

Abstract
Estrogen stimulates growth and inhibits apoptosis of breast cancer cells via genomic and non-genomic actions. However, the detailed mechanism by which estrogen inhibits the pro-apoptotic pathways that might impede the normal homeostasis and action of chemotherapeutic drugs in breast cancer cells is not well understood. Here, we report a negative regulation of a pro-apoptotic kinase, Mixed Lineage Kinase 3 (MLK3) by 17β-estradiol (E2) that hinders cytotoxic drug-induced cell death in estrogen receptor positive (ER+) breast cancer cells. MLK3 kinase activities were significantly higher in estrogen receptor negative (ER−), progesterone receptor negative (PR−) primary human breast tumors, suggesting that E2 might have a negative role in regulating MLK3 kinase activity. The kinase activities of MLK3 and its downstream target, JNK were rapidly inhibited by E2 in ER+ but not in ER− breast cancer cells. The inhibition of MLK3 kinase activity by E2 was mediated via activation of protein kinase B (PKB/AKT) because specific knockdown of AKT1/2 prevented the E2-induced inhibition of MLK3. Furthermore, E2-induced inhibition of MLK3 kinase activity involved a direct phosphorylation of MLK3 at Ser 674 site by AKT, which resulted in an attenuation of the pro-apoptotic function of MLK3. In addition, a pan-MLK inhibitor (CEP-11004) significantly attenuated Taxol-induced cell death, which was further synergized by E2. Thus, our data suggest that E2 negatively regulates the pro-apoptotic function of MLK3 during breast cancer pathogenesis and therefore MLK3 and other MLK family members might play an important role in cytotoxic drug-induced cell death in ER+ breast cancer cells.
Keywords: Estrogen, Kinase, MLKs, JNK and AKT
Introduction
Estrogens comprise a group of structurally related, hormonally active molecules that regulate cell proliferation, differentiation and homeostasis (1). Estrogen occurs naturally in several structurally related forms; however, the predominant form that binds strongly to the cognate estrogen receptor α (ERα) is 17β-estradiol (E2). E2 binds to the nuclear ERα, causing dimerization of receptors that finally drives estrogen action by regulating transcription of target genes (1, 2). It is also reported that E2 as well acts through rapid, non-genomic pathways (3, 4), where the plasma membrane localized ERα (5, 6) or G protein-coupled receptors (GPCR) superfamily, termed GPR30, acts independent of classical ERs to trigger rapid signaling by E2 (7, 8). Besides ERα, another estrogen receptor ERβ (9–11) has also been reported, whose function is still not well understood in hormonal signaling.
It is well documented that E2-mediated pathways play a central role in the survival and proliferation of breast cancer cells (1). Therefore the agents that block either E2 synthesis or antagonize E2-mediated pathways are in clinical use to control the growth of ER+ breast cancer cells. Thus the prevailing hypothesis in breast cancer field is that estrogen promotes survival of ER+ breast cancer cells by inhibiting the pro-apoptotic cellular machinery. Although, recently, it has been reported that E2 paradoxically promotes cell death in long term estrogen deprived cells (12–15), however, the detailed mechanism is not well established. The mechanism by which estrogen promotes survival of ER+ breast cancer cells is also not well understood. It is reported that the rapid action of E2 activates Src (16–18) and PI3K-AKT (19) pathways, coupled to the IGF-1 and EGF receptors, which might lead to increased cell survival. Whether E2 inhibits any pro-apoptotic kinase to provide growth advantage to E2-dependent breast cancer cells is unknown.
Here, we report a role of a pro-apoptotic kinase, MLK3 that mediates the rapid action of E2 on the survival of ER+ breast cancer cells. MLK3 is a member of MLK sub-family that belongs to the family of MAP Kinase Kinase Kinases (MAP3Ks) (20). The MLK family members are characterized by the presence of signature sequences of Ser/Thr and Tyr kinases within their catalytic domain (20). Previous work by us and others have shown that MLK family members, including MLK3 (20) activate the Jun-N-terminal kinase (JNK) (21, 22). We showed that activation of JNK was mediated via direct phosphorylation and activation of upstream kinase, SEK1/MKK4 by MLK3 (21). Furthermore, we reported that MLK3 was directly inhibited by AKT by a direct phosphorylation at a specific residue on MLK3 C-terminal regulatory domain (23). Although the detailed function of MLKs, including MLK3 is still unknown, it is reported that MLK3 behaves as a pro-apoptotic kinase, leading to cell death upon trophic factor withdrawal (24) or in response to neurotoxic assault in an animal model of Parkinson’s disease (25, 26). The in-depth mechanism by which MLK3 or other MLK family members control survival or death of breast cancer cells is yet to be determined.
In this report, we show that MLK3 kinase activity was significantly higher in ER−, PR− human breast tumors. Interestingly the kinase activity of MLK3 was inhibited in ER+ (MCF7 and ZR75-1) but not in ER− (SkBr3) cell lines by E2. Furthermore, we show that E2-induced inhibition of MLK3 kinase activity was mediated via PI3K-AKT pathway and more specifically via AKT phosphorylation of MLK3 on Ser 674. Induction of cell death mediated via MLK3 in ER+ breast cancer cells was also attenuated by E2. In addition, pharmacological inhibition of all MLK activity antagonized cytotoxic drug-induced cell death in ER+ breast cancer cells, which was completely blocked in the presence of both E2 and pan-MLK inhibitor combination. Taken together, our data demonstrate for the first time, an important role of MLK3 (and possibly other MLKs) in E2-mediated cell death pathways in breast cancer cells.
