

Abstract
Nuclear importing system and nuclear factors play important roles in mediating nuclear reprogramming and zygotic gene activation. However, the components and mechanisms that mediate nuclearly specific targeting of the nuclear proteins during nuclear reprogramming and zygotic gene activation remain largely unknown. Here, we identified a novel member of the importin-α family, AW146299(KPNA7), which is predominantly expressed in mouse oocytes and zygotes and localizes to the nucleus or spindle. Mutation of Kpna7 gene caused reproductivity reduction and sex imbalance by inducing preferential fetal lethality in females. Parthenogenesis analysis showed that the cell cycle of activated one-cell embryos is loss of control and ahead of schedule but finally failed to develop into blastocyst stage. Further RT-PCR and epigenetic modification analysis showed that knocking out of Kpna7 induced abnormalities of gene expression (dppa2, dppa4, and piwil2) and epigenetic modifications (down-regulation of histone H3K27me3). Biochemical analysis showed that KPNA7 interacts with KPNB1 (importin-β1). In summary, we identified a novel Kpna7 gene that is required for normal fertility and fecundity.
Keywords: Chromatin Histone Modification, Development, Embryo, Gene Knockout, Reproduction
Introduction
Nuclear proteins play key roles in nuclear reprogramming during fertilization and zygotic gene activation. The chromatins derived from both the female and male gametes are transcriptionally inactive and need to be remodeled to re-establish totipotency and to support embryonic development (1–7). The DNA in gametes is highly methylated and exists in a highly heterochromatization state. Upon fertilization, the paternal genome is demethylated rapidly during zygotic gene activation. The maternal genome, however, undergoes gradual demethylation (1–7). The histones and protamine that are derived from oocytes and sperm, respectively, need to be replaced with embryonic histones (4–6). Epigenetic modifications of the genome, such as covalent modification of histone residues, are asymmetric between parental genomes after fertilization. The male pronucleus is negative for H3K9me2/3,3 yet the female is positive for these residues (6). Many nuclear factors, derived from the oocyte or translated after fertilization, are necessary for the zygotic gene activation (1, 5–10). These nuclear factors need to be transported into the nucleus to execute their important roles in reprogramming the fully differentiated gametes into totipotent zygotes. We speculate that one or more stage-specific nuclear transporters are required to transport these stage-specific nuclear factors.
Karyopherins, including both importins and exportins, play key roles in mediating the bidirectional trafficking of macromolecules across the nuclear envelope (11–16). Most karyopherins, including importin-α and importin-β, bind directly to nuclear localization signal (NLS)- and nuclear export signal-containing cargos (11–16). Importin-αs are composed of a flexible N-terminal importin-b-binding (IBB) domain and a highly structured domain composed of 10 tandem armadillo (ARM) repeats. Importin-βs are composed of a flexible importin-β N-terminal domain and a highly structured domain composed of HEAT repeats. Structure analysis showed that the less structured N-terminal IBB domain containing an internal NLS bound to the NLS-binding site that formed by the large C-terminal ARM repeats domain. The structure analysis indicated one auto-inhibitory model of importin-α. Importin-α binds to classical NLS (cNLS)-containing proteins and links them to importin-β, which transports the importin-α-cargo complex through the nuclear pore complexes (11–16). The human genome encodes more than 20 karyopherins. Most of the nuclear factors contain one or more NLSs (17). The exportin CAS recycles importin-α back to the cytoplasm (18, 19). It was reported that importin-α subtype switching has a major impact on cell differentiation through the regulated nuclear import of a specific set of transcription factors (15). Because there are hundreds of nuclear proteins that need to be transported into the nucleus, the karyopherins may have target specificity and functional overlapping.
In this study, we identified a novel member of the importin-α family, AW146299. and designated it Kpna7. Our studies have shown that Kpna7 is predominantly expressed in mouse oocytes and zygotes. Mutation of Kpna7 in vivo caused reproductivity reduction and sex imbalance. Our data indicate that Kpna7 is required for normal fertility and fecundity in the mouse.
