic cells, energy production, mostly in the form of ATP, is controlled by mitochondria that link oxidative respiration with metabolism of nutrients (Figure 3). Mitochondria are compartmentalized by outer and inner membranes, and the mitochondrial respiratory chain is located in the inner membrane. Production of ATP requires 2 major steps, oxidation of NADH (or FADH2) and phosphorylation of ADP to form ATP (oxidative phosphorylation [OXPHOS]). These 2 reactions are coupled in mitochondria, and OXPHOS is an efficient and energy-conserving way of producing energy in aerobic organisms. NADH or FADH2 are generated during glucose metabolism via glycolysis and the tricarboxylic acid cycle or β-oxidation of fatty acids. NADH or FADH2 are oxidized to NAD+ or FAD while protons are pumped to the intermitochondrial membrane through respiratory complexes I, III, and IV. Electrons from NADH or FADH2 are then transferred through a series of respiratory chain complexes to O2, which finally generates H2O. A proton gradient across the membrane is the driving force of F0F1-ATPase (ATP synthase) to produce ATP from ADP. ATP is transported to the cytosol by exchanging with ADP through adenine nucleotide translocator and used for various biological events that require energy. On the other hand, mitochondria generate heat by a mechanism called “proton leak.” Proton leak from the intermembrane space to matrix (uncoupling) reduces proton-motive force and generates heat instead ATP. Uncoupling proteins (UCPs) play a major role in reducing the proton gradient.19 UCP1 is expressed almost exclusively in brown adipose tissue. UCP2 is ubiquitously expressed, and UCP3 is expressed in skeletal muscle. UCP1, up to 10% of membrane protein, regulates adaptive thermogenesis, whereas UCP2 and -3 do not appear to play a major role in thermogenesis; mice with genetic ablation of UCP2 and -3 display a normal response to cold, normal basal proton conductance, and normal body weight.20,21 Indeed, overexpression of UCP2 or -3 lowers reactive oxygen species (ROS) production,22 stimulates the metabolic rate, and protects against weight gain and insulin resistance.23 Moreover, UCP3 knockout mice show severe oxidative damage.24 Collectively, these results suggest that UCPs play an important role in mitochondrial function by regulating both heat and ROS generation. Mitochondrial function with regard to energy balance is important in normal physiology and cellular function.
Figure 3.

Mitochondrial respiratory chain and nutrient metabolism. Reducing agents (NADH or FADH2) are generated from glycolysis and Krebs cycle of glucose metabolism and β-oxidation of fatty acids. While NADH or FADH2 are oxidized to NAD+ or FAD, the electrons are carried to complex I (NADH–ubiquinone reductase), complex II (succinated ubiquinone reductase), complex III (ubiquinone–cytochrome c reductase), complex IV (cytochrome oxidase), and finally to O2, which produces H2O. Oxidation of NADH or FADH2 generates protons that are pumped to intermembrane space through complex I, III, and IV. The pumped protons increase electrochemical gradient across the membrane. This proton gradient is the driving force for F0F1-ATPase (ATP synthase) to produce ATP, which is used as an energy source in the body. On the other hand, the pumped protons can be leaked to matrix of mitochondria by UCP, which reduces proton gradient and in turn generates heat. Producing ATP or heat is controlled by energy needs in the body. ANT indicates adenine nucleotide translocator.
Mitochondrial Dysfunction
There is evidence that mitochondrial dysfunction is associated with T2DM and age-related insulin resistance (Figure 4).25,26 Genetic factors, oxidative stress, mitochondrial biogenesis, and aging may affect mitochondrial function, leading to insulin resistance and various pathological conditions.
Figure 4.

Mechanism of mitochondrial dysfunction. Excess intake of nutrients, including overloaded FFAs or hyperglycemia conditions, increases ROS production and reduces mitochondrial biogenesis, causing mitochondrial dysfunction. Mitochondrial dysfunction leads to decreased β-oxidation and ATP production and increased ROS production, resulting in insulin resistance, diabetes, and cardiovascular disease.
Genetic Factors
Mitochondrial proteins are encoded by both nuclear and mitochondrial genes. Mitochondrial genes encode 13 protein subunits of the oxidative phosphorylation complex as well as mitochondrial specific ribosomal and transfer (t)RNAs. The oxidative capacity of mitochondria is determined by the expression level of OXPHOS subunits and by the number and size of mitochondria.10 Because mitochondrial dysfunction and gene expression of mitochondrial OXPHOS genes are related to insulin resistance,26 mutations in mitochondrial genes caused by aging or cellular stress conditions may be one of the mechanisms underlying insulin resistance and other features of cardiometabolic syndrome.
It has been hypothesized that the mitochondrial genome is more susceptible to various mutagenic stressors because mitochondrial genes are more proximal to the ROS source and are not protected by histones.27 Moreover, the mitochondrial genome constitutes only coding sequences, whereas nuclear DNA contains noncoding sequences.19 Indeed, a naturally occurring thymidine-to-cytidine mutation in the mitochondrial tRNAILE gene is associated with phenotypes of hypertension, hypercholesterolemia, and hypomagnesemia.28
Another mutation, A3243G, on mitochondrial DNA that encodes tRNA (LeuUUR) causes impaired insulin secretion.29 In addition, patients with defects in acyl-coenzyme A (COA) dehydrogenase have phenotypes of cardiomyopathy, liver dysfunction, and neurological disorders.30 Furthermore, polymorphisms in the promoter of UCP2 are associated with decreased incidence of obesity, reduced insulin secretion, and a high prevalence of T2DM.31,32 Nuclear genes encoding mitochondrial proteins are also involved with insulin resistance.33 Thus, genetic factors that are inherited through nuclear or mitochondrial genes may influence the pathogenesis of the cardiometabolic syndrome and cardiovascular disease through functional impairment of mitochondria.
Mitochondrial Biogenesis
Fewer and smaller-sized mitochondria are found in skeletal muscle of insulin-resistant, obese, or T2DM subjects.34–36 The number and size of mitochondria are correlated with mitochondrial oxidative capacity.10 The decreased mitochondrial oxidative capacity accompanies the reduction in expression of mitochondrial proteins encoded by both the mitochondrial genome (cytochrome c oxidase 1) and nucleus (succinate dehydrogenase and pyruvate dehydrogenase).34 The molecular mechanism of mitochondrial biogenesis is driven, in part, through peroxisome proliferator-activated receptor (PPAR) coactivator (PGC)-1. PGC-1α was discovered as a transcriptional regulator of UCP that plays a role in thermogenesis in adipose tissue.37 The expression of PGC-1α is increased on cellular ATP demand, including exercise, cold exposure, and fasting.38–41 PGC-1 is a coactivator of nuclear transcription factors including nuclear respiratory factor (NRF)-1 and PPAR-γ and -α.37,42,43 NRF-1 regulates expression of many mitochondrial genes, including OXPHOS genes and mitochondrial transcription factor A (TFAM), that are crucial for mitochondrial gene expression and replication of the mitochondrial genome.43 Expression of PGC-1 is decreased in insulin-resistant and diabetic human subjects, and NRF-1 expression is reduced in diabetic subjects.44 Moreover, the reduction of PGC-1 expression is age dependent,45 and PGC-1α–null mice exhibit serious defects in contractility in both skeletal and cardiac muscle.42,46 Thus, insulin-resistant subjects have fewer mitochondria in their muscle, possibly because of decreased expression of PGC-1α and PGC-1β.43,47
Because expression of PGC-1 is regulated by the endothelial NO synthase (eNOS)/NO/cGMP/PGC-1 activation axis, eNOS plays an important role in mitochondria biogenesis.16,48 In fact, eNOS-deficient mice are insulin resistant and hypertensive and have defects in fatty acid metabolism and fewer mitochondria.48–50 Furthermore, exogenous NO or cGMP increase mitochondrial biogenesis.51 However, the mechanism by which cGMP activates PGC-1 is unknown.
Another important factor that regulates mitochondrial biogenesis is AMP-activated protein kinase (AMPK).52 Pharmacological drugs (β-guanidinopropionic acid [βGPA] or 5′-D-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside [AICAR]) that activate AMPK promote mitochondrial biogenesis through PGC-1α and NRFs.53,54 βGPA increases mitochondrial DNA content and expression of PGC-1α and cytochrome c in wild-type mice. However, application of βGPA to a transgenic mouse overexpressing dominant negative mutant AMPK does not have this effect. Exercise stimulates AMPK, leading to activation of PGC-1 by direct phosphorylation on threonine and serine residues. This phosphorylation event may ultimately promote mitochondrial biogenesis.55,56
Boushel et al observed that mitochondrial function that has been normalized for mitochondrial DNA content in T2DM patients is not significantly different compared with normal healthy subjects.57 Moreover, DNA microarray studies have shown that expression of PGC-1 with regard to mitochondrial biogenesis may be responsible for metabolic disorders, including T2DM and insulin resistance.44,58 These results suggest that decreased mitochondrial function is mainly attributable to the reduced number of mitochondria. Others observed that subsarcolemmal mitochondrial electron transport activity is lower in T2DM and obese subjects compared to lean active adults.36 This diminished mitochondrial electron transport activity is partly attributable to the reduced mitochondrial content, but the decrement in mitochondrial function is greater than can be explained by mitochondrial content. Moreover, mRNA expression of PGC-1α, PGC-1β, NRFs, and TFAM is not different in insulin-resistant offspring of T2DM parents compared with control groups, but the mitochondrial function is significantly decreased.34 These results suggest that reduced mitochondrial biogenesis cannot fully account for the mitochondrial dysfunction. Thus, both abnormalities in mitochondrial function, including ATP production and oxidative respiration, and mitochondrial biogenesis are associated with energy metabolism and insulin resistance.