Materials and Methods
Cell culture and treatments
Human ER+, MCF7 and ZR75-1, and ER−, SkBr3 (ATCC, Rockville, MD) breast cancer cell lines and Murine Embryonic Fibroblasts (MEFs, provided by Prof. Nissim Hay, Univ. Illinois at Chicago, IL) were maintained in DMEM containing 10% FBS, 2 mM glutamine and antibiotics (Penicillin G/streptomycin) for 2 days. To elevate the estrogenic effect of exogenously added E2, the cells were cultured in DMEM without phenol red, supplemented with 5% charcoal-dextran stripped FBS, antibiotics and 2 mM glutamine for further 2 days. For E2 treatment, cells were starved for 12 hrs in DMEM without phenol red, supplemented with 0.2 % charcoal-dextran stripped FBS and treated with or without 10 nM of E2 (Sigma, St. Louis, MO) at different time intervals, as indicated. For inhibitors treatment, the starved cells in phenol red-free DMEM medium were pre-treated, wherever indicated, with 50 μM LY294002, 100 nM Wortmannin, 20 μM AKT inhibitor VIII, isozyme-selective, AKTi-1/2 (all from Calbiochem, San Diego, CA) and 10 nM ICI 182, 780 (TOCRIS Bioscience, Ellisville, MO) for 2–3 (as indicated) hrs before E2 treatment. The pan-MLK inhibitor, CEP-11004 (kind gift from Cephalon, PA) was dissolved in DMSO and reconstituted in DMEM before treatment. To assess cell death caused by Taxol, cells were pre-treated with CEP-11004 (500 nM) for 18 hrs, and subsequently with E2 for 8 hrs and then with Paclitaxel/Taxol (1 μM) (Ivax Laboratories, Miami, FL) for 24 hrs.
Immunoblotting
Western Blot analysis was performed following protocols described previously (22–24) by incubation with primary antibodies against: phospho-AKT, total AKT, PARP (all from Cell Signaling Technology, Inc., Beverly, MA), phospho-JNK (Promega, Madison, WI) and total JNK (Santa Cruz Biotech, Santa Cruz, CA). To determine whether MLK3 is the substrate of AKT, Phospho-(Ser/Thr) AKT substrate antibody (Cell Signaling Technology, Inc., Beverly, MA) was used. The specific signals were finally detected using HRP-conjugated secondary antibodies and enhanced chemiluminescence (ECL).
Cell transfection
MCF7 cells were transiently transfected either with Flag-tagged WT-MLK3 or Flag-MLK3 S674A mutant using Lipofectaminne-2000 (Life technologies Inc., Gaithersburg, MD) following manufacturer’s protocol. The endogenous AKT1/2 in MCF7 cells were knockdown as described (27). MCF7 cells were transfected with 100 nM siRNA using Lipofectamine-2000. Forty-eight hours post transfection; cells were treated with E2 for 8 hrs, as described above.
Cell death assays
Apoptosis of cells was evaluated using a Cell Death Detection ELISAPlus (Roche Diagnostics, Indianapolis, IN) kit according to manufacturer’s instructions. Briefly, the MCF7 cells were transfected with Flag-MLK3 and then treated with E2 (10 nM) for 8 hrs. The cells were collected by centrifugation at 2000 rpm and analyzed by the cell death detection ELISA kit. Data are expressed as mean ± S.D. of three independent experiments.
Immunoprecipitation and kinase assay
The breast tumors and matching normal breast tissue samples were freshly collected and immediately snap-frozen in liquid nitrogen following IRB approved protocol. The frozen tissues were homogenized in protein extraction buffer and endogenous MLK3 (from tissues or cells) was immunoprecipitated with the appropriate antibodies. Endogenous MLK3 was immunoprecipitated by using an antibody raised against the C-terminal peptide of MLK3, developed in our laboratory (22–24). Ectopically expressed MLK3 was immunoprecipitated by using antibody against the FLAG-epitope-tag (Sigma, St. Louis, MO). In vitro kinase assay was then performed following our published protocol (22–24). The incorporation of 32P into SEK1 (K-R) was quantified by PhosphoImager (STORM 820, GE Healthcare Bio-sciences, USA) and the kinase activity was presented as arbitrary phosphoimager (PI) units.
Results
MLK3 kinase activity is significantly higher in ER−, PR− breast tumors
In order to elucidate any role of MLK3 in breast cancer cell apoptosis, we estimated the catalytic activity of MLK3 in primary human breast tumors and matching normal breast tissues. Our results showed that MLK3 kinase activity was significantly higher in some tumors (about 10 fold or more) compared to the matching normal breast tissue (Fig. 1). Further analysis was then carried out to determine any link between increased MLK3 activity with the ER and PR status in these breast tumors. Surprisingly, these results revealed that MLK3 kinase activity was about 5 fold higher, exclusively in the ER−, PR− compared to ER+, PR+ tumors (Fig. 1). Additionally, we also measured MLK3 kinase activity in five more tumors. MLK3 kinase activity was about 5–12 folds higher in the ER− tumors when compared to the ER+ breast tumors (Supplementary Fig. S1). Taken together, these results suggested the possibility that MLK3 might be negatively regulated by E2-mediated pathways in ER+ breast cancer tumors.
Figure 1. Endogenous MLK3 kinase activity is significantly higher in ER−, PR− breast tumors.
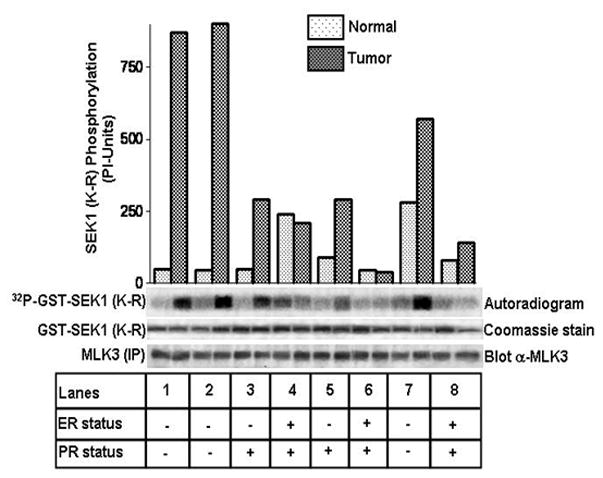
Equivalent amounts of endogenous MLK3 were immunoprecipitated from tumors and matching normal breast tissues with a specific antibody raised against MLK3. MLK3 kinase assay was performed with the immunoprecipitates using SEK1 (K-R) protein as a specific substrate. Phosphorylation of SEK1 (K-R) was detected by phosphoimager and autoradiography. The expression status of ER and PR in these tumors was provided by the department of Pathology, Scott and White Hospital, Temple, Texas.