EXPERIMENTAL PROCEDURES
Animals, Oocytes, and Embryos and Embryo Incubation in Vitro
B6D2F1 (C57BL/6J × DBA2) female mice (8–10 weeks old) were used for collection of fully grown germinal vesicle (GV) and MII oocytes. GV oocytes were collected according to the previous report (20). Zygotes were collected from the successfully mated B6D2F1 females or Kpna7 mutation mice. ICR mice were used to generate Kpna7 chimera mice. All studies adhered to procedures consistent with the National Institute of Biological Sciences Guide for the care and use of laboratory animals. For parthenogenesis and epigenetic analysis, MII oocytes were isolated from normal BDF1 mice (normal control) and Kpna7 mutation mice. MII oocytes were incubated in activation solution (CZB medium containing 10 mm SrCl2, 10 μm cytochalasin B, and 1 mm glutamine) for 6 h and further incubated in KSOM medium. For preimplantation developmental analysis, zygotes isolated from BDF1 mice and Kpna7 mutation mice were directly incubated in KSOM medium.
Kpna7 Gene Targeting
The Kpna7 mutation targeting vector was generated by sequentially subcloning Kpna7 genomic fragments (the 1.2-kb 3′ short arm and 4.6-kb 5′ long arm) into pJB1 vector. We generated the Kpna7 genomic fragments by PCR using 129/Sv mouse genomic DNA as the template. The primers used are listed (supplemental Table S1). The exon 5 and exon 6 will be deleted after targeting. The targeting vector was linearized and transfected into R1 embryonic stem cells via electroporation. Clones that survived drug selection with G418 and ganciclovir were picked up. The correctly targeted ES cells were identified and confirmed by PCR screening and sequencing. Germ line transmissible chimera mice were obtained successfully. Homozygous Kpna7 mutation mice (mut/mut) were obtained from crossing between heterozygous F1 or F2.
Cell Culture
R1 ES cells were cultured under conventional conditions. The medium was based on DMEM, containing 10% FBS (Hyclone), 103 units/ml Lif (ESGRO, Chemicon), and 1× nucleosides (Invitrogen), 1× nonessential amino acids (Invitrogen), 1× β-mercaptoethanol (Invitrogen), 2 mm glutamine (Invitrogen), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). 293T cells and PHDL cells were cultured in DMEM-based medium, which contained 10% FBS and 3% FBS (Hyclone), respectively, and 2 mm glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin.
Transient Expression Vector Construction and Transfection
The mouse Kpna7 ORF was obtained by RT-PCR from adult ovaries. The sequences were confirmed by sequencing. The DNA sequences of Kpna7 ORF reported in this paper have been deposited in the GenBankTM data base (accession numbers FJ717332 and FJ717333). The ORF and mutants were cloned into pEGFP-N1 vector for transient expression. Vigofect reagent was used according to the protocol provided by the manufacturer (Vigorous). Cells were collected 36 h after transfection. The primers used for construct cloning are listed in supplemental Table S1.
RT-PCR and Genomic PCR
Total RNA samples were prepared from adult brain, heart, kidney, liver, pancreas, skin, ovary and testis, and from oocytes and early embryos. The RNA was extracted with conventional methods for adult tissues by using TRIzol (Invitrogen). PicoPure RNA isolation kit (Arcturus) was used to extract RNA from collected oocytes and preimplantation embryos. Reverse transcription and PCR were performed as conventional methods by using M-MLV reverse transcriptase (Promega). Genomic DNA was extracted with conventional methods. The RNA was reverse-transcribed by M-MLV reverse transcriptase (Promega) and amplified by PCR for 30 or 35 cycles. Primers were selected encompassing the intronic sequences. Qualitative RT-PCR cycling was performed at 98 °C for 2 min followed by 98 °C for 15 s, 54–60 °C for 15 s, 72 °C for 40 s, and finally 72 °C for 8 min, by using the PrimeSTAR HS DNA polymerase (Takara). Quantitative RT-PCR was performed by using another set of primers. The Xist and Zfy primers were used as reported previously (21). The primer sequences used are listed in supplemental Tables S1–S3.