Oxidative Stress
Extramitochondrial oxygen consumption can occur by non-enzymatic and other enzymatic reactions, including NADPH oxidase, xanthine oxidase, uncoupled NO synthase, D-aminooxidase, p450 cytochromes, and proline hydroxylases19; however, mitochondria are the major sites of ROS production (0.2% to 2% of total oxygen taken by cells). ROS production occurs mainly at complex I (NADH CoQ reductase) and complex III (bc1 complex) in mitochondria. ROS production is increased when excess electrons are provided to mitochondrial respiratory chains. The excess electrons are transferred to oxygen, which is converted to superoxide and subsequently to hydrogen peroxide either by spontaneously or via superoxide dismutase. The highest rate of ROS production occurs when the proton gradient is high and oxygen consumption (ATP demand) is low. Excess calorie intake and low energy expenditure can cause high proton-motive force and less ATP demand. Therefore, most electron carriers are occupied by electrons, and excess electrons are transferred to oxygen without ATP production.59 When exercise increases ATP demand, electron transfers are coupled to ATP production and reduce proton-motive force. Despite intracellular protective mechanisms, including superoxide dismutase, catalase, and reduced glutathione, excess ROS is detrimental to cellular physiology. ROS generated from mitochondria damages proteins, DNA, and lipid in membrane components, which results in mitochondrial dysfunction.60,61
Aging
Aging is a process of irreversible decline in physiological function over time. Several postulated mechanisms for aging include cumulative DNA damage, mitochondrial dysfunction, telomere loss, altered gene expression, and oxidative damages.62 However, the precise molecular mechanisms of aging remain largely unknown. With increasing age, fat mass tends to gradually increase, especially visceral fat, and daily energy expenditure and physical activity tend to decrease. Because regulation of energy production is dependent on ATP needs, reduced energy expenditure with age decreases ATP needs. This leads to decreased oxidative capacity in aged skeletal muscle and heart in both animals and humans.63,64 Old mitochondria have changes in morphology in addition to increased ROS production and decreased ATP production and respiration.65 Furthermore, respiration is decreased in isolated mitochondria from elderly human subjects who have reduced mitochondrial number and function.14 Gene expression–profiling studies demonstrate that genes related to fatty acid oxidation are altered and mitochondrial density and oxidative capacity are decreased with age.63,66 In addition, mitochondrial biogenesis may be impaired by age-dependent accumulations of point mutations in human mitochondrial (mt)DNA at specific regions that control replication of mtDNA.67 Furthermore, age-dependent reduction in PGC-1 expression may account for reduced oxidative phosphorylation in elderly humans.45 This is consistent with the observation that elderly human subjects have ≈40% reduction in oxidative phosphorylation with insulin resistance.26 In addition, activation of AMPK-α2 is blunted in aged rats.55,68 Because AMPK-α2 plays an important role in mitochondrial biogenesis by activating PGC-1α,55,68 this is an additional mechanism by which the aging process may contribute to decreased mitochondrial biogenesis and insulin resistance. Thus, genetic factors, altered mitochondrial biogenesis, increased ROS production, and aging all contribute to mitochondrial dysfunction that is associated with insulin resistance.
Insulin Resistance
Insulin maintains glucose homeostasis in a dynamic relationship with both feeding (glucose uptake) and fasting (gluconeogenesis) states. Additionally, nonclassic roles of insulin associated with cardiovascular, renal, and neural functions may explain why insulin resistance is associated with the risk factors for hypertension, cardiovascular disease, nephropathy, retinopathy, and neuropathy, etc.2,3,69 Perturbation of insulin signaling by various metabolites and crosstalk with other signaling pathways leads to insulin resistance.
Pathophysiology of Insulin Resistance
Insulin resistance is characterized by a diminished ability of cells or tissues to respond to physiological levels of insulin. Genetic and environmental factors, including aging, obesity, lack of exercise, and stress, contribute to insulin resistance. Disorders of glucose and lipid metabolism cause defects in insulin signaling that are linked to various pathological conditions.3,70,71 Thus, molecular and cellular mechanisms of insulin resistance are relevant to understanding the pathogenesis of various diseases associated with insulin resistance.
Circulating free fatty acids (FFAs) are elevated by stress, lipodystrophy, or excess energy intake. Elevated plasma FFA levels lead to accumulation of FFAs, diacylglycerol (DG), and triglycerides in nonadipose tissue, including skeletal muscle, liver, heart, and β-cells. In fact, lipid infusions and high-fat feeding in human subjects and rodents reduces insulin-stimulated glucose disposal.72–74 These data suggest that defects in lipid metabolism leading to impairment of insulin signaling seem to be a major mechanism for insulin resistance.75 Impaired insulin signaling not only affects insulin-stimulated glucose metabolism in skeletal muscle but also impairs other actions of insulin in diverse tissues including, liver, adipose tissue, heart, and the vasculature.76–80
Metabolic Tissues
Insulin resistance is associated with decreased mitochondrial number, abnormal morphology, lower levels of mitochondrial oxidative enzymes, and lower ATP synthesis both in vivo36,81 and ex vivo in human muscle biopsies.82 Notably, either acute lipid infusion or chronic elevation of plasma FFAs causes hepatic insulin resistance.83,84 Elevated FFAs in the plasma leads to intracellular lipid accumulation that is associated with insulin resistance in muscle and liver.85–87 Intramyocellular lipid accumulation leads to reduction in the ratio of glycolytic to oxidative enzyme activities, which is negatively correlated with insulin sensitivity in T2DM when compared with nondiabetic subjects.35,81,88 Thus, intracellular lipid accumulation causes reduced mitochondrial oxidative capacity in skeletal muscle of T2DM subjects and offspring of patients with T2DM. Mitochondrial abnormalities including ultra-structural lesions, depletion of mtDNA, decreased activity of respiratory chain complexes,89 and impaired mitochondrial β-oxidation are also found in patients with nonalcoholic fatty liver disease (NAFLD).90 These mitochondrial abnormalities are associated with NAFLD, which leads to hepatic steatosis and other liver injury.91 Mitochondria with abnormal morphology are also observed in the liver of the Ren2, with transgenic overexpression of murine rennin, the insulin-resistant and hypertensive rat model (Figure 1). This suggests that increased tissue expression of renin causes abnormalities in mitochondria that may lead to NAFLD. Although the detailed mechanism for renin-induced mitochondrial abnormality is not understood, ROS produced through NADPH oxidase could be one potential mechanism that causes mitochondrial abnormality and hepatic dysfunction in Ren2 model (unpublished observation).
Adipose tissue that is overloaded with triglycerides in obesity, and dyslipidemia increases lipolysis and the release of FFAs that cause defects in glucose metabolism and insulin resistance in nonadipose tissues. Conversely, excess loss of adipose tissue, lipodystrophy, also leads to insulin resistance and T2DM.92 Having adipocytes that are impaired in lipid storage creates lipotoxicity in other tissues and metabolic abnormalities associated with intracellular accumulation of lipid. Moreover adipocytes release adipokines, including leptin, adiponectin, resistin, and tumor necrosis factor-α, that positively or negatively regulate metabolic pathways.93–95 Endocrine and nonendocrine roles of adipose tissue with regard to energy intake and expenditure play important roles in insulin resistance.96,97 The number of mitochondria and the expression of genes that are involved in mitochondrial biogenesis are significantly decreased in adipocytes from patients with T2DM or morbidly obese human subjects.98,99 Thus, decreased number of mitochondria, decreased mitochondrial gene expression, abnormal morphology of mitochondria, and abnormal functions in oxidative phosphorylation are commonly found in insulin-resistant metabolic tissues, including skeletal muscle, liver, and fat. These abnormalities in mitochondria are associated with intracellular lipid accumulation, insulin resistance, and pathophysiology of T2DM and NAFLD.