Estrogen negatively regulates MLK3 and its downstream target JNK in ER+ but not in ER− breast cancer cell lines
In order to understand any role of estrogen in regulating MLK3 kinase activity and its downstream events, we treated ER+ MCF7 and ER− SkBr3 cell lines with 10 nM of E2 for different time intervals (Fig. 2). The optimal dose of E2 (i.e. 10 nM) to inhibit MLK3 kinase activity was determined by dose course (data not included). Our results showed that MLK3 kinase activity was rapidly inhibited by E2 within 30 min of treatment with maximal inhibition, (about 4.5 fold) after about 8 hrs of E2 treatment in the ER+ MCF7 (Fig. 2A) but not in ER−SkBr3 cells (Fig. 2B). To prove that inhibitory action of E2 on MLK3 kinase activity was not cell type dependent, we treated another ER+ breast cancer cell line, ZR75-1 with E2 and estimated MLK3 kinase activity. Endogenous MLK3 kinase activity in ER+ ZR75-1 cells was also inhibited in a similar fashion as in MCF7 cells (Fig. 2C). To confirm that the inhibition of MLK3 kinase activity in ER+ breast cancer cell lines was due to specific action of estrogen, we utilized the anti-estrogen compound, ICI 182,780 (28) to block the effect of E2 in MCF7 cell line. The inhibitory effect of E2 on MLK3 kinase activity was completely abrogated by ICI compound (Fig. 2D). Our earlier studies showed that MLK3 mediates downstream JNK activation via SEK1/MKK4 (21). Therefore, in order to determine, whether E2-mediated inhibition of MLK3 activity subsequently also leads to inhibition of its downstream JNK kinase activity, we determined the activation status of JNK by using phospho-antibody directed against the activation sites of JNK. Inhibition of MLK3 by E2 also resulted in a corresponding inhibition of JNK activation in ER+ (Supplementary Fig. S2A) but not in ER− (Supplementary Fig. S2B) cell lines. These results demonstrated that E2 indeed inhibits the kinase activities of MLK3 and its downstream JNK in ER+ breast cancer cells.
Figure 2. E2 inhibits endogenous MLK3 in ER+ but not in ER− cell lines.
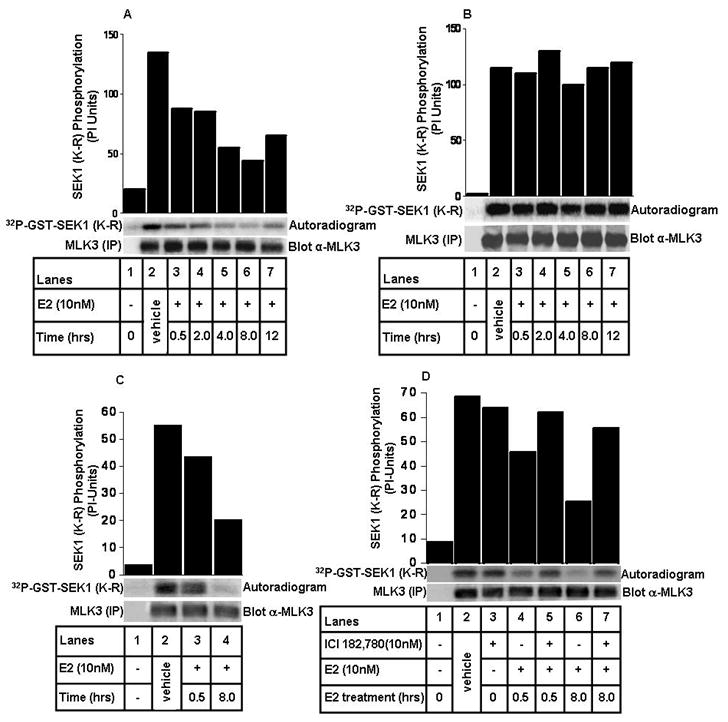
(A) MCF7 cells were treated with 10 nM of E2 at different time points, as indicated. MLK3 was immunoprecipitated and subjected to in vitro kinase assays. Equal expression of MLK3, utilized for kinase assays, was determined by anti-MLK3 immunoblotting. (B) SkBr3 cells were treated with E2 similarly as described in A. MLK3 was immunoprecipitated and subjected to kinase assay as described in Fig. 1. (C) ZR75-1 cells were treated with 10 nM of E2 at indicated time points. MLK3 kinase activity was measured as described above. (D) MCF7 cells were treated with 10 nM of ICI 182,780 for 3 hrs prior to E2 (10 nM) treatment and MLK3 kinase activity was measured. These data represent one of three similar experiments (for panel A and B) and one of two (for panel C and D).
Estrogen inhibits MLK3 and JNK kinase activities via PI3K-AKT pathway
Next, we wanted to know the mechanism by which E2 inhibits MLK3 and JNK kinase activities. Our previous results showed that upon incubation with insulin, AKT can phosphorylate and inhibit MLK3 kinase activity (23). Since E2 is reported to activate PI3K-AKT pathway in breast cancer cells (19), it was conceivable that the inhibitory effect of E2 on MLK3 kinase activity might be mediated via PI3K-AKT pathway. To understand the contribution of PI3K-AKT in these events, we first determined whether E2 activates AKT in an ER-dependent manner. Both ER+ MCF7 and ER− SkBr3 cells were treated with E2 for different time intervals and equal amounts of total cell lysates were Western Blotted with phospho-AKT antibody. In the MCF7 cells, E2 was able to activate AKT in a time dependent manner and the maximal activation was observed in 8 hrs of E2 treatment (Fig. 3A), which corresponded with maximal MLK3 inhibition (Fig. 2A). However, E2 was unable to activate AKT in the ER− SkBr3 cells (Fig. 3B); suggesting that E2-mediated activation of AKT involves ER. To examine any role of PI3K-AKT pathway on E2-mediated inhibition of MLK3 activity, the ER+ MCF7 cells were pre-treated with two different pharmacological inhibitors of PI3K-AKT pathway (LY2949002 or Wortmanin) and then treated with 10 nM of E2 for 8 hrs. MLK3 kinase assays performed following these treatments showed that E2-induced inhibition of MLK3 kinase activity was antagonized by both the inhibitors of PI3K-AKT pathway (Fig. 3C). These results suggested that E2 inhibits MLK3 kinase activity via activation of PI3K-AKT pathway. We also observed that E2-induced inhibition of JNK, (a downstream target of MLK3) was regulated similarly by PI3K-AKT inhibitors (Fig. 3D). Taken together, these results clearly suggest that E2 effects on MLK3 and its downstream target JNK are mediated via PI3K-AKT pathway. However, these results do not prove whether it is PI3K or AKT that ultimately causes E2-induced MLK3 inhibition.
