

Abstract
Egg-laying behavior of the Caenorhabditis elegans hermaphrodite is regulated by G-protein signaling pathways. Here we show that the egg-laying–defective mutant egl-6(n592) carries an activating mutation in a G-protein-coupled receptor that inhibits C. elegans egg-laying motor neurons in a Go-dependent manner. Ligands for EGL-6 are Phe-Met-Arg-Phe-NH2 (FMRFamide)-related peptides encoded by the genes flp-10 and flp-17. flp-10 is expressed in both neurons and non-neuronal cells. The major source of flp-17 peptides is a pair of presumptive sensory neurons, the BAG neurons. Genetic analysis of the egl-6 pathway revealed that the EGL-6 neuropeptide signaling pathway functions redundantly with acetylcholine to inhibit egg-laying. The retention of embryos in the uterus of the C. elegans hermaphrodite is therefore under the control of a presumptive sensory system and is coordinately regulated by neuropeptides and the small-molecule neurotransmitter acetylcholine.
Network connectivity and the biophysical properties of component neurons generate motor programs that are the bases of innate behaviors. How motor programs are modulated by sensory systems and how they couple to other neural circuits to create natural behaviors remain major questions in neuroscience. The egg-laying behavior of the C. elegans hermaphrodite is a simple behavior that is stimulated by the detection of a bacterial food source1,2. The study of C. elegans mutants defective in egg-laying behavior offers the opportunity to identify genetic, molecular and cellular pathways that control a modulated behavior.
A simple neuromuscular circuit is required for C. elegans egg-laying behavior. Sixteen vulval and uterine muscles, all connected by gap junctions, contract during egg-laying. Vulval muscles receive synaptic input from two classes of motor neurons: VC neurons in the ventral nerve cord and sublateral HSN motor neurons3. HSN neurons provide excitatory serotonergic input to vulval muscles1,2 and might also provide cholinergic input4. The VC neurons are cholinergic4 and might also contain serotonin5. VC neurons have been reported to inhibit or promote egg-laying behavior under different experimental conditions6,7.
Molecular characterization of genes required for normal egg-laying by C. elegans has shown that G-protein signaling pathways antagonistically regulate egg-laying behavior. C. elegans Gq and Go stimulate and inhibit, respectively, egg-laying behavior8-10, as do Gq and Go effectors11-13. Metabotropic serotonin receptors, which are likely to couple to a Gq signaling pathway, act on the vulval muscles14-16. The prohormone convertase EGL-3 and the carboxypeptidase EGL-21 promote egg laying, suggesting the existence of excitatory peptidergic signals17,18 that might also act through Gq signaling. Neurochemical signals that activate the inhibitory Go pathway have not been defined, although acetylcholine acting through the muscarinic receptor GAR-2 and unidentified ligands for the orphan G-protein-coupled receptor (GPCR) EGL-47 are candidates7,19.
Here we show that egl-6, defined by the egg-laying defective mutant egl-6(n592), encodes a GPCR for FMRFamide-related peptides (FaRPs) that inhibit the HSN motor neurons in a Go-dependent manner. One source of peptide ligands for EGL-6 is a pair of presumptive sensory neurons, the BAG neurons, suggesting that FaRP signaling integrates a sensory system with the egg-laying motor program. Loss of function in both the egl-6 pathway and acetylcholine signaling causes a strong, synthetic hyperactive egg-laying phenotype. FaRP neuropeptides therefore act redundantly with acetylcholine to inhibit egg-laying, indicating that inhibition in a motor program can arise from the convergence of two distinct neurochemical signals.
RESULTS
egl-6(n592) causes increased activity of an orphan GPCR
The mutation n592, which defines the gene egl-6, causes a semidominant egg-laying defect that is bypassed by drugs that act directly on egg-laying muscles2. We hypothesized that egl-6(n592) activates a pathway that inhibits egg-laying, and we cloned the affected gene.
We mapped egl-6(n592) to a 167-kb interval on linkage group X. PCR products spanning this interval derived from wild-type genomic DNA were unable rescue the egg-laying–defective phenotype of egl-6(n592) mutants. We tested comparable PCR products from egl-6(n592) DNA for the ability to phenocopy the n592 mutation. A PCR product predicted to contain only the gene C46F4.1 caused a severe egg-laying–defective phenotype (Fig. 1a). The comparable PCR product derived from wild-type DNA also caused an egg-laying–defective phenotype (Fig. 1a).
Figure 1.

egl-6 encodes two isoforms of a seven-pass transmembrane receptor. (a) The effects of egl-6 mutations on egg-laying. The number of transgenic lines with egg-laying defects as a fraction of lines generated is shown for each mutation. Mutated sites are indicated in b as open arrowheads. (b) egl-6 intron-exon structure and mutations. Coding sequences are depicted as solid boxes. Closed arrowhead indicates the position of the n592 mutation, which is predicted to alter both isoforms by changing Ala135 to Thr (EGL-6A sequence coordinates). Sequences deleted in alleles n4536 and n4537, which remove most of the egl-6 coding sequence, are indicated. Open arrowheads indicate the positions of amber nonsense mutations introduced into an egl-6 transgene, and a frameshift was created at the BglII site indicated. (c) Predicted secondary structure of EGL-6A. Both predicted egl-6 protein products have seven transmembrane domains. The first 32 residues of EGL-6A, which are replaced by 30 different residues in EGL-6B, are shaded gray. Ala135, predicted to be mutated to threonine by the n592 mutation, is shaded black.
We determined the structure of C46F4.1 transcripts by RT-PCR and 3′ and 5′ rapid amplification of cDNA ends (RACE) experiments. C46F4.1 generates two transcripts with alternative translational starts (Fig. 1b), each predicted to encode an orphan GPCR (Fig. 1c). Disrupting both open reading frames, either by introducing stop codons after both predicted translational start sites or by introducing a frameshift in a shared exon, abrogated the ability of C46F4.1 transgenes to phenocopy n592 (Fig. 1a); disrupting either open reading frame alone did not have this effect. We identified a missense mutation, predicted to change an alanine in the third transmembrane domain to threonine, in the C46F4.1 coding sequence in n592 mutants (Fig. 1b,c). Given our mapping results, the ability of C46F4.1 transgenes to phenocopy egl-6(n592) and the identification of a mutation in the coding sequence of C46F4.1, we concluded that egl-6 is C46F4.1.
We used PCR to screen a library of mutagenized worms for mutants carrying deletions in egl-6 and found additional alleles of egl-6, n4536 and n4537. Both deletions remove the translational start site of the egl-6b transcript and most of the coding sequence of both egl-6 transcripts (Fig. 1b) and are likely to be null alleles of egl-6.