Cardiovascular Tissues
Cardiovascular diseases, including coronary artery disease, hypertension, heart failure, and stroke, are associated with insulin resistance and endothelial dysfunction.4,100 As described above, FFAs, but not glucose infusion, contributes to insulin resistance and reduces mitochondrial oxidative capacity, cardiac efficiency, and ATP production and increases myocardial oxygen consumption in obese and insulin-resistant ob/ob mice.101 In addition, intramyocardial lipid accumulation induces lipotoxic injury and cardiac dysfunction, including diastolic dysfunction, left ventricular hypertrophy, and impaired septal contractility in rodent and human obesity.102,103 These results suggest that reduced mitochondrial oxidative capacity contribute to cardiac dysfunction. Mitochondria occupy 20% to 30% of the cell volume in cardiac myocytes, compared with ≈67% in oxidative skeletal muscle such as the soleus and only 2% to 3% in glycolytic muscle such as the gastrocnemius muscle.104 Depressed mitochondrial transcription factors and oxidative capacity in rat contribute to failing cardiac function. The heart possesses a relatively low endogenous antioxidant capacity, as contributed by both enzymatic and nonenzymatic free radical scavengers and antioxidants, making it susceptible to oxidative stress with attendant structural and functional abnormalities.7 Increased oxidative stress in the heart has been linked to ventricular hypertrophy, systolic and diastolic functional abnormalities, and abnormal insulin metabolic signaling.7,105 Several studies have shown an association between mitochondrial oxidative energy and alterations in mitochondrial morphology and function, including decreased production of ATP and impaired activation of mitochondrial ATP-activated potassium channels.106–108 Transmission electron microscopic analysis of myocardial tissue in insulin-resistant rodent models has demonstrated the presence of increased numbers of morphologically abnormal mitochondria (Figure 2).5,7,42,109 Increased numbers of mitochondria have been associated with hypertrophied hearts displaying oxidative stress.5,7,110 Accordingly, these changes may represent an adaptive response to greater energy requirements as well as oxidative stress. However, in pathological hypertrophy, which can be observed at the later stage of pathogenesis of cardiac dysfunction, the number of mitochondria and DNA contents are reduced.111
Another important insulin action in the heart is to increase myocardial blood flow and reduce coronary vascular resistance.112 Impairment of endothelium-dependent vasodilation and glucose intolerance accompany intramyocardial lipid accumulation, and this precedes T2DM and heart failure.113 Thus, the role of mitochondria in cardiac function is important, and cardiac mitochondrial dysfunction may contribute to various cardiovascular diseases, including coronary heart disease, hypertension, cardiomyopathy, and heart failure.
Although the association between insulin resistance and endothelial dysfunction has been emphasized,114 the role of mitochondrial function in endothelium is not clearly understood. Endothelial cells produce 75% of ATP from glycolysis115; thus, oxidative metabolism does not seem to be important in vascular endothelial cells. However, mitochondria in endothelial cells may play an important role in cellular signaling as sensors for local oxygen concentration and as regulators of intracellular [Ca2+] and NO production.116 Moreover, Brownlee suggested that mitochondrial dysfunction occurs as a “unifying mechanism” for microvascular and macrovascular complications through ROS production.18 Hyperglycemia induced by endothelial dysfunction is inhibited by blocking ROS production from mitochondria and by overexpression of UCP1, or manganese superoxide dismutase.18,117 Furthermore, eNOS in vascular endothelial cells seems to play an important role in insulin-stimulated NO production and vasodilation,118 as well as in mitochondrial biogenesis.48,51 Indeed, eNOS-null mice are insulin resistant and hypertensive and have dyslipidemia.48,119,120 Thus, insulin resistance decreases eNOS activity, and associated mitochondrial dysfunction impairs various cardiac functions that may contribute to heart failure, cardiomyopathy, and coronary artery disease.
Pancreatic β-Cells
Insulin-resistant patients can develop overt T2DM when pancreatic β-cells cannot produce enough insulin to maintain euglycemia. Pancreatic β-cells in T2DM cannot sense glucose properly, contributing to impaired insulin secretion. Glucose oxidation by mitochondria produces ATP, which increases the ATP/ADP ratio. ATP/ADP ratio is primarily regulated by mitochondrial function, and the increased ATP/ADP ratio contributes to inhibition of potassium channel (KATP), which leads to plasma membrane depolarization, voltage-gated calcium channel opening, calcium influx, and secretion of insulin. Thus, mitochondrial function may be correlated with β-cell function because of the importance of the ATP/ADP ratio.121 In addition, when mitochondrial genes are removed from β-cells, insulin secretion is impaired, and pancreatic β-cell function is restored when the cells are replenished with normal mitochondria.122 Furthermore, tissue-specific knockout of TFAM, a nuclear encoded mitochondrial protein, results in reduced β-cell mass, impaired insulin secretion, and development of overt diabetes with severe mtDNA depletion by 5 weeks after birth.123 The results of studies support the notion that mitochondrial function is important for healthy β-cell function, whereas mitochondrial dysfunction may contribute to the pathogenesis of T2DM by affecting insulin secretion as well as insulin action.
Signaling Between Mitochondrial Dysfunction and Insulin Resistance
Insulin Signaling
Canonical insulin signaling pathways are initiated by insulin binding to the extracellular α subunit of the insulin receptor (IR) (Figure 5).71,124 This causes a conformational change in the β subunit of IR, which has intrinsic tyrosine kinase activity, resulting in autophosphorylation of IR tyrosine residues (eg, NPEY motif of the receptor) and enhanced tyrosine kinase activity of the receptor. The activated receptor phosphorylates insulin receptor substrate (IRS) family members and activates phosphatidylinositol 3-kinase (PI3K), a lipid kinase that phosphorylates phosphatidylinositol 4,5-bisphosphate generating phosphatidylinositol 3,4,5-triphosphate. Receptor tyrosine kinases including insulin receptor activate class IA isotypes of PI3K that are composed of heterodimer with catalytic (p110α, p110β, p110δ) and regulatory (p85α, p50α, p55α, p55γ, p85β) subunits.125,126 Activation of PI3K leads to stimulation of various downstream serine kinases, including phosphoinositide dependent kinase-1, protein kinase B (Akt), atypical protein kinase C (PKC), and other serine kinases, which culminates in the pleiotropic biological actions and metabolic functions of insulin. Similarly, SH2 domain of Grb-2 binds to Shc and activates GTP exchange factor Sos, which subsequently activates the small GTP protein Ras. Activation of Ras leads to the activation of downstream signaling that activates Raf and mitogen-activated protein (MAP)/extracellular signal-regulated kinase kinase (MEK), and MAP kinase (MAPK). This MAPK branch of the insulin signaling pathway regulates growth, mitogenesis, and differentiation. Insulin signaling constitutes a highly complex network with multiple feedback loops and crosstalk between major signaling branches, as well as signaling from heterologous receptors.127 It is noteworthy that in addition to the classic role of insulin signaling, the roles of insulin signaling in cardiac growth, vasodilation, and maintenance of vascular tone is similar but the biological responses are distinct.2,3,114,128 Thus, the complexity of insulin signaling gives rise to the specificities of insulin signaling and actions based on the context, ie, the tissue type and the particular physiological or pathophysiological conditions.
Figure 5.

Insulin signaling pathway. The metabolic PI3K branch of insulin signaling pathway and tissue-specific actions of insulin are shown. PI3K branch of insulin signaling pathway plays a major role in gluconeogenesis in the liver, enhances NO production in the endothelium and heart, and glucose uptake in skeletal muscle, adipose tissue, and heart.
Molecular Mechanism of Insulin Resistance
Defects at multiple sites in insulin signaling pathway have been suggested as mechanisms underlying insulin resistance71: (1) increased serine phosphorylation of IRS proteins129; (2) increased degradation of IRS proteins130; (3) increased activity of phosphatases including (src homology 2 domain containing inositol 5′-phosphatase 2 [SHIP2], phosphatase tensin homolog deleted on chromosome ten [PTEN], and phosphotyrosine phosphatase 1B [PTP-1B])131–133; (4) decreased activation of insulin receptor downstream signaling molecules including Akt and atypical PKC.134
Reduction in tyrosine phosphorylation of IRS family members has been observed in insulin-resistant animal models and human subjects, including those who are obese or made insulin resistant by lipid infusion.135–139 One possible mechanism to explain decreased tyrosine phosphorylation of IRS-1 and -2 is activation of serine/threonine kinases that can phosphorylate IRS family members at multiple serine sites.70,129 Phosphorylation of IRS proteins at particular serine residues inhibits the interaction of IRS proteins with the IR. This leads to reduction in tyrosine phosphorylation of IRS and subsequently decreases activation of PI3K.129 Interestingly, high-fat diet–induced insulin resistance is ameliorated when specific serine/threonine kinases are genetically ablated or pharmacologically inhibited.140–143 Increased proinflammatory signaling is another potential mechanism underlying insulin resistance. FFAs stimulate receptor (Toll-like receptor)-mediated proinflammatory signaling, which activates IκB kinase (IKK)β and c-Jun N-terminal kinase (JNK) and stimulates production of other cytokines, including tumor necrosis factor-α, interleukin-1β, and interleukin-6.144–146 IKKβ and JNK are well-known serine kinases that phosphorylate IRS-1 at serine residues, leading to decreased metabolic signaling. Inhibition of IKKβ or JNK with antiinflammatory drugs or gene knockout improves insulin sensitivity contemporaneously with reductions in serine phosphorylation of IRS proteins.142,143,147,148 Endoplasmic reticulum (ER) stress is another mechanism contributing to insulin resistance through activation of serine kinases. JNK activation, as a consequence of ER stress, increases serine phosphorylation of IRS proteins. Chemical chaperones including 4-phenyl butyric acid and taurine-conjugated ursodeoxycholic acids (TUDCA) significantly reduce ER stress, resulting in improved insulin sensitivity.149 This treatment also decreases fatty liver in animal models and is associated with a reduction in hepatic JNK activity as well as IRS-1 serine phosphorylation.149 Lastly, mitochondrial dysfunction and consequent increases in ROS, in turn, activate various serine kinases that phosphorylate IRS proteins, leading to insulin resistance.34 Furthermore, ROS stimulates proinflammatory signaling by activation of IKKβ that phosphorylates IRS-1 at serine residues.150 Although, the detailed mechanism for serine kinase activation mediated by ROS is not clearly understood, decreased ROS production by antioxidants or increased expression of UCP2/3 improves both mitochondrial function and insulin sensitivity. Mitochondrial dysfunction results in accumulation of fatty acid metabolites, DG, and long-chain fatty acyl-CoA (LCFA-CoA).151 Intracellular accumulation of DG, allosteric activator of PKCs, activates PKCs, including PKC-β, δ, and θ, that increase serine phosphorylation of IRS proteins, leading to inhibition of insulin signaling and insulin resistance (Figure 6).139,151,152 In fact, the PKCθ-deficient mouse is protected from fat-induced insulin resistance.140 This suggests that activation of PKCs attributable to the mitochondrial dysfunction may cause insulin resistance. Thus, lipid-induced mitochondrial dysfunction impairs insulin signaling both directly and indirectly through generation of excess ROS.