Figure 3. E2 inhibits MLK3 and its downstream target, JNK kinase activities via PI3K-AKT pathway.
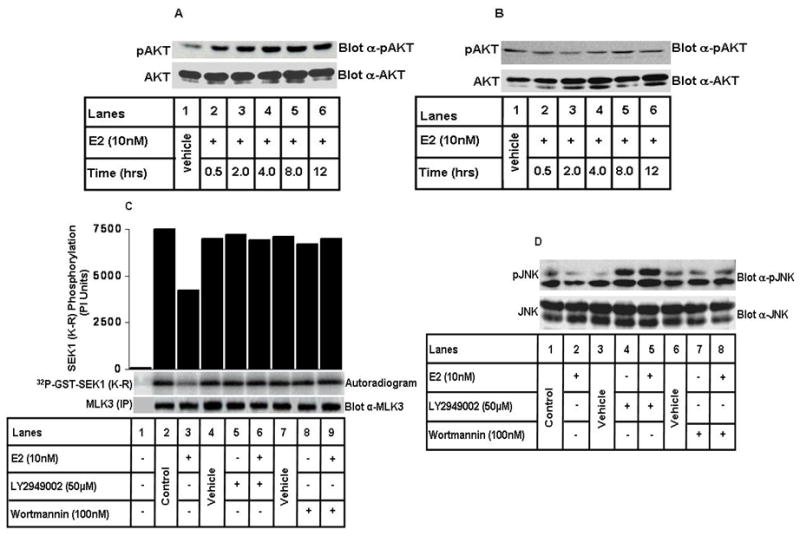
(A) MCF7 cells were treated with 10 nM of E2 for different time intervals, as indicated and western blotting was done using anti-phospho AKT and anti-AKT antibodies. (B) SkBr3 cells were treated with E2 and western blotting was done as described in A. (C) MCF7 cells were pretreated with PI3-kinase inhibitors, either LY294002 (50 μM) or Wortmannin (100 nM) for 2 hrs and then with E2 (10 nM) for 8 hrs. MLK3 kinase assay was performed as described in Fig. 1. (D) Cell lysates from C were immunoblotted for anti-phospho JNK and anti-JNK antibodies. These data represent one of four similar experiments.
Protein Kinase B (PKB/AKT) mediates the MLK3 inhibition by Estrogen
In order to understand whether the inhibitory effect of E2 is mediated directly via AKT in ER+ MCF7 cells, we took advantage of AKT specific inhibitor, AKTi-1/2, which is reported to inhibit AKT specifically (29). The cells were pre-treated with AKT inhibitor followed by treatment with E2. As observed earlier with the PI3K inhibitors, E2 was unable to inhibit MLK3 kinase activity when the cells were pre-treated with AKT inhibitor (Fig. 4A), suggesting that AKT does mediate the E2’s inhibitory effect on MLK3 kinase activity. To determine whether the AKT inhibitor used actually inhibits AKT activity, the inhibitor pre-treated cell lysates were also blotted with p-AKT antibody, which showed a complete inhibition of AKT phosphorylation following inhibitor pre-treatment (Supplementary Fig. S3). Pharmacological inhibitors of PI3K and AKT have been used extensively to demonstrate direct role of PI3K and AKT in cell signaling. However, use of these inhibitors does not rule out their potential non-specific effects on other pathways. To demonstrate specific role of AKT in E2-induced inhibition of MLK3 kinase activity, we first examined the expression of various AKT isoforms in MCF7 cells. We observed that AKT1 and AKT2 are both expressed in MCF7 cell line (Supplementary Fig. S4). To conclusively determine the involvement of AKT, we then knocked down AKT1 and AKT2 in MCF7 cells by using specific siRNA that was reported to knockdown both AKT isoforms (27). Subsequently, we examined whether E2 was able to inhibit MLK3 kinase activity in the absence of endogenous AKT1/2. As shown in Fig. 4B, while E2 was able to inhibit MLK3 kinase activity in the parental cells; but failed to do so in the absence of endogenous AKT1/2 expression in MCF7 cells. As stated before, we have shown earlier that insulin can inhibit MLK3 kinase activity via direct phosphorylation of MLK3 by AKT (23). In order to know whether AKT phosphorylates MLK3 upon E2 treatment, we performed studies utilizing a phospho-AKT substrate antibody that recognizes the phosphorylated AKT substrates (30). Endogenous MLK3 was immunoprecipitated from cell lysates as described in Fig. 4A and the immunoprecipitates were blotted with phospho-AKT substrate antibody. These results showed that endogenous MLK3 was phosphorylated upon E2 treatment and this phosphorylation was blocked by AKT inhibitor (Fig. 4C), suggesting that AKT does phosphorylate MLK3 upon E2 treatment. To further define that the inhibitory effect of E2 on MLK3 was via AKT-mediated phosphorylation, we used an MLK3 mutant, where the AKT phosphorylation site was mutated (23). This site on MLK3 was initially identified as the insulin regulated phosphorylation site by AKT (23). The MCF7 cells were transfected with either Flag-tagged-MLK3-Wild Type (WT) or with MLK3 Ser 674 Ala (MLK3 S674A) mutant and then treated either with E2 or vehicle. In vitro kinase assays performed following immunoprecipitation of ectopic MLK3 using the anti-FLAG antibody showed that 10 nM of E2 was able to inhibit ectopically expressed wild type MLK3, reasonably to a lesser extent compared to endogenous MLK3 (Fig. 4D). Interestingly, E2 was unable to inhibit the activity of MLK3 S674A mutant, suggesting that E2-induced AKT does inhibit MLK3 via direct phosphorylation of Ser 674 site. Taken together these results convincingly prove that the inhibitory effect of E2 in ER+ breast cancer cells is mediated via phosphorylation of MLK3 by AKT.