To determine how the n592 mutation affects egl-6, we tested the effects of egl-6 gene dosage on egg-laying. We quantified C. elegans egg-laying behavior using the developmental stage of newly laid eggs, which reflects the time embryos spent in utero2 (Fig. 2a). Statistical comparisons between strains analyzed in this manner are presented in Supplementary Table 1 online. Wild-type worms retained most eggs until the embryos developed to a multicellular but premorphogenic stage (Fig. 2b). egl-6(n592) mutants laid embryos that had developed to the twofold stage or later, reflecting slower rates of egg-laying and longer retention of embryos. egl-6 deletion (egl-6(Δ)) mutants had wild-type egg-laying behavior, indicating that the n592 mutation does not reduce egl-6 gene function. By contrast, transgenic worms with extra copies of wild-type egl-6 laid later-stage eggs than did the wild-type worms. These observations indicate that n592 increases egl-6 activity and that egl-6 inhibits egg-laying. Because activation of GPCRs is accompanied by allosteric changes in the relative positions of transmembrane domains20, n592 might increase receptor function by stabilizing an active conformer of the receptor.
Figure 2.

egl-6(gf) inhibition of egg-laying requires Go signaling. (a) Stages of embryonic development used to assay the retention time of freshly laid eggs. (b) The n592 mutation increases egl-6 function. Distributions of the developmental stages of eggs laid by worms carrying the n592 mutation and by worms with normal, decreased or increased egl-6 gene dosage are shown. egl-6(n592) mutants and worms overexpressing (OE) egl-6 laid later-stage embryos than did wild-type worms, indicating increased retention time in utero. The egl-6 deletion allele used was n4536. The transgene used for egl-6 overexpression was nIs181. (c) Go signaling is required for EGL-6 gain-of-function mutation to inhibit egg-laying. Shown are distributions of the developmental stages of eggs laid by worms carrying a goa-1 loss-of-function mutation together with mutations or transgenes that increase or decrease egl-6 function. goa-1; egl-6(gf) double mutants were strongly suppressed for the egg-laying defect caused by egl-6(gf) but were still significantly different from goa-1 single mutants (p < 10-6) . The egl-6 deletion allele used was n4536. The goa-1 allele used was n1134. The transgene used for egl-6 overexpression was nIs181.
Go signaling inhibits C. elegans egg-laying behavior8,9. Because egl-6 encodes a presumptive GPCR that inhibits egg-laying, we tested whether the egg-laying–defective phenotype caused by a gain-of-function (gf) mutation in egl-6 requires Go signaling. Loss-of-function (lf) mutations in the gene encoding C. elegans Goα, goa-1, increase rates of egg-laying8,9. We observed a corresponding change in the stage of eggs newly laid by goa-1 mutants, with a majority having eight cells or fewer (Fig. 2c). goa-1(lf) also strongly suppressed the egg-laying defects caused by either the n592 mutation or overexpression of wild-type egl-6. egl-6 deletion did not modify the egg-laying behavior of goa-1 mutants. We observed similar results with a different goa-1(lf) allele, n3055 (data not shown). Our data show that egl-6 function strongly depends on goa-1, suggesting that EGL-6 couples to a Go signaling pathway. However, some EGL-6 signaling might be independent of goa-1, as both the egl-6(n592gf) gain-of-function mutation and extra copies of wild-type egl-6 slightly delayed the egg-laying behavior of goa-1 mutants (Fig. 2c).
EGL-6 inhibits the HSN motor neurons
To determine where egl-6 functions, we constructed a green fluorescent protein (GFP) reporter transgene, egl-6∷gfp. This transgene caused an egg-laying defect that strongly depended on goa-1 function (data not shown), suggesting that it encodes a functional receptor. We detected egl-6∷gfp expression using an antibody to GFP and observed strong and consistent expression in HSN motor neurons (Fig. 3a) and in GLR cells, glia-like cells in the head. We consistently observed weaker staining of the DVA tail interneuron and occasionally observed staining of the lateral interneurons SDQL and SDQR.
Figure 3.

egl-6 expression in the HSN motor neurons inhibits egg-laying. (a) egl-6 is expressed in the HSN motor neurons and the GLR cells. Fainter expression in the DVA interneuron is not visible in this exposure. Occasional expression was observed in the neurons SDQL and SDQR. D, dorsal; V, ventral; A, anterior; P, posterior; L, left; R, right. (b) Expression of egl-6 isoforms in the HSN motor neurons inhibits egg-laying. egl-6a and egl-6b cDNAs were under the control of tph-1 promoter variants that drive transgene expression either in both the HSN motor neurons and the NSM pharyngeal neurons (tph-1L) or in the NSM pharyngeal neurons only (tph-1S). The number of transgenic lines with egg-laying defects as a fraction of lines generated is shown for each transgene.
HSN motor neurons innervate vulval muscles and are required for normal egg-laying2,3. To test whether egl-6 expression in HSN neurons suffices to inhibit egg-laying, we used transgenes with regulatory elements from the tryptophan hydroxylase gene tph-1 (ref. 21) to express egl-6. One promoter, tph-1L, drives gene expression in HSN neurons and pharyngeal serotonergic NSM neurons, and a truncated variant, tph-1S, drives gene expression primarily in NSM neurons19. Expression of either egl-6 isoform under the control of the tph-1L promoter caused egg-laying defects, whereas the same cDNAs under the control of the NSM-specific promoter tph-1S did not affect egg-laying (Fig. 3b). Both isoforms of EGL-6 can, therefore, inhibit egg-laying. These data, together with the detection of the egl-6 reporter in HSN motor neurons, suggest that EGL-6 receptors mediate inhibition of the HSN neurons.
EGL-6 is a receptor for FLP-10 and FLP-17 FaRPs
We searched protein sequence databases using BLAST22 and found that EGL-6 has similarity to insect receptors for FaRPs23,24. We postulated that EGL-6 is a neuropeptide receptor and sought ligands for EGL-6 in two ways: by screening neuropeptide-encoding transgenes for the ability to inhibit egg-laying in an egl-6-dependent manner, and by screening a library of synthetic peptides for peptides that activate EGL-6 in vitro.
Our first approach to identify EGL-6 ligands was based on the hypothesis that overexpression of a ligand-encoding gene would inhibit egg-laying as does egl-6 overexpression. We generated transgenic worms carrying extra copies of neuropeptide genes, including flp-1 through flp-23, which are predicted to encode FMRFamide-like peptides, and ten nlp genes, which are predicted to encode neuropeptide-like peptides25,26. flp-10 and flp-17 transgenes inhibited egg-laying by wild-type worms but not by egl-6(Δ) mutants (see below), suggesting that flp-10 and flp-17 encode ligands for EGL-6. The introduction of stop codons or frameshift mutations into the flp-10 and flp-17 coding sequences abrogated their ability to inhibit egg-laying behavior (data not shown). To quantify the egg-laying defects caused by these transgenes and the dependence of these defects on the EGL-6 receptor, we generated strains carrying stably integrated versions of flp-10 and flp-17 transgenes. Stably integrated flp-10 or flp-17 transgenes inhibited egg-laying in wild-type worms, and these defects required the egl-6 gene (Fig. 4a,b).