Figure 6.

Proposed molecular mechanism for insulin resistance caused by mitochondrial dysfunction. FFAs activate inflammatory signaling and reduce ATP production that contributes to mitochondrial dysfunction and accumulation of LCFA-CoA and DG. Accumulation of lipid metabolite activates PKCs (β, δ, and θ). ROS produced by NADPH oxidase by angiotensin II causes mitochondrial dysfunction. Conversely, mitochondrial dysfunction increases ROS production, which causes activation of serine/threonine kinases, including IKKβ, JNK, and PKCs, which increases serine phosphorylation of IRS proteins and subsequently results in insulin resistance. Increased serine phosphorylation of IRS-1/2 leads to decreased activity of insulin downstream signaling pathways, including PI3K, Akt, and PKCζ, which culminates in decreased glucose uptake, increased glucose production, and reduced vasodilation and insulin secretion. The reduced insulin responsiveness (insulin resistance) causes diabetes and cardiovascular diseases. PDK-1 indicates 3′-phosphoinositide-dependent protein kinase 1.
Therapeutic Intervention
Pharmacological Intervention
Because mitochondrial function is associated with mitochondrial biogenesis, stimulation of mitochondrial biogenesis may have beneficial effects in both metabolic and cardiovascular diseases. In fact, beneficial effects of thiazolidinediones (synthetic PPAR-γ ligands) have been reported to improve insulin resistance in liver, adipocytes, and heart, as well as β-cell function and endothelial dysfunction in studies using animal models and human intervention studies.153–158 The molecular mechanism of insulin-sensitizing activity for thiazolidinediones may be, in part, through increased mitochondrial biogenesis.98 Metformin is used as another insulin sensitizer, which is reported to reduce ROS production, increase expression of PGC-1α, and stimulate AMPK.68,159,160 Metformin may improve mitochondrial function by reducing oxidative stress and stimulating mitochondrial biogenesis through activation of AMPK/PGC-1/NRFs axis pathway. Studies from our group have demonstrated that increases in tissue angiotensin II increase NADPH oxidase activity and tissue ROS and that this is associated with abnormalities in mitochondrial structure and function. Thus, drugs that reduce actions of angiotensin II also increase insulin sensitivity, reduce ROS production, and improve mitochondrial function. For example, treatment with angiotensin-converting enzyme inhibitor has been reported to increase insulin sensitivity, reduce ROS production, and increase mitochondrial biogenesis.8,158,161 Angiotensin receptor blockers also significantly improve insulin resistance and block angiotensin-induced oxidative stress in both human and animal models.6,162–164 Excessive tissue angiotensin causes insulin resistance, cardiomyopathy, hypertension, and renal failure, possibly because of mitochondrial dysfunction; thus, angiotensin receptor blockers or angiotensin-converting enzyme inhibitors may also exert beneficial effects to various pathological conditions by enhancing mitochondrial biogenesis and function.3,5–7,164–168 The detailed molecular mechanism for the actions of these drugs targeting excess tissue angiotensin in mitochondrial dysfunction is not well defined. Reducing angiotensin receptor–mediated ROS production may improve mitochondrial function and improve insulin-mediated metabolic actions. However, further investigation is needed to better understand the detailed mechanisms of these and other therapeutic drugs with regard to mitochondrial functions and biogenesis.
Mitochondrial dysfunction is induced by increased ROS, whereas impairment of mitochondrial function, in turn, produces more ROS and lipid byproducts, including LCFA-CoA and DG. Thus, therapeutic intervention using antioxidant supplements may be beneficial to interrupt this vicious cycle. In fact, α-lipoic acid reduces hyperglycemia and increased GLUT4 content in rat skeletal muscle tissue.169 Moreover, α-lipoic acid opposes ROS-induced inhibition of insulin signaling.170 Although oral vitamin C supplementation does not significantly affect insulin resistance or endothelial function in T2DM,171 intraarterial vitamin C improves endothelial-dependent vasodilation in T2DM.172 Tempol, a superoxide scavenger, is able to ameliorate cardiac and vascular dysfunction, normalize angiotensin II–induced insulin resistance, and improve mitochondrial morphology and function.6,7,164 Pharmacological targets that can stimulate mitochondrial biogenesis (eg, thiazolidinediones) or reducing ROS production (eg, metformin, angiotensin receptor blockers, and antioxidants) may have beneficial effects on cardiometabolic syndrome partly by improving mitochondrial function.
Exercise
Large-scale epidemiological studies demonstrate that low aerobic exercise correlates with increased mortality and cardiovascular disease.173,174 Impaired mitochondrial function may be an important mechanism for low aerobic capacity and cardiovascular risk factors that accompany the cardiometabolic syndrome.175 Exercise improves insulin action and glucose tolerance in insulin-resistant subjects and animal models.176,177 Substantial evidence indicates that aerobic exercise stimulates mitochondrial biogenesis by increasing gene expression of PGC-1, NRF-1, and TFAM.178,179 Endurance exercise training increases mitochondrial size, number, and oxidative activity contributing to improved whole-body glucose metabolism.180 Increased expression of eNOS by physical activity may stimulate mitochondrial biogenesis.49,181 Moderate-intensity physical activity combined with weight loss improves insulin sensitivity through increasing skeletal muscle electron transport chain activity and increasing mitochondrial cristae (without changing mtDNA content).182 Age-associated reduction in expression of mitochondrial genes and mitochondrial biogenesis is restored with aerobic exercise.63,183 Thus, exercise can improve glucose and lipid metabolism by activation of AMPK and PGC-1α that increase mitochondrial biogenesis and function.
Calorie Restriction
Calorie restriction increases lifespan in organisms ranging from yeast to mammals.184,185 Calorie restriction ameliorates many of the pathophysiological conditions associated with the cardiometabolic syndrome related to glucose and lipid metabolism.186 One study using oligonucleotide-based array techniques showed that expression of stress response genes and oxidative stress–inducible genes are enhanced, whereas gene expression of energy metabolism is decreased with aging. Interestingly, in the same study, calorie restriction reversed these changes in gene expression patterns.66 Calorie restriction increases expression of UCP2 and -3 in human subjects.187 Calorie restriction increases mitochondrial biogenesis, oxygen consumption, ATP production, and expression of SIRT1 (NAD+-dependent deacetylase) through eNOS expression. SIRT family members play a central role in the physiological effect of calorie restriction. The specific roles of SIRT1, -3, and -4 in metabolism have been described in various tissues.186 SIRT1 activates PGC-1α, which may lead to mitochondrial biogenesis. Furthermore, eNOS-null mice do not respond appropriately to calorie restriction,50 suggesting that calorie restriction improves mitochondrial biogenesis and function, in part, through increases in NO production. Another study has shown that calorie restriction induces mitochondrial biogenesis, reduced ROS production.188 Mitochondrial biogenesis or improvement of mitochondrial function may be a major mechanism for the beneficial effects of calorie restriction. Thus, the beneficial effects of calorie restriction mediated by enhancement of mitochondrial biogenesis and function may lead to improvement of glucose and lipid metabolism, as well as insulin resistance. Therapeutic interventions to improve mitochondrial function or to stimulate mitochondrial biogenesis may ameliorate insulin resistance and other components of the cardiometabolic syndrome and improve cardiovascular function and outcomes (Figure 7).
Figure 7.
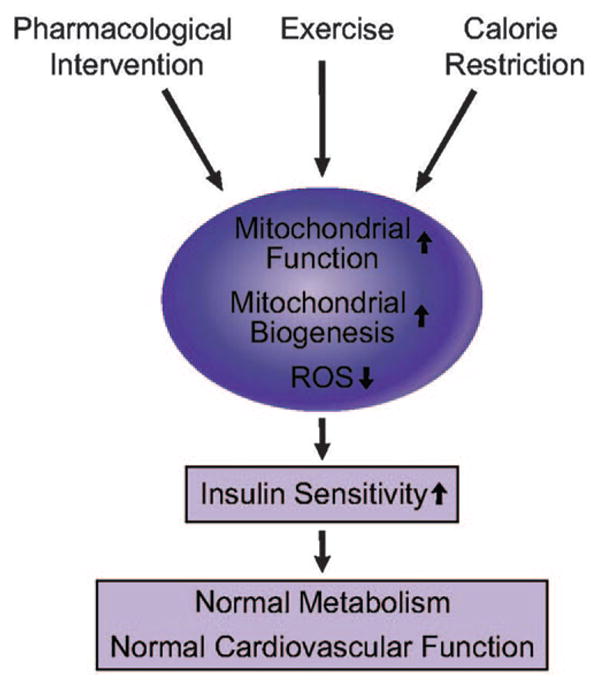
Improvement of mitochondrial function by pharmacological intervention, exercise, and calorie restriction can improve insulin sensitivity, which leads to normal metabolism and cardiovascular function.