Figure 4. Protein kinase AKT mediates the MLK3 inhibition by E.
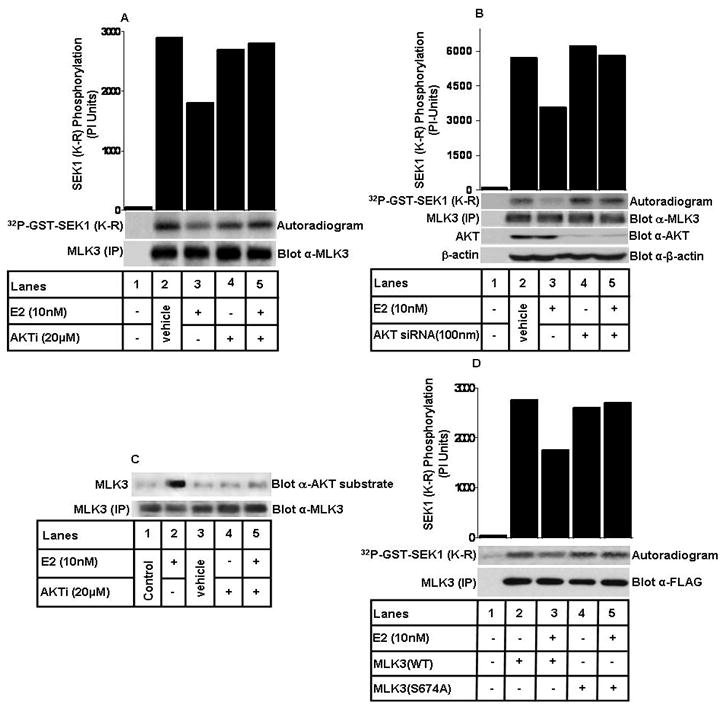
2. (A) MCF7 cells were pretreated with 20 μM of AKT specific inhibitor (AKTi-1/2) for 2 hrs and then treated with E2 (10 nM) for 8 hrs. MLK3 kinase assay was performed as described above in Fig. 1. (B) MCF7 cells were transfected with AKT1/2 specific siRNA for 48 hrs and then treated with 10 nM of E2 for 8 hrs. The cell lysates were first used to judge the knockdown of total AKT and then used to measure MLK3 kinase activity. (C) MLK3 was immunoprecipitated with a specific antibody and immunoblotted for anti-phospho-AKT substrate antibody. (D) MCF7 cells were transfected either with Flag-epitope-tagged MLK3 WT or MLK3 S674A mutant expression vectors and then treated with E2 (10 nM) for 8 hrs. MLK3 was immunoprecipitated using anti-FLAG antibody and kinase assay was performed as described above in Fig. 1. These data represent one of three similar experiments (for panel A, C and D) and one of two (for panel B).
Estrogen inhibits the pro-apoptotic activity of MLK3
MLK3 has been shown to cause cell death in neuronal and non neuronal cells (23, 24), whereas E2 in most breast cancer cells, especially in ER+ ones causes survival (31, 32). E2 has also been shown to have trophic effects on neuronal cells (33–35). Since MLK3 and E2-mediated pathways have opposing effects, we wanted to know whether pro-apoptotic activity of MLK3 is antagonized by E2 in ER+ breast cancer cells. To understand the effect of E2 on pro-apoptotic activity of MLK3, MCF7 cells were transfected with wild type MLK3 (which was shown earlier to be constitutive active) (21), and then treated either with E2 or vehicle for 8 hrs. The cell lysates were blotted with anti-PARP antibody to determine the effect of E2 on cell death induced by MLK3 expression. We observed that MLK3 overexpression resulted in PARP cleavage, which was significantly attenuated following E2 treatment (Fig. 5A), suggesting that E2 attenuates the cell death activity of MLK3. We also measured the extent of cell death in these cells by using the apoptosome assay. In this assay, E2 again antagonized the cell death function of MLK3 (Fig. 5B). Collectively, these results suggest that E2 down-regulates the pro-apoptotic function of MLK3 in ER+ breast cancer cells.
Figure 5. Pro-apoptotic activity of MLK3 is inhibited by E.
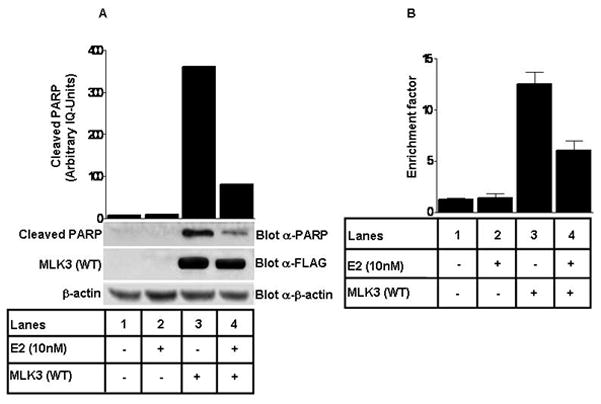
2. (A) MCF7 cells were transfected with Flag-epitope-tagged MLK3 (WT) and then treated with E2 (10 nM) for 8 hrs. Cell lysates were immunoblotted with anti-PARP antibody. Equal expression of MLK3 was detected by anti-FLAG antibody and β-actin was used as a loading control. (B) Apoptosis was further confirmed in the same extracts using cell death detection ELISA kit (Roche) following manufacture’s protocol. These data represent one of three similar experiments.