Figure 4.
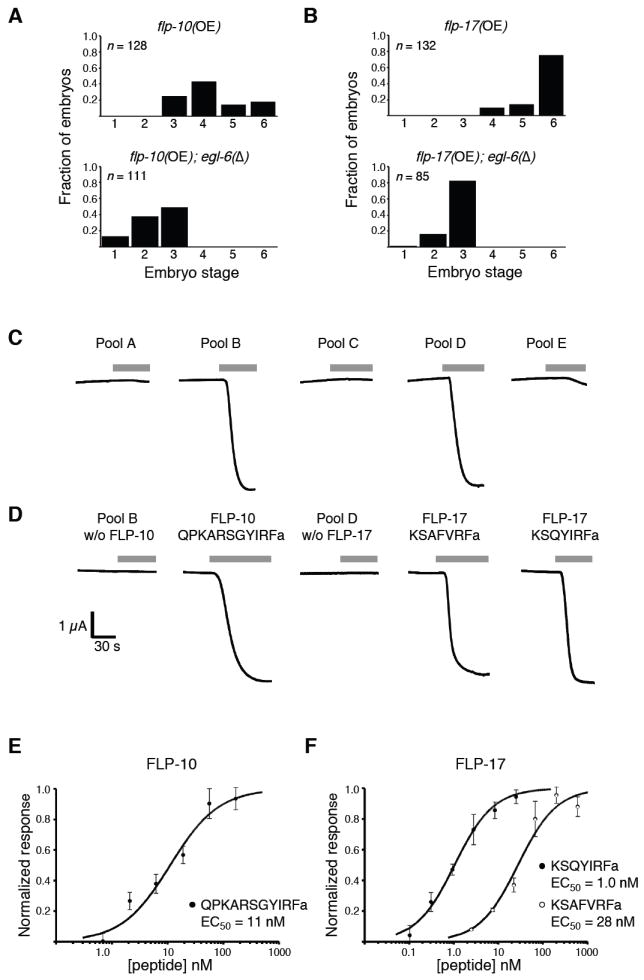
flp-10 and flp-17 encode ligands for the EGL-6 receptor. (a,b) Overexpression of flp-10 or flp-17 inhibits egg-laying behavior of wild-type worms but not of egl-6 mutants. Shown are distributions of developmental stages of eggs laid by transgenic worms carrying extra copies of flp-10 (a) or flp-17 (b) in the presence or absence of an egl-6 deletion allele. flp-10 and flp-17 overexpression increased egg retention times compared to wild type, and deletion of egl-6 suppressed the egg-laying defects caused by flp-10 and flp-17 overexpression. The transgene used for flp-10 overexpression was nIs209. The transgene used for flp-17 overexpression was nIs211. The egl-6 deletion allele used was n4536. (c,d) Identification of EGL-6 ligands by screening pools of synthetic peptides. Pools of synthetic peptides (see Supplementary Table 1) were applied to X. laevis oocytes coexpressing EGL-6 and GIRK channels. The final concentration of each peptide in the pools was 1 μM. Whole-cell currents were monitored using a two-electrode voltage clamp. Gray bars indicate the periods during which peptide pools were applied. (e,f) Peptides encoded by flp-10 (e) and flp-17 (f) activate EGL-6 at nanomolar concentrations. Dilution series of the indicated peptides were applied to X. laevis oocytes coexpressing EGL-6 and GIRK channels. The response was calculated as the ratio of the peptide-evoked inward K+ current and the peptide-independent inward K+ current. Data were fitted to the Hill equation and normalized to the calculated maximum current. The means of at least three experiments ± s.e.m. are plotted. EC50, half-maximal effective concentration.
We also tested peptides for EGL-6 agonist activity in vitro. The coupling of GPCRs to G protein–activated inwardly rectifying K+ (GIRK) channels in heterologous expression systems has been used to match ligands to orphan GPCRs (for example, in ref. 27). We coexpressed EGL-6 and GIRKs in Xenopus laevis oocytes and tested pools of synthetic peptides corresponding to predicted and observed C. elegans neuropeptides25,26 for the ability to activate GIRK conductance using a two-electrode voltage clamp. We tested 55 peptides in five pools (Supplementary Table 2 online). Two pools, B and D, evoked inward currents (Fig. 4c) that were observed only in oocytes coexpressing EGL-6 and GIRKs (data not shown). These pools therefore contained EGL-6 agonists. Pool B contained the predicted flp-10 peptide product QPKARSGYIRFamide, and pool D contained both predicted flp-17 peptide products, KSAFVRFamide and KSQYIRFamide. Omitting these peptides from the active pools eliminated agonist activity; furthermore, each of these peptides could function as agonists when applied individually (Fig. 4d). flp-10 and flp-17 peptides were effective at nanomolar concentrations in this assay (Fig. 4e,f).
We concluded that flp-10 and flp-17 encode ligands for the EGL-6 receptor, because (i) the egg-laying defects caused by flp-10 and flp-17 overexpression required the egl-6 receptor gene in vivo, and (ii) flp-10 and flp-17 peptides activated the EGL-6 receptor in vitro.
Activated mutant EGL-6 receptor is ligand-dependent
We isolated deletion alleles of flp-10 and flp-17 (Fig. 5a). The n4543 deletion removes the translational start site of flp-10 and most of the coding sequence, including sequences encoding the active peptide. The n4894 mutation removes the translational start site of flp-17 and is likely to eliminate the production of both FaRPs encoded by flp-17.
Figure 5.

The EGL-6 receptor is regulated by flp-10 and flp-17 in vivo. (a) flp-10 and flp-17 intron-exon structure. Coding sequences are depicted as solid boxes. The flp-10 and flp-17 loci are both on linkage group IV, but in opposing orientations. Sequences deleted in flp-10(n4543) and flp-17(n4894) are indicated. The open arrowhead indicates the position of an amber nonsense mutation that abrogated the ability of the flp-10 transgene to inhibit egg-laying. A frameshift that abrogated the ability of the flp-17 transgene to inhibit egg-laying was introduced at the indicated BamHI site. Closed squares indicate the presence of 21U RNA-encoding genes within the flp-17 locus. The predicted peptide products of flp-10 and flp-17 are shown beneath the corresponding gene models. (b) flp-10 and flp-17 mutants lay normally staged embryos. The distributions of developmental stages of embryos laid by wild-type worms, flp-10 mutants, flp-17 mutants and flp-10 flp-17 double mutants are shown. (c) flp-10 and flp-17 mutations partially suppress the egg-laying defect caused by the egl-6 gain-of-function mutation. Shown are the distributions of developmental stages of embryos laid by egl-6(gf) mutants and egl-6(gf) mutants with mutations that delete the ligand-encoding genes flp-10 and flp-17. The egl-6 gain-of-function allele used was n592. The flp-10 and flp-17 deletion alleles used were n4543 and n4894, respectively.