Antibody Preparation
Polyclonal antibody against mouse KPNA7 protein was prepared from rabbits by using the C-terminal fragment (Glu-234 to Val-499 of the predominant isoform). The cDNA sequences containing this encoding region was cloned into pGEX-4T vector and expressed in Escherichia coli. The antigen fragment was purified by using His tag beads and was performed by subcutaneous immunization. Finally, the antibody was purified by protein-A affinity.
Immunofluorescent Staining, Confocal Microscopy, and Adobe Photoshop Analysis
In brief, the samples were permeabilized with 0.5% Triton X-100 and blocked with 5% normal horse serum for 2 h. The first antibodies were incubated overnight at 4 °C. All primary antibodies against epigenetic markers were purchased from Upstate or Abcam and were used at the recommended concentration. The Alexa-594-conjugated secondary antibodies were incubated for 1 h at room temperature. Samples were further counterstained with 100 ng/ml DAPI. Images were obtained with an Olympus IX 71 microscope (Olympus, Japan) equipped with a CCD camera (DVC) or LSM510 Meta confocal microscope (Zeiss). The staining intensity was analyzed by histogram signal intensity using Adobe photoshop.
Immunoprecipitation and Western Blotting
293T cells were transfected to transiently express KPNA7-FLAG, KPNA7-HIS, KPNA7-deIBB-HIS, KPNA7mut-HIS, KPNA2-HIS, KPNB1-FLAG, KPNB1-FLAG-EGFP, and KPNB1-HIS singly or combinationally. Samples were collected 36 h after transfection. Immunoprecipitations were performed according to the protocol provided by the manufacturer (FLAGIPT-1; Sigma). Proteins were separated in 10% SDS-polyacrylamide gels and transblotted to PVDF membrane. Immunoblot analysis was performed with anti-FLAG and anti-HIS monoclonal antibody. Blots were detected by using ECL (Amersham Biosciences) according to the protocol provided by the manufacturer.
Silver Staining and Mass Spectrometry Analysis
Proteins were separated in 10% SDS-polyacrylamide gels. Silver staining was performed according to the protocol provided by the manufacturer (PROTSIL2-1KT, Sigma). The bands of interest were analyzed by LTQ linear ion trap mass spectrometer (Thermo Electron).
RESULTS
Mouse Kpna7 Gene Is Predominantly Expressed in Oocytes, Zygotes, and Two-cell Stage Embryos, and the KPNA7 Protein Is Localized to the Nucleus
The AW146299 gene (GeneID, 381686; LOCUS, NM_001013774) was identified in a search for genes that are preferentially expressed in oocytes and early embryos (6, 8, 10). To analyze the expression pattern, we performed RT-PCR analysis. The results showed that mouse AW146299 is specifically expressed in the adult ovary (Fig. 1A). Moreover, it is predominantly expressed in oocytes, zygotes, and two-cell stage embryos (Fig. 1B). The AW146299 ORF encodes for an importin-α subfamily protein (499 amino acids). We have designated the gene Kpna7. Through our cloning and sequencing, we found that there also existed a longer isoform of KPNA7 (520 amino acids). Cloning and sequencing analysis showed that the 1500-bp ORF encoding the 499-amino acid-long protein is the predominant transcript (17/20). Sequence alignment showed that mouse KPNA7 had 60.1% identity with human KPNA7 in protein level (supplemental Fig. S1). The amino acids that play key roles in a forming groove for nuclear localization signal binding are well conserved (supplemental Fig. S1). To analyze the endogenous expression of KPNA7 in protein level, we generated a polyclonal antibody against its C-terminal ARM repeats. Immunostaining with the polyclonal KPNA7 antibody showed that mouse KPNA7 protein localized to the nucleus in GV stage oocytes (Fig. 1C), zygotes (Fig. 1, E–H), and two-cell stage embryos (Fig. 1, I and J). Interestingly, KPNA7 protein localized to the spindle in MII oocytes (Fig. 1D). Our data also showed that the KPNA7 intensity significantly decreased from zygotes to late two-cell embryos. To confirm the specificity of the KPNA7 antibody, we stained GV and MII oocytes with preimmune serum, and we could not observe any specific positive signals (Fig. 1, K–N). We further transfected the KPNA7-EGFP construct to mouse pancreatic PHDL epithelial cells and stained with the KPNA7 antibody. The result showed that immunostaining signals could only be detected in the KPNA7-EGFP-positive cells (Fig. 1, O–R, solid arrows) but not in the KPNA7-EGFP-negative cells (Fig. 1R, open arrows). These results indicated that the antibody was KPNA7-specific.