MLK3-mediated cell death by cytotoxic drug is attenuated by Estrogen
Cytotoxic drugs, such as Taxol is generally used to induce cell death in cancer cells, including breast cancer cells (36). Our previous studies have shown that Taxol and other cytotoxic drugs induce MLK3 kinase activity (unpublished results). We thus wanted to know whether Taxol-induced cell death in ER+ MCF7 cells is also antagonized by E2, and any possible link of endogenous MLKs in mediating this effect. To perform these studies, we utilized the available pan-MLK inhibitor, CEP-11004 that inhibits the activities of all MLK family members, including MLK3 (25, 26, 37). The cells were pre-treated with CEP-11004 for 18 hrs followed by treatment with E2 for 8 hrs, and then with Taxol for 24 hrs, as indicated in Fig. 6A. The cell death in these cells was determined by examining the cleavage of PARP. Taxol was able to induce cell death in these cells as indicated by increased PARP cleavage (Fig. 6A, lane 4), which was partially blocked by CEP-11004 (Fig. 6A, lane 6), suggesting the participation of MLK group of kinases. Interestingly, Taxol-induced cell death was significantly reduced by E2 (Fig. 6A, lane 5) and completely blocked by E2 and CEP-11004 combination (Fig. 6A, lane 7). These results suggest that MLK members, including MLK3 mediate breast cancer cell death by Taxol, and this effect of Taxol is down modulated by inhibitory effect of E2 on MLK3 kinase activity.
Figure 6. Cytotoxic drug-induced breast cancer cell death via MLKs is inhibited by E.
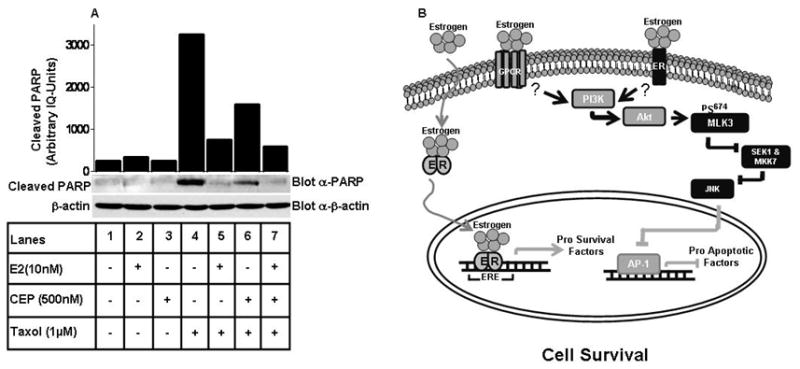
2 and a model of E2-mediated inhibition of MLK3 and its downstream effect on cell death. (A) MCF7 cells were pre-treated with MLKs inhibitor, CEP-11004 for 18 hrs and subsequently treated first, with E2 for 8 hrs and then Taxol for 24 hrs. Cell lysates were immunoblotted with anti-PARP and anti-β-actin antibodies. (B) Model of MLK3 kinase activity inhibition by E2 that promote ER+ breast cancer cell survival. The details are described within the text. The data presented here represent one of three similar experiments.
Discussion
The overall goal of chemo- or radio-therapies for breast cancer treatment is to promote cell death in the tumors, so that they can undergo regression and not metastasize to different organs. Most of the modalities currently used for breast cancer treatment, result in cell death by modulating various cell death pathways (38). Thus, it is clear that any impediment of the normal cell death pathways might lead to drug-resistance and breast cancer progression. It is well documented that the steroidal hormone, estrogen promotes cell survival and proliferation of breast cancer cells by regulating the downstream signaling pathways via genomic and non-genomic actions of the hormone (1, 3, 39). Therefore in estrogen receptor positive breast cancer cells, agents that antagonize ER signaling pathway or block estrogen synthesis are expected to be effective in promoting cell death (40). Detailed understanding of the pathway by which estrogen promotes cell survival and identification of potential targets are necessary to design rational therapeutic approaches to combat estrogen-dependent breast cancer.
Here, we demonstrate that the pro-apoptotic kinase MLK3 is a target of ER and define the mechanism by which ER antagonizes MLK3 signaling. In order to define the role of MLK3 in breast cancer cells, we measured MLK3 kinase activities in primary breast tumors, as a first step. We were perplexed initially to observe that in some tumors; the MLK3 kinase activities were very high compared to others, and also to the matching normal tissues (Figs. 1 & S1). These results suggested that MLK3 might have some yet to be identified role in breast tumors. Further analysis and comparison of the MLK3 kinase activities with the ER and PR status of the breast tumors analyzed showed that there was a direct correlation between ER and PR expression with MLK3 kinase activities in these tumors. Our data clearly showed that MLK3 kinase activities were significantly higher in ER−, PR− tumors (Figs. 1 & S1). These results suggested the possibility that estrogen might antagonize MLK3 activity and prompted us to examine the role of estrogen in the regulation of MLK3 kinase activities. The treatment of ER+ breast cancer cells with 10 nM of E2 significantly inhibited MLK3 kinase activities within 30 min (Fig. 2A); however, it was unable to inhibit the MLK3 kinase activity in ER− breast cancer cells (Fig. 2B). These results, combined with those obtained from the tumor samples (Figs. 1 & S1), clearly showed that E2 negatively regulates MLK3 kinase activity in an estrogen receptor expression- dependent manner. Interestingly, our data also suggested a non-genomic effect of E2 on initial MLK3 inhibition, because MLK3 was inhibited within 30 min of E2 treatment (Fig. 2A); however, the MLK3 was maximally inhibited at 8 hrs of E2 treatment, which might be due to genomic action of E2 for sustained inhibition. Nonetheless, irrespective of genomic or non-genomic action of E2 on MLK3 kinase activity, our data clearly suggest that the E2 effect was specific because anti-estrogen compound, ICI 182,780 blocked the inhibitory action of E2 on MLK3 kinase activity (Fig. 2D).
Activation of JNK in neuronal and non-neuronal cells has been shown to cause cell death (23, 24). It has also been reported that MLK3 causes cell death via JNK activation (41, 42) and thus one possible downstream effect of E2-mediated MLK3 inhibition in breast cancer cells might be to antagonize MLK3/JNK-induced cell death. We also observed that inhibition of MLK3 in ER+ breast cancer cells leads to inhibition of JNK (Fig. S2), suggesting that MLK3 regulates JNK activity in these cells. Whether MLK3 causes cell death in breast cancer cells via JNK activation is yet to be determined. It is also reported that pro-survival kinase, AKT is over expressed (43, 44) in breast tumors and is activated by E2 (45). Since earlier, we reported that AKT directly phosphorylates MLK3 and inhibits its activity (23), it was tempting to determine whether estrogen regulates MLK3 activity via AKT pathway. It is possible that during breast cancer pathogenesis, the pro-survival pathways regulated by E2 down modulate MLK3’s pro-apoptotic function via AKT activation. We observed that AKT indeed phosphorylated MLK3 following E2 treatment, which was determined by phospho-AKT substrate antibody (Fig. 4C). This conclusion was further confirmed by using the phospho-deficient MLK3 mutant that lacks the AKT phosphorylation site (Fig. 4D).