Deletion of flp-10 and flp-17 did not grossly affect egg-laying behavior, either individually or in combination (Fig. 5b). Deletion of flp-10, however, suppressed the egg-laying defect caused by the n592gf mutation of egl-6, and this suppression was enhanced by deletion of flp-17 (Fig. 5c). These data show that the activity of both the wild-type receptor and the mutant receptor encoded by egl-6(n592gf) is regulated by flp-10 and flp-17 peptides in vivo.
flp-10 and flp-17 can function in different cells
To identify the sources of flp-10 and flp-17 peptides, we generated flp-10∷gfp and flp-17∷gfp reporters and detected their expression using antibody to GFP (Fig. 6). We observed consistent flp-10∷gfp expression in ASIL, ASIR, DVB, PVCL, PVCR and PVR neurons and in vulval tissue, uterine cells, spermathecae and the head mesodermal cell (Fig. 6a). We also observed faint and inconsistent staining of a small number of unidentified neurons. The pattern of flp-10∷gfp expression we observed was similar but not identical to a flp-10 expression pattern previously observed using a transcriptional reporter28. Specifically, we did not observe flp-10 expression in the BAG neurons, and the previous study did not note flp-10 expression in the uterus, spermathecae or head mesodermal cell28. We observed expression of flp-17∷gfp expression primarily in a pair of anterior sensory neurons, BAGL and BAGR (Fig. 6b). The flp-17∷gfp expression we observed was similar to that previously reported28. We also observed faint but consistent expression of flp-17∷gfp in three pairs of unidentified head neurons.
Figure 6.

flp-10 and flp-17 are expressed in different cells. (a,b) Expression of flp-10∷gfp and flp-17∷gfp was detected by immunocytochemistry. (a) flp-10∷gfp expression was observed in the ASI sensory neurons, the DVB motor neuron, the PVC interneurons and the PVR neuron. flp-10∷gfp expression was also observed in the head mesoderm cell (open arrowhead), vulval cells, uterus and spermathecae. The ASI dendrites contained high levels of FLP-10∷GFP (closed arrowhead). Both micrographs are from the same worm. (b) flp-17∷gfp expression was detected primarily in a pair of anterior sensory neurons, BAGL and BAGR. Both micrographs are from the same worm. (c–e) BAG neurons are required for flp-17 function. Neurons expressing flp-10 and flp-17 reporter transgenes were ablated with a laser microbeam in wild-type animals (c) and in animals overexpressing flp-10 (d) or flp-17 (e). The distributions of developmental stages of eggs laid by operated animals are shown. The transgene used for flp-10 overexpression was nIs209[flp-10(+)]. The transgene used for flp-17 overexpression was nIs211. Asterisks denote the presence of the gcy-33∷gfp transgene, nIs242, used in some strains to facilitate identification of BAG neurons.
To test whether expression of flp-10 and flp-17 in specific neurons is required for these genes to inhibit egg-laying, we laser-ablated neurons in transgenic worms overexpressing flp-10 or flp-17. Ablation of BAG neurons or of six neurons expressing flp-10∷gfp together had no marked effect on egg-laying by wild-type worms (Fig. 6c). We then ablated six neurons expressing flp-10∷gfp in worms overexpressing flp-10 to test whether these neurons were required for the egg-laying defect conferred by the flp-10 transgene. Ablation of ASIL, ASIR, DVB, PVCL, PVCR and PVR together did not strongly modify the egg-laying phenotype of worms overexpressing flp-10 (Fig. 6d), although the small change we observed in the distribution of egg stage might be significant (P = 0.0193). The six neurons we identified as expressing flp-10∷gfp expressing were not, therefore, strictly required for flp-10 function. By contrast, laser ablation of the BAG neurons strongly suppressed the egg-laying defect conferred by flp-17 overexpression (Fig. 6e), suggesting that the BAG neurons are a principal site of flp-17 expression. Ablation of BAG cells also partially suppressed the egg-laying defects conferred by flp-10 overexpression (Fig. 6d).
That the flp-10 transgene functioned in the absence of the six neurons that expressed flp-10∷gfp suggested that non-neuronal—possibly vulval or gonadal—expression of flp-10 inhibits egg laying. It is also possible that our flp-10∷gfp reporter was expressed in only a subset of flp-10-expressing neurons, in which case we would not have ablated all flp-10-expressing neurons. To test whether non-neuronal cells are competent to express and process flp-10 peptide, we expressed flp-10 specifically in vulval cells and spermathecae, non-neuronal tissues that expressed the flp-10∷gfp reporter. We expressed flp-10 in vulval cells, in spermathecae and pan-neuronally using heterologous promoters fused to the flp-10 cDNA (see Supplementary Methods online). Egg-laying defects were observed in three of four transgenic lines carrying the vulval expression construct, five of five lines carrying the spermathecal expression construct and three of six lines carrying the pan-neuronal expression construct. Non-neuronal cells can therefore express functional FLP-10 peptide. Together with our laser ablation studies, these data suggest that non-neuronal cells are one source of endogenous FLP-10 peptide.
The BAG neurons inhibit C. elegans egg-laying behavior
egl-6 overexpression caused an egg-laying defect that was partially suppressed by flp-17 deletion (Fig. 7a,b). The egg-laying defect of worms overexpressing egl-6 therefore partly depends on flp-17. To whether ablation of BAG cells would phenocopy flp-17 deletion and whether flp-17 has a BAG cell–independent function, we ablated the BAG cells in worms overexpressing egl-6, with or without a flp-17 deletion allele (Fig. 7a). BAG cell ablation suppressed the egg-laying defect of worms overexpressing egl-6, but this suppression was not enhanced by flp-17 deletion—that is, deleting flp-17 had no effect in the absence of BAG cells (Fig. 7b). The suppression by flp-17 deletion of the egg-laying defect caused by egl-6 overexpression was enhanced by flp-10 deletion (Fig.7c).
Figure 7.

The BAG sensory neurons inhibit egg-laying. (a,b) The effect of BAG neuron ablation is not enhanced by a flp-17 mutation. The BAG neurons were ablated with a laser microbeam in egl-6-overexpressing animals that were (a) otherwise wild-type or (b) mutant for flp-17. The distributions of developmental stages of eggs laid by operated worms are shown. (c) flp-10 mutation enhances the effects of flp-17 mutation more strongly than does BAG cell ablation. The distributions of developmental stages of eggs laid by transgenic worms overexpressing egl-6 and lacking flp-10 and flp-17 are shown. The transgene for egl-6 overexpression used was nIs181. The flp-17 allele used was n4894.