Summary and Perspectives
Insulin resistance plays a central role in the pathogenesis of the cardiometabolic syndrome, T2DM, and attendant cardiovascular complications. Excess nutrients or sedentary lifestyle cause various pathological conditions that are associated with mitochondrial dysfunction. Genetic factors, reduced mitochondrial biogenesis, increased oxidative stress, and aging may be causal factors for abnormalities in mitochondrial dysfunction. The resultant mitochondrial dysfunction, in turn, increases ROS production, resulting in a vicious cycle. Antioxidants protect from both mitochondrial dysfunction and insulin resistance by scavenging free radicals. However, both lipid accumulation and excess ROS further stimulate various serine/threonine kinases and inflammatory signaling pathways that inhibit insulin signaling. Although several serine/threonine kinases that are responsible for insulin resistance have been identified, the detailed molecular mechanisms by which ROS can activate these kinases are unknown.
Blockade of the AT1R and scavengers of ROS prevent cardiac, vascular, and hepatic dysfunction as well as mitochondrial dysfunction in rodents with tissue overexpression of angiotensin II.2,7 These observations suggest that angiotensin II contributes to mitochondrial dysfunction through a mechanism that is distinct from elevated FFAs. Studies from our laboratory suggest that excess tissue angiotensin II results in enhanced NADPH oxidase activity, leading to increased ROS and mitochondrial abnormalities associated with insulin resistance, cardiac dysfunction, vascular inflammation, and reduced NO-mediated relaxation.6,7,165,168,189 ROS produced by angiotensin II activation of NADPH oxidase may damage cellular proteins or transduce signaling that influences mitochondrial biogenesis and function. However, the precise mechanism by which ROS can affect mitochondrial biogenesis and function remains to be clarified.
Intracellular lipid accumulation is observed in insulin-resistant skeletal muscle.13 Lipid infusion decreases glucose transport and ATP synthesis.9 It is not clear whether reduced ATP synthesis causes insulin resistance or whether reduced insulin sensitivity leads to reduction in generation of ATP. The intramyocellular triglyceride contents in lipid-infused skeletal muscle are not different from controls.9 Thus, the accumulation of lipid metabolites, including LCFA-CoA and DG, may not be attributable to the increased de novo synthesis of lipid but attributable to reduction in β-oxidation of fatty acid in the mitochondria. The cause-and-effect relationship between reduced mitochondrial function and excess intramyocellular lipid content is an area ripe for investigation.
Decreased number of mitochondria may be a mechanism for insulin resistance. Regulatory mechanisms for mitochondrial biogenesis and expression of OXPHOS genes related to energy status are not completely defined and require further investigation. Nonetheless, mitochondrial dysfunction seems to play a central role in metabolic and cardiovascular disorders. Thus, new therapeutic strategies that regulate mitochondrial function and mitochondrial biogenesis may have therapeutic potential for decreased insulin action and pancreatic β-cell production, lipid accumulation in liver (eg, NAFLD), skeletal muscle impairments, endothelial-mediated vasorelaxation, and both systolic and diastolic myocardial function, all components of the cardiometabolic syndrome.
Acknowledgments
We acknowledge Dr Michael J. Quon, Dr Adam T. Whaley-Connell, Elaine Rehmer, and Brenda Hunter for help in preparation of this manuscript and Dr Melvin Hayden for providing electron microscopic data.
Sources of Funding
This work was supported by NIH grant R01 HL73101-01A1 to (J.R.S.) and the Veterans Affairs Merit System grant 0018 (to J.R.S.).
Footnotes
Disclosures
None.
References
- 1.Smith SC., Jr Multiple risk factors for cardiovascular disease and diabetes mellitus. Am J Med. 2007;120:S3–S11. doi: 10.1016/j.amjmed.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Cooper SA, Whaley-Connell A, Habibi J, Wei Y, Lastra G, Manrique CM, Stas S, Sowers JR. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol. 2007;293:H2009–H2023. doi: 10.1152/ajpheart.00522.2007. [DOI] [PubMed] [Google Scholar]
- 3.Sowers JR. Insulin resistance and hypertension. Am J Physiol Heart Circ Physiol. 2004;286:H1597–H1602. doi: 10.1152/ajpheart.00026.2004. [DOI] [PubMed] [Google Scholar]
- 4.Reaven GM, Chen YD. Insulin resistance, its consequences, and coronary heart disease. Must we choose one culprit? Circulation. 1996;93:1780–1783. doi: 10.1161/01.cir.93.10.1780. [DOI] [PubMed] [Google Scholar]
- 5.Stas S, Whaley-Connell A, Habibi J, Appesh L, Hayden MR, Karuparthi PR, Qazi M, Morris EM, Cooper SA, Link CD, Stump C, Hay M, Ferrario C, Sowers JR. Mineralocorticoid receptor blockade attenuates chronic overexpression of the renin-angiotensin-aldosterone system stimulation of reduced nicotinamide adenine dinucleotide phosphate oxidase and cardiac remodeling. Endocrinology. 2007;148:3773–3780. doi: 10.1210/en.2006-1691. [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Whaley-Connell AT, Chen K, Habibi J, Uptergrove GM, Clark SE, Stump CS, Ferrario CM, Sowers JR. NADPH oxidase contributes to vascular inflammation, insulin resistance, and remodeling in the transgenic (mRen2) rat. Hypertension. 2007;50:384–391. doi: 10.1161/HYPERTENSIONAHA.107.089284. [DOI] [PubMed] [Google Scholar]
- 7.Whaley-Connell A, Govindarajan G, Habibi J, Hayden MR, Cooper SA, Wei Y, Ma L, Qazi M, Link D, Karuparthi PR, Stump C, Ferrario C, Sowers JR. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic (mRen2) 27 Ren2 rat. Am J Physiol Endocrinol Metab. 2007;293:E355–E363. doi: 10.1152/ajpendo.00632.2006. [DOI] [PubMed] [Google Scholar]
- 8.de Cavanagh EM, Piotrkowski B, Basso N, Stella I, Inserra F, Ferder L, Fraga CG. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. FASEB J. 2003;17:1096–1098. doi: 10.1096/fj.02-0063fje. [DOI] [PubMed] [Google Scholar]
- 9.Brehm A, Krssak M, Schmid AI, Nowotny P, Waldhausl W, Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006;55:136–140. [PubMed] [Google Scholar]
- 10.Ritz P, Berrut G. Mitochondrial function, energy expenditure, aging and insulin resistance. Diabetes Metab. 2005;31(Spec No 2):5S67–5S73. doi: 10.1016/s1262-3636(05)73654-5. [DOI] [PubMed] [Google Scholar]
- 11.Frisard M, Ravussin E. Energy metabolism and oxidative stress: impact on the metabolic syndrome and the aging process. Endocrine. 2006;29:27–32. doi: 10.1385/ENDO:29:1:27. [DOI] [PubMed] [Google Scholar]
- 12.Befroy DE, Petersen KF, Dufour S, Mason GF, de Graaf RA, Rothman DL, Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 14.Mogensen M, Sahlin K, Fernstrom M, Glintborg D, Vind BF, Beck-Nielsen H, Hojlund K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–1599. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- 15.Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116:434–448. doi: 10.1161/CIRCULATIONAHA.107.702795. [DOI] [PubMed] [Google Scholar]
- 16.Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res. 2007;100:795–806. doi: 10.1161/01.RES.0000259591.97107.6c. [DOI] [PubMed] [Google Scholar]
- 17.Wiederkehr A, Wollheim CB. Minireview: implication of mitochondria in insulin secretion and action. Endocrinology. 2006;147:2643–2649. doi: 10.1210/en.2006-0057. [DOI] [PubMed] [Google Scholar]
- 18.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 19.Harper ME, Bevilacqua L, Hagopian K, Weindruch R, Ramsey JJ. Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol Scand. 2004;182:321–331. doi: 10.1111/j.1365-201X.2004.01370.x. [DOI] [PubMed] [Google Scholar]
- 20.Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 21.Vidal-Puig AJ, Grujic D, Zhang CY, Hagen T, Boss O, Ido Y, Szczepanik A, Wade J, Mootha V, Cortright R, Muoio DM, Lowell BB. Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem. 2000;275:16258–16266. doi: 10.1074/jbc.M910179199. [DOI] [PubMed] [Google Scholar]
- 22.Lee KU, Lee IK, Han J, Song DK, Kim YM, Song HS, Kim HS, Lee WJ, Koh EH, Song KH, Han SM, Kim MS, Park IS, Park JY. Effects of recombinant adenovirus-mediated uncoupling protein 2 overexpression on endothelial function and apoptosis. Circ Res. 2005;96:1200–1207. doi: 10.1161/01.RES.0000170075.73039.5b. [DOI] [PubMed] [Google Scholar]
- 23.Clapham JC, Arch JR, Chapman H, Haynes A, Lister C, Moore GB, Piercy V, Carter SA, Lehner I, Smith SA, Beeley LJ, Godden RJ, Herrity N, Skehel M, Changani KK, Hockings PD, Reid DG, Squires SM, Hatcher J, Trail B, Latcham J, Rastan S, Harper AJ, Cadenas S, Buckingham JA, Brand MD, Abuin A. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature. 2000;406:415–418. doi: 10.1038/35019082. [DOI] [PubMed] [Google Scholar]