As prevailing clinical and non-clinical data supports the pro-survival functions of estrogen pathway, it was expected that probably inhibition of MLK3 by E2 will promote survival of breast cancer cells. The inhibition of MLK3 kinase activity by E2 attenuated the pro-apoptotic functions of MLK3 (Fig. 5), suggesting that under normal conditions, MLK3 and perhaps other MLKs might regulate the normal cell death pathway in breast epithelial cells. The biggest challenge for any cancer treatment, including breast cancer is to overcome the resistance that these cells acquire towards induction of cell death, in response to cytotoxic drugs. Based on the facts that (i) MLK3 or other MLK members are pro-apoptotic kinases, and (ii) that cytotoxic drugs activate MLK3 kinase activity (our unpublished observations), it is conceivable that inhibition of these protein kinases by E2 might antagonize cytotoxic drug-induced cell death. In fact, pre-treatment of ER+ cells with a combination of pan-MLK inhibitor CEP-11004 and E2 completely abrogated cytotoxic drug-induced cell death (Fig. 6A), thus confirming that MLK3 or some yet to be identified MLK family member(s) play an important role in modulating breast cancer cell death.
Based on our current data and published results, we propose a model of MLK3 inhibition by E2 that finally leads to down modulation of cell death pathway in ER+ breast cancer cells (Fig. 6B). E2 can bind to both ERα and G-protein coupled receptor at the membrane or to the nuclear ERαs resulting in their dimerization. The binding of E2 to membrane localized ERα or GPCR initiates the non-genomic action of E2 that causes activation of AKT via PI3K. Once AKT is activated, it phosphorylates MLK3 at Ser 674 site, resulting in inhibition of MLK3 kinase activity. Inhibition of MLK3 attenuates JNK activity and finally blocks the transcription of pro-apoptotic genes via AP-1 transcription factor. At the same time, nuclear ERα binds to estrogen response elements (ERE) and promotes transcription of pro-survival genes. Therefore a combination of non-genomic action of E2 on AKT-MLK3-JNK pathway and genomic action via up-regulation of pro-survival factors finally shifts the balance towards cell survival.
In conclusion, our data provide an insight towards the role of MLK3 in ER+ breast cancer cell survival. Our biochemical data clearly demonstrate that E2 inhibits the MLK3 kinase activities and by doing so down regulates its pro-apoptotic function. This E2 effect is physiological because MLK3 was highly active in ER−, PR− breast tumors but not in ER+, PR+ tumors. The detail role of MLK3 in ER−, PR− breast cancer pathogenesis is a challenging one and is one of our focus for future studies. Interestingly our data also show that inhibition of MLK3 by E2 is mediated via AKT phosphorylation of MLK3 on Ser 674 site. We have also shown earlier that MLK3 is activated by ceramide (22) and it is reported that ceramide inhibits AKT (46). Therefore it seems likely that agents that can activate MLK3 and inhibit AKT simultaneously might prove beneficial to promote cell death in ER+ breast cancer cells.
Supplementary Material
Acknowledgments
This work was supported in part by Komen Breast Cancer Foundation Grant BCTR0503916, National Institutes of Health (NIH) Grant GM55835 and Veterans Affairs Merit Award (to AR). BR is supported through NIH support (CA121221) and Veterans Affairs Merit Award.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–76. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–9. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levin ER. Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol. 2001;91:1860–7. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- 4.Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol. 2005;19:1951–9. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson CS, Campbell CH, Gametchu B. Membrane oestrogen receptors on rat pituitary tumour cells: immuno-identification and responses to oestradiol and xenoestrogens. Exp Physiol. 1999;84:1013–22. doi: 10.1111/j.1469-445x.1999.01903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zivadinovic D, Watson CS. Membrane estrogen receptor-alpha levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res. 2005;7:R130–44. doi: 10.1186/bcr959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–60. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 8.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 9.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93:5925–30. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tremblay GB, Tremblay A, Copeland NG, et al. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol Endocrinol. 1997;11:353–65. doi: 10.1210/mend.11.3.9902. [DOI] [PubMed] [Google Scholar]
- 11.Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 12.Lewis JS, Meeke K, Osipo C, et al. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst. 2005;97:1746–59. doi: 10.1093/jnci/dji400. [DOI] [PubMed] [Google Scholar]
- 13.Santen R, Jeng MH, Wang JP, et al. Adaptive hypersensitivity to estradiol: potential mechanism for secondary hormonal responses in breast cancer patients. J Steroid Biochem Mol Biol. 2001;79:115–25. doi: 10.1016/s0960-0760(01)00151-0. [DOI] [PubMed] [Google Scholar]
- 14.Song RX, Mor G, Naftolin F, et al. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17beta-estradiol. J Natl Cancer Inst. 2001;93:1714–23. doi: 10.1093/jnci/93.22.1714. [DOI] [PubMed] [Google Scholar]
- 15.Lewis JS, Osipo C, Meeke K, Jordan VC. Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. J Steroid Biochem Mol Biol. 2005;94:131–41. doi: 10.1016/j.jsbmb.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 16.Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci U S A. 2002;99:14783–8. doi: 10.1073/pnas.192569699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Migliaccio A, Castoria G, Di Domenico M, De Falco A, Bilancio A, Auricchio F. Src is an initial target of sex steroid hormone action. Ann N Y Acad Sci. 2002;963:185–90. doi: 10.1111/j.1749-6632.2002.tb04109.x. [DOI] [PubMed] [Google Scholar]