These data, together with the expression of flp-17∷gfp in BAG cells and the requirement of BAG cells for the function of the flp-17 transgene, indicate that BAG cells are the principal source of endogenous FLP-17 peptides. That the effect of BAG cell ablation on egg-laying behavior was stronger than that of deleting flp-17 indicates that BAG cells provide one or more inhibitory signals in addition to FLP-17 peptides. Although some inhibition provided by BAG cells might require flp-10, we observed that flp-10 deletion has a larger effect than does BAG cell ablation (Fig. 7), suggesting that flp-10 can function in cells in addition to BAG cells. Our observation that BAG cell ablation only partially suppressed the egg-laying defect caused by flp-10 overexpression (Fig. 6d) is consistent with this hypothesis.
EGL-6 and acetylcholine redundantly modulate egg laying
Although activation of the egl-6 pathway by mutation or overexpression inhibited egg-laying, mutants lacking egl-6 or the ligand-encoding genes flp-10 and flp-17 had grossly normal egg-laying behavior (Figs. 2b and 4c). In addition to using the developmental stage of newly laid eggs to assay egg-laying behavior, we analyzed the timing of egg-laying events by egl-6(Δ) mutants (Supplementary Fig. 1 online) and the modulation of egg-laying behavior of egl-6(Δ) mutants by the presence or absence of a bacterial food source (data not shown). These experiments did not reveal notable differences between egl-6(Δ) mutants and wild-type worms.
We also tested whether redundancy between the egl-6 signaling pathway and another signaling pathway might explain our observation that mutants defective in the egl-6 pathway have grossly normal egg-laying behavior. We constructed strains with multiple mutations in egl-6 and genes required for the synthesis or storage of specific neurotransmitters (GABA, dopamine, glutamate, tyramine, octopamine, serotonin or acetylcholine) and examined these strains for synthetic egg-laying defects. Genes tested for interactions with the egl-6 pathway were unc-25, which is required for GABA synthesis29; cat-2, which is required for dopamine synthesis30; eat-4, which encodes a vesicular glutamate transporter31; tdc-1, which is required for tyramine and octopamine synthesis32; tph-1, which is required for serotonin synthesis21; and unc-17 and cha-1, which encode a vesicular acetylcholine transporter and a choline acetyltransferase required for acetylcholine synthesis, respectively33,34.
We observed that strains carrying mutations in the egl-6 pathway and genes required for acetylcholine signaling had a strong synthetic hyperactive egg-laying phenotype (Fig. 8a). unc-17 and cha-1, which are required for acetylcholine synthesis and storage, are in an operon. The unc-17(e113) mutation reduces the function of both unc-17 and cha-1 (ref. 35). The egg-laying behavior of unc-17(e113) mutants (Fig. 8a) was similar to that of wild-type worms and egl-6 deletion mutants (Fig. 2b). By contrast, unc-17(e113); egl-6(Δ) double mutants laid early-stage embryos with eight or fewer cells at a high frequency, reflecting decreased time spent in utero. Deletion of flp-10, flp-17 or both affected the egg-laying behavior of unc-17(e113) mutants similarly. We saw comparable interactions between another deletion allele of egl-6 and unc-17(e113) and between both deletion alleles of egl-6 and three other mutations affecting the unc-17 cha-1 operon: e876, p1152 and n2421 (data not shown). We also ablated BAG cells, which are a principal site of flp-17 expression, in unc-17(e113) mutants and observed a higher frequency of early-stage embryos laid (Fig. 8a). Given that potentiation of acetylcholine signaling by mutation of the acetylcholinesterase-encoding genes ace-1 and ace-2 causes locomotion defects36 and inhibits egg-laying behavior7, we also tested whether deletion of the EGL-6 signaling pathway could modify the egg-laying defect of acetylcholinesterase mutants. The egg-laying defect of ace-2; ace-1 double mutants was suppressed by deletion of egl-6, flp-10 and flp-17 (Fig. 8b). These data show that EGL-6 signaling and cells required for the production of EGL-6 ligands function redundantly with acetylcholine to inhibit egg-laying.
Figure 8.

EGL-6 signaling inhibits egg-laying behavior redundantly with acetylcholine. (a) Loss of function in both EGL-6 signaling and acetylcholine signaling decreases egg retention time. The distribution of developmental stages of embryos laid by unc-17 mutants (defective in vesicular acetylcholine transport) is shown together with the distributions of stages of eggs laid by worms mutant for unc-17 and genes in the egl-6 pathway or unc-17 mutants lacking BAG neurons. BAG neurons were ablated with a laser microbeam in unc-17 mutants. An asterisk denotes the presence of the gcy-33∷gfp transgene nIs242, used in some strains to facilitate identification of BAG neurons. The unc-17 allele used was e113. The egl-6, flp-10, and flp-17 alleles used were n4536, n4543 and n4894, respectively. (b) Loss of function in the egl-6 pathway suppresses the egg-laying defect of mutants with excess acetylcholine signaling. Shown are distributions of developmental stages of eggs laid by ace-2; ace-1 double mutants (defective in acetylcholinesterase function) and by worms multiply mutant for acetylcholinesterase-encoding genes and genes in the egl-6 pathway. The ace-1 allele used was p1000. The ace-2 allele used was g72. The egl-6, flp-10, and flp-17 alleles used were n4536, n4543 and n4894, respectively.
Interactions between mutations that affect egl-6 signaling and mutations that alter acetylcholine signaling are consistent with the hypothesis that EGL-6 functions upstream of or in parallel to an acetylcholine signal to inhibit egg-laying. The egg-laying defect of ace-2; ace-1 double mutants was suppressed by deletion of the egl-6 pathway, but the egg-laying behavior of suppressed worms was still distinguishable from that of wild-type worms, suggesting that acetylcholine does not inhibit egg-laying behavior by acting strictly upstream of egl-6 signaling. The locomotion defects of mutants with reduced acetylcholine signaling (unc-17 and cha-1 mutants) or excess acetylcholine signaling (ace-2; ace-1 double mutants) were not modified by mutations in egl-6 or in the flp-10 and flp-17 ligand-encoding genes (data not shown), suggesting that EGL-6 signaling does not function in all cholinergic circuits.
DISCUSSION
Through a molecular genetic analysis of C. elegans mutants defective in egg-laying behavior, we identified a FaRP signaling pathway that inhibits motor neurons in the C. elegans egg-laying system in a Go-dependent manner. This pathway consists of the EGL-6 GPCR and two classes of peptide ligands encoded by the genes flp-10 and flp-17. We found that the flp-17 peptides KSAFVRFamide and KSQYIRFamide are provided by a pair of presumptive sensory neurons, the BAG cells. The flp-10 peptide QPKARSGYIRFamide is expressed by both neuronal and non-neuronal cell types, and our data suggest that non-neuronal cells are a source of this inhibitory peptide. We conclude that the EGL-6 receptor transduces inhibitory signals to the HSN motor neurons from multiple cell types.