- 24.Brand MD, Pamplona R, Portero-Otin M, Requena JR, Roebuck SJ, Buckingham JA, Clapham JC, Cadenas S. Oxidative damage and phospholipid fatty acyl composition in skeletal muscle mitochondria from mice underexpressing or overexpressing uncoupling protein 3. Biochem J. 2002;368:597–603. doi: 10.1042/BJ20021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stump CS, Short KR, Bigelow ML, Schimke JM, Nair KS. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci U S A. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 28.Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF, Toka HR, Nelson-Williams C, Raja KM, Kashgarian M, Shulman GI, Scheinman SJ, Lifton RP. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306:1190–1194. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maassen JA, ‘T Hart LM, Van Essen E, Heine RJ, Nijpels G, Jahangir Tafrechi RS, Raap AK, Janssen GM, Lemkes HH. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53(suppl 1):S103–S109. doi: 10.2337/diabetes.53.2007.s103. [DOI] [PubMed] [Google Scholar]
- 30.He M, Rutledge SL, Kelly DR, Palmer CA, Murdoch G, Majumder N, Nicholls RD, Pei Z, Watkins PA, Vockley J. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am J Hum Genet. 2007;81:87–103. doi: 10.1086/519219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esterbauer H, Schneitler C, Oberkofler H, Ebenbichler C, Paulweber B, Sandhofer F, Ladurner G, Hell E, Strosberg AD, Patsch JR, Krempler F, Patsch W. A common polymorphism in the promoter of UCP2 is associated with decreased risk of obesity in middle-aged humans. Nat Genet. 2001;28:178–183. doi: 10.1038/88911. [DOI] [PubMed] [Google Scholar]
- 32.Sesti G, Cardellini M, Marini MA, Frontoni S, D’Adamo M, Del Guerra S, Lauro D, De Nicolais P, Sbraccia P, Del Prato S, Gambardella S, Federici M, Marchetti P, Lauro R. A common polymorphism in the promoter of UCP2 contributes to the variation in insulin secretion in glucose-tolerant subjects. Diabetes. 2003;52:1280–1283. doi: 10.2337/diabetes.52.5.1280. [DOI] [PubMed] [Google Scholar]
- 33.Muller YL, Bogardus C, Pedersen O, Baier L. A Gly482Ser missense mutation in the peroxisome proliferator-activated receptor gamma coactivator-1 is associated with altered lipid oxidation and early insulin secretion in Pima Indians. Diabetes. 2003;52:895–898. doi: 10.2337/diabetes.52.3.895. [DOI] [PubMed] [Google Scholar]
- 34.Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 36.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 37.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 38.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 41.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 42.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 44.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ling C, Poulsen P, Carlsson E, Ridderstrale M, Almgren P, Wojtaszewski J, Beck-Nielsen H, Groop L, Vaag A. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J Clin Invest. 2004;114:1518–1526. doi: 10.1172/JCI21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 47.St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, Spiegelman BM. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 48.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 49.Le Gouill E, Jimenez M, Binnert C, Pierre-Yves J, Thalmann S, Nicod P, Scherrer U, Vollenweider P. eNOS knock-out mice have defective mitochondrial beta-oxidation. Diabetes. 2007;56:2690–2696. doi: 10.2337/db06-1228. [DOI] [PubMed] [Google Scholar]
- 50.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 51.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, Cantoni O, Carruba MO, Moncada S, Clementi E. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci U S A. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33–39. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–E1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 54.Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- 55.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47:1369–1373. doi: 10.2337/diab.47.8.1369. [DOI] [PubMed] [Google Scholar]
- 57.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 59.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choksi KB, Boylston WH, Rabek JP, Widger WR, Papaconstantinou J. Oxidatively damaged proteins of heart mitochondrial electron transport complexes. Biochim Biophys Acta. 2004;1688:95–101. doi: 10.1016/j.bbadis.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 61.Murphy MP. Induction of mitochondrial ROS production by electrophilic lipids: a new pathway of redox signaling? Am J Physiol Heart Circ Physiol. 2006;290:H1754–H1755. doi: 10.1152/ajpheart.00040.2006. [DOI] [PubMed] [Google Scholar]
- 62.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 63.Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526(pt 1):203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J. 2003;17:714–716. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- 65.McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin CS, Jan YN, Kenyon C, Bargmann CI, Li H. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet. 2004;36:197–204. doi: 10.1038/ng1291. [DOI] [PubMed] [Google Scholar]
- 66.Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 67.Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 68.Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T, Araki E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–127. [PubMed] [Google Scholar]
- 69.Strachan MW. Insulin and cognitive function. Lancet. 2003;362:1253. doi: 10.1016/S0140-6736(03)14615-6. [DOI] [PubMed] [Google Scholar]
- 70.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(suppl 2):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 72.Boden G, Jadali F. Effects of lipid on basal carbohydrate metabolism in normal men. Diabetes. 1991;40:686–692. doi: 10.2337/diab.40.6.686. [DOI] [PubMed] [Google Scholar]
- 73.Han DH, Hansen PA, Host HH, Holloszy JO. Insulin resistance of muscle glucose transport in rats fed a high-fat diet: a reevaluation. Diabetes. 1997;46:1761–1767. doi: 10.2337/diab.46.11.1761. [DOI] [PubMed] [Google Scholar]
- 74.Kim JK, Wi JK, Youn JH. Metabolic impairment precedes insulin resistance in skeletal muscle during high-fat feeding in rats. Diabetes. 1996;45:651–658. doi: 10.2337/diab.45.5.651. [DOI] [PubMed] [Google Scholar]
- 75.Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ouwens DM, Boer C, Fodor M, de Galan P, Heine RJ, Maassen JA, Diamant M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia. 2005;48:1229–1237. doi: 10.1007/s00125-005-1755-x. [DOI] [PubMed] [Google Scholar]
- 77.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 78.Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97:2601–2610. doi: 10.1172/JCI118709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, Ye J. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3–L1 adipocytes. Mol Endocrinol. 2004;18:2024–2034. doi: 10.1210/me.2003-0383. [DOI] [PubMed] [Google Scholar]
- 80.Kim F, Tysseling KA, Rice J, Pham M, Haji L, Gallis BM, Baas AS, Paramsothy P, Giachelli CM, Corson MA, Raines EW. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol. 2005;25:989–994. doi: 10.1161/01.ATV.0000160549.60980.a8. [DOI] [PubMed] [Google Scholar]
- 81.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279:E1039–E1044. doi: 10.1152/ajpendo.2000.279.5.E1039. [DOI] [PubMed] [Google Scholar]
- 83.Boden G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr Diab Rep. 2006;6:177–181. doi: 10.1007/s11892-006-0031-x. [DOI] [PubMed] [Google Scholar]
- 84.Lam TK, Yoshii H, Haber CA, Bogdanovic E, Lam L, Fantus IG, Giacca A. Free fatty acid-induced hepatic insulin resistance: a potential role for protein kinase C-delta. Am J Physiol Endocrinol Metab. 2002;283:E682–E691. doi: 10.1152/ajpendo.00038.2002. [DOI] [PubMed] [Google Scholar]
- 85.Boden G, Lebed B, Schatz M, Homko C, Lemieux S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes. 2001;50:1612–1617. doi: 10.2337/diabetes.50.7.1612. [DOI] [PubMed] [Google Scholar]
- 86.Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med. 2006;119:S10–S16. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cherrington AD. Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48:1198–1214. doi: 10.2337/diabetes.48.5.1198. [DOI] [PubMed] [Google Scholar]
- 88.Simoneau JA, Veerkamp JH, Turcotte LP, Kelley DE. Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. FASEB J. 1999;13:2051–2060. doi: 10.1096/fasebj.13.14.2051. [DOI] [PubMed] [Google Scholar]
- 89.Perez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 90.Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol. 2005;42:928–940. doi: 10.1016/j.jhep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 91.Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67:101–154. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- 92.Oseid S, Beck-Nielsen H, Pedersen O, Sovik O. Decreased binding of insulin to its receptor in patients with congenital generalized lipodystrophy. N Engl J Med. 1977;296:245–248. doi: 10.1056/NEJM197702032960503. [DOI] [PubMed] [Google Scholar]
- 93.Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, Steppan CM, Ahima RS, Obici S, Rossetti L, Lazar MA. Regulation of fasted blood glucose by resistin. Science. 2004;303:1195–1198. doi: 10.1126/science.1092341. [DOI] [PubMed] [Google Scholar]