- 18.Kousteni S, Bellido T, Plotkin LI, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–30. [PubMed] [Google Scholar]
- 19.Greger JG, Fursov N, Cooch N, et al. Phosphorylation of MNAR promotes estrogen activation of phosphatidylinositol 3-kinase. Mol Cell Biol. 2007;27:1904–13. doi: 10.1128/MCB.01732-06. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Gallo KA, Johnson GL. Signalling: Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–72. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 21.Rana A, Gallo K, Godowski P, et al. The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J Biol Chem. 1996;271:19025–8. doi: 10.1074/jbc.271.32.19025. [DOI] [PubMed] [Google Scholar]
- 22.Sathyanarayana P, Barthwal MK, Kundu CN, et al. Activation of the Drosophila MLK by Ceramide Reveals TNF-alpha and Ceramide as Agonists of Mammalian MLK3. Mol Cell. 2002;10:1527–33. doi: 10.1016/s1097-2765(02)00734-7. [DOI] [PubMed] [Google Scholar]
- 23.Barthwal MK, Sathyanarayana P, Kundu CN, et al. Negative regulation of mixed lineage kinase 3 (MLK3) by protein kinase B (AKT)leads to cell survival. J Biol Chem. 2002 doi: 10.1074/jbc.M211598200. [DOI] [PubMed] [Google Scholar]
- 24.Mishra R, Barthwal MK, Sondarva G, et al. Glycogen synthase kinase-3beta induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. J Biol Chem. 2007;282:30393–405. doi: 10.1074/jbc.M705895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons In vivo. J Pharmacol Exp Ther. 1999;288:421–7. [PubMed] [Google Scholar]
- 26.Wang LH, Besirli CG, Johnson EM., Jr Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu Rev Pharmacol Toxicol. 2004;44:451–74. doi: 10.1146/annurev.pharmtox.44.101802.121840. [DOI] [PubMed] [Google Scholar]
- 27.Katome T, Obata T, Matsushima R, et al. Use of RNA interference-mediated gene silencing and adenoviral overexpression to elucidate the roles of AKT/protein kinase B isoforms in insulin actions. J Biol Chem. 2003;278:28312–23. doi: 10.1074/jbc.M302094200. [DOI] [PubMed] [Google Scholar]
- 28.Robertson JF. Faslodex (ICI 182, 780), a novel estrogen receptor downregulator--future possibilities in breast cancer. J Steroid Biochem Mol Biol. 2001;79:209–12. doi: 10.1016/s0960-0760(01)00138-8. [DOI] [PubMed] [Google Scholar]
- 29.Logie L, Ruiz-Alcaraz AJ, Keane M, et al. Characterization of a protein kinase B inhibitor in vitro and in insulin-treated liver cells. Diabetes. 2007;56:2218–27. doi: 10.2337/db07-0343. [DOI] [PubMed] [Google Scholar]
- 30.Karlsson HK, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes. 2005;54:1692–7. doi: 10.2337/diabetes.54.6.1692. [DOI] [PubMed] [Google Scholar]
- 31.Rodrik V, Zheng Y, Harrow F, Chen Y, Foster DA. Survival signals generated by estrogen and phospholipase D in MCF-7 breast cancer cells are dependent on Myc. Mol Cell Biol. 2005;25:7917–25. doi: 10.1128/MCB.25.17.7917-7925.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weldon CB, Elliott S, Zhu Y, et al. Regulation of estrogen-mediated cell survival and proliferation by p160 coactivators. Surgery. 2004;136:346–54. doi: 10.1016/j.surg.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 33.Lebesgue D, Chevaleyre V, Zukin RS, Etgen AM. Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids. 2009;74:555–61. doi: 10.1016/j.steroids.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee ES, Yin Z, Milatovic D, Jiang H, Aschner M. Estrogen and tamoxifen protect against Mn-induced toxicity in rat cortical primary cultures of neurons and astrocytes. Toxicol Sci. 2009;110:156–67. doi: 10.1093/toxsci/kfp081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson ME, Dubal DB, Wise PM. Estradiol protects against injury-induced cell death in cortical explant cultures: a role for estrogen receptors. Brain Res. 2000;873:235–42. doi: 10.1016/s0006-8993(00)02479-3. [DOI] [PubMed] [Google Scholar]
- 36.Saunders DE, Lawrence WD, Christensen C, Wappler NL, Ruan H, Deppe G. Paclitaxel-induced apoptosis in MCF-7 breast-cancer cells. Int J Cancer. 1997;70:214–20. doi: 10.1002/(sici)1097-0215(19970117)70:2<214::aid-ijc13>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 37.Saporito MS, Hudkins RL, Maroney AC. Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem. 2002;40:23–62. doi: 10.1016/s0079-6468(08)70081-x. [DOI] [PubMed] [Google Scholar]
- 38.Schlotter CM, Vogt U, Allgayer H, Brandt B. Molecular targeted therapies for breast cancer treatment. Breast Cancer Res. 2008;10:211. doi: 10.1186/bcr2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheskis BJ, Greger JG, Nagpal S, Freedman LP. Signaling by estrogens. J Cell Physiol. 2007;213:610–7. doi: 10.1002/jcp.21253. [DOI] [PubMed] [Google Scholar]
- 40.Conzen SD. Minireview: nuclear receptors and breast cancer. Mol Endocrinol. 2008;22:2215–28. doi: 10.1210/me.2007-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mota M, Reeder M, Chernoff J, Bazenet CE. Evidence for a role of mixed lineage kinases in neuronal apoptosis. J Neurosci. 2001;21:4949–57. doi: 10.1523/JNEUROSCI.21-14-04949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713–24. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun M, Wang G, Paciga JE, et al. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431–7. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kucab JE, Lee C, Chen CS, et al. Celecoxib analogues disrupt Akt signaling, which is commonly activated in primary breast tumours. Breast Cancer Res. 2005;7:R796–807. doi: 10.1186/bcr1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee YR, Park J, Yu HN, Kim JS, Youn HJ, Jung SH. Up-regulation of PI3K/Akt signaling by 17beta-estradiol through activation of estrogen receptor-alpha, but not estrogen receptor-beta, and stimulates cell growth in breast cancer cells. Biochem Biophys Res Commun. 2005;336:1221–6. doi: 10.1016/j.bbrc.2005.08.256. [DOI] [PubMed] [Google Scholar]
- 46.Zhou H, Summers SA, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem. 1998;273:16568–75. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.