Because presumptive sensory neurons are one source of EGL-6 ligands, EGL-6 signaling might modulate egg-laying behavior in response to environmental cues. BAG cells have specialized cilia characteristic of sensory neurons37,38 but have not been associated with a sensory modality. BAG cell processes have goblet-shaped termini surrounding a region of hypodermal tissue, but the processes are not exposed to the external environment37,38 and might therefore function as receptors for factors that can permeate the cuticle or are generated internally. Synapses between the BAG cells and the HSN motor neurons have not been observed by electron microscopic reconstruction of the C. elegans nervous system3. Therefore, flp-17-encoded FaRPs are likely to function as paracrine or endocrine signals in the control of egg-laying behavior.
Our data suggest that non-neuronal expression of flp-10 peptides inhibits egg-laying behavior. Most non-neuronal expression of flp-10 that we observed was in components of the hermaphrodite’s reproductive system: vulval, uterine and spermathecal cells. We further found that expression of flp-10 in vulval and spermathecal cells using heterologous promoters suffices to inhibit egg-laying. It is not fully understood how somatic components of the hermaphrodite’s reproductive system might modulate egg-laying behavior. Others have proposed that mechanical stimuli trigger release of neuromodulators from neuroendocrine cells at the junction of the vulva and uterus39. Oocytes passing through the spermathecae, distension of the uterus and mechanical stimulation of vulval tissue might similarly trigger FLP-10 release and allow ovulation, egg retention and the act of egg-laying to alter the activity of HSN motor neurons.
EGL-6 signaling is dispensable for the egg-laying behavior of wild-type worms under normal laboratory conditions. By contrast, the egg-laying behavior of acetylcholine signaling mutants was strongly affected by perturbations of the EGL-6 pathway (Fig. 8). Loss of egl-6 function or of the function of one or both genes encoding EGL-6 ligands in unc-17 mutants resulted in the expulsion of immature embryos from the uterus. The egg-laying behavior of acetylcholinesterase-deficient mutants, which are egg-laying defective, was also sensitive to mutation of one or both genes that encode EGL-6 ligands. These data indicate that even partial loss of function in EGL-6 signaling can alter the egg-laying behavior of mutants with abnormal acetylcholine signaling.
What is the circuit that provides inhibitory cholinergic input to the egg-laying system? The interactions between the EGL-6 pathway and acetylcholine signaling support models in which FLP-10 and FLP-17 FaRPs work either in parallel to or upstream of acetylcholine to inhibit egg laying. We have tested whether EGL-6 signaling functions redundantly with the GAR-2 metabotropic acetylcholine receptor, which is expressed on HSN motor neurons and has been proposed to negatively regulate HSN function7,40; we did not observe egg-laying defects in gar-2; egl-6 double mutants. Similarly, ablation of the cholinergic VC4 and VC5 egg-laying motor neurons did not modify the egg-laying behavior of egl-6 mutants (data not shown). It is therefore likely that the cholinergic pathway that functions redundantly with EGL-6 signaling requires cells and receptors previously not implicated in the control of egg-laying behavior. We observed that the hyperactive egg-laying phenotype of unc-17; egl-6 double mutants is comparable to that of goa-1 mutants (Figs. 2 and 8). If cholinergic inhibition of egg-laying is mediated by metabotropic acetylcholine receptors, a significant fraction of inhibitory Go signaling in the egg-laying system might be controlled by these receptors together with EGL-6. Three metabotropic acetylcholine receptors have been found in C. elegans40-42. Other receptors that might mediate inhibitory cholinergic signaling are acetylcholine-gated chloride channels43.
Our data and those of others show that the C. elegans egg-laying system is regulated by multiple neurotransmitters, including serotonin, acetylcholine and FaRPs. Interactions among these neurotransmitter systems are critical for the generation and regulation of egg-laying behavior. There is evidence that acetylcholine signaling is required for the stimulatory effects of serotonin on egg-laying, as if these two neurochemical signals constitute a logical AND gate in the control of egg-laying6,44. We have described a different relationship between neurotransmitter systems in the C. elegans egg-laying system: cholinergic and peptidergic signals synergistically inhibit egg-laying, constituting a logical OR gate. Cholinergic and peptidergic inhibition of egg-laying might be independently invoked by sensory systems or other cellular circuits to stop egg-laying by C. elegans in unfavorable conditions. The logical relationships among neurotransmitter systems that control C. elegans egg-laying behavior might have general implications for the study and manipulation of neural circuits. We suggest that even if inactivation of individual neurochemical pathways has little or no effect on the function of a target circuit, the combinatorial perturbation of pathways might cause strong and specific alterations of circuit function.
METHODS
Strains and transgenes
A detailed description of strains used and construction of transgenes can be found in Supplementary Methods.
Mapping and cloning of egl-6(n592)
We isolated egg-laying–defective Mec non-Lon and Mec non-Lon recombinants from the strain MT16027 lon-2(e678) egl-6(n592) mec-7(e1506) after crossing with the polymorphic wild-type strain CB4856 and determined crossover sites as described45. We mapped n592 to a 167-kb region between SNPs on the cosmids F13D11 and F22F4. PCR products spanning this interval were derived from wild-type and n592 genomic DNA, injected into the wild-type strain N2 and scored for the ability to phenocopy the n592 mutation. A transgene containing only the gene C46F4.1 phenocopied n592. The predicted gene structure of C46F4.1 was confirmed by RT-PCR. We used 5′ RACE (Invitrogen) to identify a transcript with an alternative start and an SL1 trans-spliced leader sequence. The 3′ sequences of egl-6 transcripts were amplified using PCR from a C. elegans cDNA library (Stratagene) using gene-specific primers and primers directed against the cloning vector. Sequences of PCR-amplified regions of genomic DNA comprising all exons and splice junctions of C46F4.1 from n592 mutants and wild-type worms were determined using an ABI Prism 3100 Genetic Analyzer.
Isolation of deletion alleles
Libraries of mutagenized animals were constructed and screened by PCR for deletion alleles of egl-6, flp-10 and flp-17 essentially as described46. Deletion mutants were isolated from frozen stocks and backcrossed to wild-type worms at least four times. Sequences deleted in each allele are described in Supplementary Methods.
Immunocytochemistry
Worms were fixed, permeabilized and stained as previously described47 using a 1:50 dilution of monoclonal antibody to GFP (Millipore) and a 1:100 dilution of Alexa488-conjugated goat antibody to mouse (Invitrogen). Samples were viewed using a Zeiss Axiophot epifluorescence microscope, and images were acquired with a Hamamatsu Orca charge-coupled device camera and OpenLabs image acquisition software.