- 94.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001:7941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 95.Saltiel AR. You are what you secrete. Nat Med. 2001;7:887–888. doi: 10.1038/90911. [DOI] [PubMed] [Google Scholar]
- 96.Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 97.Gnudi L, Tozzo E, Shepherd PR, Bliss JL, Kahn BB. High level overexpression of glucose transporter-4 driven by an adipose-specific promoter is maintained in transgenic mice on a high fat diet, but does not prevent impaired glucose tolerance. Endocrinology. 1995;136:995–1002. doi: 10.1210/endo.136.3.7867610. [DOI] [PubMed] [Google Scholar]
- 98.Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005;54:1392–1399. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- 99.Semple RK, Crowley VC, Sewter CP, Laudes M, Christodoulides C, Considine RV, Vidal-Puig A, O’Rahilly S. Expression of the thermogenic nuclear hormone receptor coactivator PGC-1alpha is reduced in the adipose tissue of morbidly obese subjects. Int J Obes Relat Metab Disord. 2004;28:176–179. doi: 10.1038/sj.ijo.0802482. [DOI] [PubMed] [Google Scholar]
- 100.Kostis JB, Sanders M. The association of heart failure with insulin resistance and the development of type 2 diabetes. Am J Hypertens. 2005;18:731–737. doi: 10.1016/j.amjhyper.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 101.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 102.Szczepaniak LS, Dobbins RL, Metzger GJ, Sartoni-D’Ambrosia G, Arbique D, Vongpatanasin W, Unger R, Victor RG. Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med. 2003;49:417–423. doi: 10.1002/mrm.10372. [DOI] [PubMed] [Google Scholar]
- 103.Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144:3483–3490. doi: 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- 104.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol. 2003;551:491–501. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, Rosenzweig A, Kahn CR, Abel ED, Walsh K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem. 2002;277:37670–37677. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 106.Ozcan C, Bienengraeber M, Dzeja PP, Terzic A. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H531–H539. doi: 10.1152/ajpheart.00552.2001. [DOI] [PubMed] [Google Scholar]
- 107.Santos DL, Palmeira CM, Seica R, Dias J, Mesquita J, Moreno AJ, Santos MS. Diabetes and mitochondrial oxidative stress: a study using heart mitochondria from the diabetic Goto-Kakizaki rat. Mol Cell Biochem. 2003;246:163–170. [PubMed] [Google Scholar]
- 108.Katakam PV, Jordan JE, Snipes JA, Tulbert CD, Miller AW, Busija DW. Myocardial preconditioning against ischemia-reperfusion injury is abolished in Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2007;292:R920–R926. doi: 10.1152/ajpregu.00520.2006. [DOI] [PubMed] [Google Scholar]
- 109.Nishio Y, Kanazawa A, Nagai Y, Inagaki H, Kashiwagi A. Regulation and role of the mitochondrial transcription factor in the diabetic rat heart. Ann N Y Acad Sci. 2004;1011:78–85. doi: 10.1007/978-3-662-41088-2_9. [DOI] [PubMed] [Google Scholar]
- 110.Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, Hayashidani S, Shiomi T, Kubota T, Hamasaki N, Takeshita A. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003;107:1418–1423. doi: 10.1161/01.cir.0000055318.09997.1f. [DOI] [PubMed] [Google Scholar]
- 111.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–2548. doi: 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- 112.Lautamaki R, Airaksinen KE, Seppanen M, Toikka J, Harkonen R, Luotolahti M, Borra R, Sundell J, Knuuti J, Nuutila P. Insulin improves myocardial blood flow in patients with type 2 diabetes and coronary artery disease. Diabetes. 2006;55:511–516. doi: 10.2337/diabetes.55.02.06.db05-1023. [DOI] [PubMed] [Google Scholar]
- 113.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 114.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 115.Culic O, Gruwel ML, Schrader J. Energy turnover of vascular endothelial cells. Am J Physiol. 1997;273:C205–C213. doi: 10.1152/ajpcell.1997.273.1.C205. [DOI] [PubMed] [Google Scholar]
- 116.Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. 2007;100:1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- 117.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 118.Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179) J Biol Chem. 2001;276:30392–30398. doi: 10.1074/jbc.M103702200. [DOI] [PubMed] [Google Scholar]
- 119.Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- 120.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 121.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001;414:807–812. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 122.Soejima A, Inoue K, Takai D, Kaneko M, Ishihara H, Oka Y, Hayashi JI. Mitochondrial DNA is required for regulation of glucose-stimulated insulin secretion in a mouse pancreatic beta cell line, MIN6. J Biol Chem. 1996;271:26194–26199. doi: 10.1074/jbc.271.42.26194. [DOI] [PubMed] [Google Scholar]
- 123.Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- 124.Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106:165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- 126.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 127.Sedaghat AR, Sherman A, Quon MJ. A mathematical model of metabolic insulin signaling pathways. Am J Physiol Endocrinol Metab. 2002;283:E1084–E1101. doi: 10.1152/ajpendo.00571.2001. [DOI] [PubMed] [Google Scholar]
- 128.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 129.Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE. 2005;2005:pe4. doi: 10.1126/stke.2682005pe4. [DOI] [PubMed] [Google Scholar]
- 130.Zhande R, Mitchell JJ, Wu J, Sun XJ. Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Mol Cell Biol. 2002;22:1016–1026. doi: 10.1128/MCB.22.4.1016-1026.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE, Donner DB. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci U S A. 2001;98:640–4645. doi: 10.1073/pnas.051042298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Egawa K, Maegawa H, Shimizu S, Morino K, Nishio Y, Bryer-Ash M, Cheung AT, Kolls JK, Kikkawa R, Kashiwagi A. Protein-tyrosine phosphatase-1B negatively regulates insulin signaling in l6 myocytes and Fao hepatoma cells. J Biol Chem. 2001;276:10207–10211. doi: 10.1074/jbc.M009489200. [DOI] [PubMed] [Google Scholar]
- 133.Vinciguerra M, Foti M. PTEN and SHIP2 phosphoinositide phosphatases as negative regulators of insulin signalling. Arch Physiol Biochem. 2006;112:89–104. doi: 10.1080/13813450600711359. [DOI] [PubMed] [Google Scholar]
- 134.Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem. 2004;279:36608–36615. doi: 10.1074/jbc.M406499200. [DOI] [PubMed] [Google Scholar]
- 135.Pratipanawatr W, Pratipanawatr T, Cusi K, Berria R, Adams JM, Jenkinson CP, Maezono K, DeFronzo RA, Mandarino LJ. Skeletal muscle insulin resistance in normoglycemic subjects with a strong family history of type 2 diabetes is associated with decreased insulin-stimulated insulin receptor substrate-1 tyrosine phosphorylation. Diabetes. 2001;50:2572–2578. doi: 10.2337/diabetes.50.11.2572. [DOI] [PubMed] [Google Scholar]
- 136.Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Goodyear LJ, Giorgino F, Sherman LA, Carey J, Smith RJ, Dohm GL. Insulin receptor phosphorylation, insulin receptor substrate-1 phosphorylation, and phosphatidylinositol 3-kinase activity are decreased in intact skeletal muscle strips from obese subjects. J Clin Invest. 1995;95:2195–2204. doi: 10.1172/JCI117909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Anai M, Funaki M, Ogihara T, Kanda A, Onishi Y, Sakoda H, Inukai K, Nawano M, Fukushima Y, Yazaki Y, Kikuchi M, Oka Y, Asano T. Enhanced insulin-stimulated activation of phosphatidylinositol 3-kinase in the liver of high-fat-fed rats. Diabetes. 1999;48:158–169. doi: 10.2337/diabetes.48.1.158. [DOI] [PubMed] [Google Scholar]
- 139.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 140.Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, Liu ZX, Soos TJ, Cline GW, O’Brien WR, Littman DR, Shulman GI. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004;114:823–827. doi: 10.1172/JCI22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 142.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 143.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 144.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. 2007;100:1589–1596. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- 146.Senn JJ. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem. 2006;281:26865–26875. doi: 10.1074/jbc.M513304200. [DOI] [PubMed] [Google Scholar]
- 147.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, Shoelson SE, Shulman GI. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108:437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nishikawa T, Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2007;9:343–353. doi: 10.1089/ars.2006.1458. [DOI] [PubMed] [Google Scholar]