EGL-6 expression in X. laevis oocytes
egl-6 cDNAs were flanked with EcoRI sites by PCR and cloned into the pGEMHE vector48. egl-6, GIRK1 and GIRK4 cRNAs were prepared using the mMessage Machine kit (Ambion). X. laevis oocytes were injected with 50 ng of egl-6 receptor sense cRNA and 0.5 ng of GIRK1 and GIRK4 sense cRNA. Injected oocytes were incubated at 18 °C in ND96 medium (96 mM NaCl, 2.5 mM KCl, 1 mM MgCl2 and 5 mM HEPES, pH 7.6) for 2–5 d before recording.
Whole-cell current recordings were made using the two-electrode voltage-clamp technique at a holding potential of −80 mV as described27. The recording chamber was continuously perfused with ND96 medium. To assay activation of GIRK channels, we equilibrated oocytes in high-K+ medium (96 mM KCl and 2.5 mM NaCl instead of 2.5 mM KCl and 96 mM NaCl) to reverse the K+ gradient and measured inward currents before, during and after addition of test peptide. Data were acquired with Clampex 8.0 software (Molecular Devices) and analyzed offline with Clampfit (Molecular Devices). All experiments using X. laevis oocytes were done according to guidelines of the Committee on Animal Care at MIT. Peptides were synthesized by the MIT Biopolymers Laboratory.
Behavioral assays and neuron ablation
Transgenic lines were scored as egg-laying defective if young adults (24 h after late L4) had, on average, greater than 30 eggs in utero. To score the developmental stages of newly laid eggs, young adults were transferred to fresh nematode growth medium plates with bacteria, five worms per plate, for 1 h at room temperature and then removed. Operated worms were assayed individually, and a single operated worm was assayed two and four times.
Eggs on the plate were examined under a high-power dissecting microscope and categorized as described in Figure 2.
Distributions of the developmental stages of eggs laid by worms of different genotypes were analyzed with the Wilcoxon Mann-Whitney rank-sum test, a nonparametric test of statistical significance, as implemented in the Coin package of the R statistical analysis program49. The results of statistical tests of significance can be found in Supplementary Table 1. Egg-laying defects of many strains were also independently quantified by measuring the number of retained eggs in staged adults as previously described19, and we observed similar interactions between receptor- and ligand-encoding genes, receptor- and G protein-encoding genes, and genes in the egl-6 signaling pathway and acetylcholine biosynthesis genes (data not shown).
Laser microsurgeries were performed on L2-stage larvae as described50. Cell identifications were made on the basis of nuclear position and cell morphology. A gcy-33∷gfp transgene was used in some experiments to identify BAG neurons (see Supplementary Methods).
Supplementary Material
Acknowledgments
We thank Y. Kohara for flp-10 and flp-17 cDNAs; N. Dascal for GIRK1 and GIRK4 expression constructs; J. Rand for unc-17 cha-1 strains; J. Moresco and M. Koelle for tph-1 promoter constructs; A. Fire for expression vectors; A. Hellman, S. McGonagle, B. Castor, and N. An for technical assistance; N. Abe and R. O’Hagan for help with Xenopus oocyte electrophysiology; J. Chung for help with data analysis; and B. Galvin and A. Saffer for critical reading of the manuscript. N.R. received support from the Howard Hughes Medical Institute, the Life Sciences Research Foundation, and the Medical Foundation. H.R.H. is David H. Koch Professor of Biology and an Investigator of the Howard Hughes Medical Institute. This work was supported by National Institutes of Health grant GM24663.
Footnotes
AUTHOR CONTRIBUTIONS
N.R. performed all experiments. N.R. and H.R.H. designed the experiments and wrote the manuscript.
Note: Supplementary information is available on the Nature Neuroscience website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Horvitz HR, Chalfie M, Trent C, Sulston JE, Evans PD. Serotonin and octopamine in the nematode Caenorhabditis elegans. Science. 1982;216:1012–1014. doi: 10.1126/science.6805073. [DOI] [PubMed] [Google Scholar]
- 2.Trent C, Tsung N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Phil Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 4.Rand JB, Nonet ML. In: Neurotransmitter assignments for specific neurons in C. elegans II. Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1997. pp. 1049–1052. [Google Scholar]
- 5.Duerr JS, Gaskin J, Rand JB. Identified neurons in C. elegans coexpress vesicular transporters for acetylcholine and monoamines. Am J Physiol Cell Physiol. 2001;280:C1616–C1622. doi: 10.1152/ajpcell.2001.280.6.C1616. [DOI] [PubMed] [Google Scholar]
- 6.Waggoner LE, Zhou GT, Schafer RW, Schafer WR. Control of alternative behavioral states by serotonin in Caenorhabditis elegans. Neuron. 1998;21:203–214. doi: 10.1016/s0896-6273(00)80527-9. [DOI] [PubMed] [Google Scholar]
- 7.Bany IA, Dong MQ, Koelle MR. Genetic and cellular basis for acetylcholine inhibition of Caenorhabditis elegans egg-laying behavior. J Neurosci. 2003;23:8060–8069. doi: 10.1523/JNEUROSCI.23-22-08060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mendel JE, et al. Participation of the protein Go in multiple aspects of behavior in C. elegans. Science. 1995;267:1652–1655. doi: 10.1126/science.7886455. [DOI] [PubMed] [Google Scholar]
- 9.Segalat L, Elkes DA, Kaplan JM. Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science. 1995;267:1648–1651. doi: 10.1126/science.7886454. [DOI] [PubMed] [Google Scholar]
- 10.Brundage L, et al. Mutations in a C. elegans Gqalpha gene disrupt movement, egg laying, and viability. Neuron. 1996;16:999–1009. doi: 10.1016/s0896-6273(00)80123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- 12.Miller KG, Emerson MD, Rand JB. Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron. 1999;24:323–333. doi: 10.1016/s0896-6273(00)80847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nurrish S, Segalat L, Kaplan JM. Serotonin inhibition of synaptic transmission: Galpha(0) decreases the abundance of UNC-13 at release sites. Neuron. 1999;24:231–242. doi: 10.1016/s0896-6273(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 14.Carnell L, Illi J, Hong SW, McIntire SL. The G-protein-coupled serotonin receptor SER-1 regulates egg laying and male mating behaviors in Caenorhabditis elegans. J Neurosci. 2005;25:10671–10681. doi: 10.1523/JNEUROSCI.3399-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hobson RJ, et al. SER-7, a Caenorhabditis elegans 5-HT7-like receptor, is essential for the 5-HT stimulation of pharyngeal pumping and egg laying. Genetics. 2006;172:159–169. doi: 10.1534/genetics.105.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shyn SI, Kerr R, Schafer WR. Serotonin and Go modulate functional states of neurons and muscles controlling C. elegans egg-laying behavior. Curr Biol. 2003;13:1910–1915. doi: 10.1016/j.cub.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 17.Kass J, Jacob TJ, Kim P, Kaplan JM. The EGL-3 prohormone convertase regulates mechanosensory responses of Caenorhabditis elegans. J Neurosci. 2001;21:9265–9272. doi: 10.1523/JNEUROSCI.21-23-09265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacob TC, Kaplan JM. The EGL-21 carboxypeptidase E facilitates acetylcholine release at Caenorhabditis elegans neuromuscular junctions. J Neurosci. 2003;23:2122–2130. doi: 10.1523/JNEUROSCI.23-06-02122.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moresco JJ, Koelle MR. Activation of EGL-47, a Galpha(o)-coupled receptor, inhibits function of hermaphrodite-specific motor neurons to regulate Caenorhabditis elegans egg-laying behavior. J Neurosci. 2004;24:8522–8530. doi: 10.1523/JNEUROSCI.1915-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 21.Sze JY, Victor M, Loer C, Shi Y, Ruvkun G. Food and metabolic signaling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature. 2000;403:560–564. doi: 10.1038/35000609. [DOI] [PubMed] [Google Scholar]
- 22.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 23.Cazzamali G, Grimmelikhuijzen CJ. Molecular cloning and functional expression of the first insect FMRFamide receptor. Proc Natl Acad Sci USA. 2002;99:12073–12078. doi: 10.1073/pnas.192442799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meeusen T, et al. Identification in Drosophila melanogaster of the invertebrate G protein-coupled FMRFamide receptor. Proc Natl Acad Sci USA. 2002;99:15363–15368. doi: 10.1073/pnas.252339599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C. The ever-expanding neuropeptide gene families in the nematode Caenorhabditis elegans. Parasitology. 2005;131(Suppl):S109–S127. doi: 10.1017/S0031182005009376. [DOI] [PubMed] [Google Scholar]
- 26.Nathoo AN, Moeller RA, Westlund BA, Hart AC. Identification of neuropeptide-like gene families in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2001;98:14000–14005. doi: 10.1073/pnas.241231298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogers C, et al. Inhibition of Caenorhabditis elegans social feeding by FMRFamide-related peptide activation of NPR-1. Nat Neurosci. 2003;6:1178–1185. doi: 10.1038/nn1140. [DOI] [PubMed] [Google Scholar]
- 28.Kim K, Li C. Expression and regulation of an FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. J Comp Neurol. 2004;475:540–550. doi: 10.1002/cne.20189. [DOI] [PubMed] [Google Scholar]
- 29.Jin Y, Jorgensen E, Hartwieg E, Horvitz HR. The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J Neurosci. 1999;19:539–548. doi: 10.1523/JNEUROSCI.19-02-00539.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sulston J, Dew M, Brenner S. Dopaminergic neurons in the nematode C. elegans. J Comp Neurol. 1975;163:215–226. doi: 10.1002/cne.901630207. [DOI] [PubMed] [Google Scholar]
- 31.Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L. EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J Neurosci. 1999;19:159–167. doi: 10.1523/JNEUROSCI.19-01-00159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alkema MJ, Hunter-Ensor M, Ringstad N, Horvitz HR. Tyramine functions independently of octopamine in the Caenorhabditis elegans nervous system. Neuron. 2005;46:247–260. doi: 10.1016/j.neuron.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 33.Alfonso A, Grundahl K, Duerr JS, Han HP, Rand JB. The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter. Science. 1993;261:617–619. doi: 10.1126/science.8342028. [DOI] [PubMed] [Google Scholar]
- 34.Alfonso A, Grundahl K, McManus JR, Rand JB. Cloning and characterization of the choline acetyltransferase structural gene (cha-1) from C. elegans. J Neurosci. 1994;14:2290–2300. doi: 10.1523/JNEUROSCI.14-04-02290.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rand JB. Genetic analysis of the cha-1-unc-17 gene complex in Caenorhabditis. Genetics. 1989;122:73–80. doi: 10.1093/genetics/122.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Culotti JG, Von Ehrenstein G, Culotti MR, Russell RL. A second class of acetylcholinesterase-deficient mutants of the nematode Caenorhabditis elegans. Genetics. 1981;97:281–305. doi: 10.1093/genetics/97.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward S, Thomson N, White JG, Brenner S. Electron microscopical reconstruction of the anterior sensory anatomy of the nematode Caenorhabditis elegans. J Comp Neurol. 1975;160:313–337. doi: 10.1002/cne.901600305. [DOI] [PubMed] [Google Scholar]
- 38.Ware RW, Clark D, Crossland K, Russell RL. The nerve ring of the nematode Caenorhabditis elegans: sensory input and motor output. J Comp Neurol. 1975;162:71–110. [Google Scholar]
- 39.Jose AM, Bany IA, Chase DL, Koelle MR. A specific subset of transient receptor potential vanilloid-type channel subunits in Caenorhabditis elegans endocrine cells function as mixed heteromers to promote neurotransmitter release. Genetics. 2007;175:93–105. doi: 10.1534/genetics.106.065516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee YS, et al. Characterization of GAR-2, a novel G protein-linked acetylcholine receptor from Caenorhabditis elegans. J Neurochem. 2000;75:1800–1809. doi: 10.1046/j.1471-4159.2000.0751800.x. [DOI] [PubMed] [Google Scholar]
- 41.Hwang JM, et al. Cloning and functional characterization of a Caenorhabditis elegans muscarinic acetylcholine receptor. Receptors Channels. 1999;6:415–424. [PubMed] [Google Scholar]
- 42.Lee YS, et al. Cloning and expression of a G protein-linked acetylcholine receptor from Caenorhabditis elegans. J Neurochem. 1999;72:58–65. doi: 10.1046/j.1471-4159.1999.0720058.x. [DOI] [PubMed] [Google Scholar]
- 43.Putrenko I, Zakikhani M, Dent JA. A family of acetylcholine-gated chloride channel subunits in Caenorhabditis elegans. J Biol Chem. 2005;280:6392–6398. doi: 10.1074/jbc.M412644200. [DOI] [PubMed] [Google Scholar]
- 44.Weinshenker D, Garriga G, Thomas JH. Genetic and pharmacological analysis of neurotransmitters controlling egg laying in C. elegans. J Neurosci. 1995;15:6975–6985. doi: 10.1523/JNEUROSCI.15-10-06975.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wicks SR, Yeh RT, Gish WR, Waterston RH, Plasterk RH. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat Genet. 2001;28:160–164. doi: 10.1038/88878. [DOI] [PubMed] [Google Scholar]
- 46.Jansen G, Hazendonk E, Thijssen KL, Plasterk RH. Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nat Genet. 1997;17:119–121. doi: 10.1038/ng0997-119. [DOI] [PubMed] [Google Scholar]
- 47.Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- 48.Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 49.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2006. URL http://www.R-project.org. [Google Scholar]
- 50.Bargmann CI, Avery L. Laser killing of cells in Caenorhabditis elegans. Methods Cell Biol. 1995;48:225–250. doi: 10.1016/s0091-679x(08)61390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.