- 151.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 152.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 153.Garcia-Ruiz I, Rodriguez-Juan C, Diaz-Sanjuan T, Martinez MA, Munoz-Yague T, Solis-Herruzo JA. Effects of rosiglitazone on the liver histology and mitochondrial function in ob/ob mice. Hepatology. 2007;46:414–423. doi: 10.1002/hep.21687. [DOI] [PubMed] [Google Scholar]
- 154.Stuhlinger MC, Abbasi F, Chu JW, Lamendola C, McLaughlin TL, Cooke JP, Reaven GM, Tsao PS. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA. 2002;287:1420–1426. doi: 10.1001/jama.287.11.1420. [DOI] [PubMed] [Google Scholar]
- 155.Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest. 2004;114:1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lautamaki R, Airaksinen KE, Seppanen M, Toikka J, Luotolahti M, Ball E, Borra R, Harkonen R, Iozzo P, Stewart M, Knuuti J, Nuutila P. Rosiglitazone improves myocardial glucose uptake in patients with type 2 diabetes and coronary artery disease: a 16-week randomized, double-blind, placebo-controlled study. Diabetes. 2005;54:2787–2794. doi: 10.2337/diabetes.54.9.2787. [DOI] [PubMed] [Google Scholar]
- 157.Matsui J, Terauchi Y, Kubota N, Takamoto I, Eto K, Yamashita T, Komeda K, Yamauchi T, Kamon J, Kita S, Noda M, Kadowaki T. Pioglitazone reduces islet triglyceride content and restores impaired glucose-stimulated insulin secretion in heterozygous peroxisome proliferator-activated receptor-gamma-deficient mice on a high-fat diet. Diabetes. 2004;53:2844–2854. doi: 10.2337/diabetes.53.11.2844. [DOI] [PubMed] [Google Scholar]
- 158.Potenza MA, Marasciulo FL, Tarquinio M, Quon MJ, Montagnani M. Treatment of spontaneously hypertensive rats with rosiglitazone and/or enalapril restores balance between vasodilator and vasoconstrictor actions of insulin with simultaneous improvement in hypertension and insulin resistance. Diabetes. 2006;55:3594–3603. doi: 10.2337/db06-0667. [DOI] [PubMed] [Google Scholar]
- 159.Cleasby ME, Dzamko N, Hegarty BD, Cooney GJ, Kraegen EW, Ye JM. Metformin prevents the development of acute lipid-induced insulin resistance in the rat through altered hepatic signaling mechanisms. Diabetes. 2004;53:3258–3266. doi: 10.2337/diabetes.53.12.3258. [DOI] [PubMed] [Google Scholar]
- 160.Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WG, 4th, Schlattner U, Neumann D, Brownlee M, Freeman MB, Goldman MH. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem. 2004;279:43940–43951. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]
- 161.de Cavanagh EM, Inserra F, Toblli J, Stella I, Fraga CG, Ferder L. Enalapril attenuates oxidative stress in diabetic rats. Hypertension. 2001;38:1130–1136. doi: 10.1161/hy1101.092845. [DOI] [PubMed] [Google Scholar]
- 162.Shiuchi T, Iwai M, Li HS, Wu L, Min LJ, Li JM, Okumura M, Cui TX, Horiuchi M. Angiotensin II type-1 receptor blocker valsartan enhances insulin sensitivity in skeletal muscles of diabetic mice. Hypertension. 2004;43:1003–1010. doi: 10.1161/01.HYP.0000125142.41703.64. [DOI] [PubMed] [Google Scholar]
- 163.Koh KK, Quon MJ, Han SH, Chung WJ, Ahn JY, Seo YH, Kang MH, Ahn TH, Choi IS, Shin EK. Additive beneficial effects of losartan combined with simvastatin in the treatment of hypercholesterolemic, hypertensive patients. Circulation. 2004;110:3687–3692. doi: 10.1161/01.CIR.0000143085.86697.13. [DOI] [PubMed] [Google Scholar]
- 164.Blendea MC, Jacobs D, Stump CS, McFarlane SI, Ogrin C, Bahtyiar G, Stas S, Kumar P, Sha Q, Ferrario CM, Sowers JR. Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression. Am J Physiol Endocrinol Metab. 2005;288:E353–E359. doi: 10.1152/ajpendo.00402.2004. [DOI] [PubMed] [Google Scholar]
- 165.Wei Y, Sowers JR, Nistala R, Gong H, Uptergrove GM, Clark SE, Morris EM, Szary N, Manrique C, Stump CS. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J Biol Chem. 2006;281:35137–35146. doi: 10.1074/jbc.M601320200. [DOI] [PubMed] [Google Scholar]
- 166.Hayden MR, Chowdhury N, Govindarajan G, Karuparthi PR, Habibi J, Sowers JR. Myocardial myocyte remodeling and fibrosis in the cardiometabolic syndrome. J Cardiometab Syndr. 2006;1:326–333. doi: 10.1111/j.1559-4564.2006.05626.x. [DOI] [PubMed] [Google Scholar]
- 167.Hayden MR, Sowers JR. Isletopathy in type 2 diabetes mellitus: implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antioxid Redox Signal. 2007;9:891–910. doi: 10.1089/ars.2007.1610. [DOI] [PubMed] [Google Scholar]
- 168.Whaley-Connell AT, Chowdhury NA, Hayden MR, Stump CS, Habibi J, Wiedmeyer CE, Gallagher PE, Tallant EA, Cooper SA, Link CD, Ferrario C, Sowers JR. Oxidative stress and glomerular filtration barrier injury: role of the renin-angiotensin system in the Ren2 transgenic rat. Am J Physiol Renal Physiol. 2006;291:F1308–F1314. doi: 10.1152/ajprenal.00167.2006. [DOI] [PubMed] [Google Scholar]
- 169.Khamaisi M, Potashnik R, Tirosh A, Demshchak E, Rudich A, Tritschler H, Wessel K, Bashan N. Lipoic acid reduces glycemia and increases muscle GLUT4 content in streptozotocin-diabetic rats. Metabolism. 1997;46:763–768. doi: 10.1016/s0026-0495(97)90120-7. [DOI] [PubMed] [Google Scholar]
- 170.Rudich A, Tirosh A, Potashnik R, Khamaisi M, Bashan N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia. 1999;42:949–957. doi: 10.1007/s001250051253. [DOI] [PubMed] [Google Scholar]
- 171.Chen H, Karne RJ, Hall G, Campia U, Panza JA, Cannon RO, III, Wang Y, Katz A, Levine M, Quon MJ. High-dose oral vitamin C partially replenishes vitamin C levels in patients with type 2 diabetes and low vitamin C levels but does not improve endothelial dysfunction or insulin resistance. Am J Physiol Heart Circ Physiol. 2006;290:H137–H145. doi: 10.1152/ajpheart.00768.2005. [DOI] [PubMed] [Google Scholar]
- 172.Ting HH, Timimi FK, Boles KS, Creager SJ, Ganz P, Creager MA. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 1996;97:22–28. doi: 10.1172/JCI118394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kavanagh T, Mertens DJ, Hamm LF, Beyene J, Kennedy J, Corey P, Shephard RJ. Prediction of long-term prognosis in 12 169 men referred for cardiac rehabilitation. Circulation. 2002;106:666–671. doi: 10.1161/01.cir.0000024413.15949.ed. [DOI] [PubMed] [Google Scholar]
- 174.Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346:793–801. doi: 10.1056/NEJMoa011858. [DOI] [PubMed] [Google Scholar]
- 175.Wisloff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, Fernstrom M, Rezaei K, Lee SJ, Koch LG, Britton SL. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. 2005;307:418–420. doi: 10.1126/science.1108177. [DOI] [PubMed] [Google Scholar]
- 176.Hughes VA, Fiatarone MA, Fielding RA, Kahn BB, Ferrara CM, Shepherd P, Fisher EC, Wolfe RR, Elahi D, Evans WJ. Exercise increases muscle GLUT-4 levels and insulin action in subjects with impaired glucose tolerance. Am J Physiol. 1993;264:E855–E862. doi: 10.1152/ajpendo.1993.264.6.E855. [DOI] [PubMed] [Google Scholar]
- 177.Henriksen EJ. Invited review: effects of acute exercise and exercise training on insulin resistance. J Appl Physiol. 2002;93:788–796. doi: 10.1152/japplphysiol.01219.2001. [DOI] [PubMed] [Google Scholar]
- 178.Toledo FG, Menshikova EV, Ritov VB, Azuma K, Radikova Z, DeLany J, Kelley DE. Effects of physical activity and weight loss on skeletal muscle mitochondria and relationship with glucose control in type 2 diabetes. Diabetes. 2007;56:2142–2147. doi: 10.2337/db07-0141. [DOI] [PubMed] [Google Scholar]
- 179.Short KR, Vittone JL, Bigelow ML, Proctor DN, Rizza RA, Coenen-Schimke JM, Nair KS. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes. 2003;52:1888–1896. doi: 10.2337/diabetes.52.8.1888. [DOI] [PubMed] [Google Scholar]
- 180.Constable SH, Favier RJ, McLane JA, Fell RD, Chen M, Holloszy JO. Energy metabolism in contracting rat skeletal muscle: adaptation to exercise training. Am J Physiol. 1987;253:C316–C322. doi: 10.1152/ajpcell.1987.253.2.C316. [DOI] [PubMed] [Google Scholar]
- 181.Suvorava T, Lauer N, Kojda G. Physical inactivity causes endothelial dysfunction in healthy young mice. J Am Coll Cardiol. 2004;44:1320–1327. doi: 10.1016/j.jacc.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 182.Menshikova EV, Ritov VB, Toledo FG, Ferrell RE, Goodpaster BH, Kelley DE. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am J Physiol Endocrinol Metab. 2005;288:E818–E825. doi: 10.1152/ajpendo.00322.2004. [DOI] [PubMed] [Google Scholar]
- 183.Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 185.Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- 186.Guarente L. Sirtuins as potential targets for metabolic syndrome. Nature. 2006;444:868–874. doi: 10.1038/nature05486. [DOI] [PubMed] [Google Scholar]
- 187.Millet L, Vidal H, Andreelli F, Larrouy D, Riou JP, Ricquier D, Laville M, Langin D. Increased uncoupling protein-2 and -3 mRNA expression during fasting in obese and lean humans. J Clin Invest. 1997;100:2665–2670. doi: 10.1172/JCI119811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lopez-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, de Cabo R. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]