FIGURE 1.

Gene expression pattern and subcellular localization of mouse Kpna7. A and B, gene expression pattern of Kpna7 in tissues (A) and oocytes and preimplantation embryos (B). Mouse Kpna7 gene was specifically expressed in oocytes, zygotes, and two-cell stage embryos. Its expression was dramatically down-regulated after the two-cell stage. C–J, Kpna7 protein expression and subcellular localization in preimplantation stages. Mouse Kpna7 protein was highly expressed in GV oocytes (C), MII oocytes (D, localizes to the spindle (solid arrow) but not the chromatin (open arrow)), and zygotes (E–H, from early to late zygote stage). Its expression was dramatically down-regulated in two-cell stage embryos (I and J). K–N, negative control. No significant and specific staining signal had been observed in GV and MII oocytes with the preimmune serum. O–R, specificity analysis of Kpna7 antibody in mouse pancreatic epithelial cells. Kpna7 antibody only recognizes the ectopically expressed mouse Kpna7 protein (arrows) but not other importins (open arrows). (bars, C–J and O–R, 20 μm; K–N, 10 μm). Br, brain; He, heart; Ki, kidney; Li, liver; Pa, pancreas; Sk, skeleton; Ov, ovary; Te, testis; GV, germinal vesicle stage oocyte; MII, metaphase-II stage oocyte; Zy, zygote; 2c, two-cell; 4c, four-cell; 8c, eight cell; Mo, morula; Bl, blastocyst.
Construct Design and Sequence Analysis of the kpna7 Mutation
We sought to understand the functions of endogenous Kpna7 in vivo in germ line and embryonic development. A targeting construct was designed to delete exon 5 and 6, which should result in a splicing problem or frameshift in the truncated Kpna7 mRNA (Fig. 2A). The heterozygous and homozygous Kpna7 targeted mice were screened by genomic PCR and sequencing (Fig. 2C) and finally confirmed by ovary RT-PCR (Fig. 2D) and sequencing (supplemental Fig. S2). Sequencing results demonstrated that deletion of exon 5 and 6 resulted in loss of the short but predominant transcript (1500-bp variant). However, because of the existence of an unpredicted new exon 7, a predicted 1215-bp truncated ORF (in-frame reading by fusing exon 4 and the new exon 7) was found (Fig. 2B). Thus, we supposed that the predominant transcript should have been destroyed and the corresponding 499-amino acid-long protein should be absent, whereas the truncated longer transcript might encode a mutant KPNA7 that lacked the ARM1-3 domain.
FIGURE 2.

Kpna7 targeting design and sequence analysis. A, original design of the Kpna7 gene targeting. Exons 5 and 6 were designed to be deleted, and finally the truncated transcript should be destroyed because it caused incorrect splicing and wrong encoding. B, existence of a previously unexpected new exon after exon 6 caused in-frame encoding between exon 4 and the new exon 7, which generated one 1215-bp long truncated Kpna7 mRNA. The truncated Kpna7 mRNA encodes a protein that lacks the exon 5- and exon 6-encoded polypeptide fragment but with a new exon 7-encoded polypeptide fragment. C and D, genomic PCR (C) and RT-PCR (D) analysis of the wild type (wt1-3), Kpna7 heterozygous (F1-162, F2-14, and F2-101), and homozygous (F2-42, F2-46, F3-163, and F3-164) mutation mice.
To detect the KPNA7 expression in protein level, we stained GV and MII oocytes, as well as zygotes and two-cell embryos that were isolated from wild type and Kpna7 mutation mice, respectively. Because the identity of the polypeptide sequences was used for antibody preparation, the antibody should also identify the predicted KPNA7 mutant. Surprisingly, we could not detect any specific positive signal with the KPNA7-specific antibody in the Kpna7 mutation oocytes and early embryos (Fig. 3, A–D). In contrast, the specific positive signals were easily detected in the wild type oocytes and embryos (Fig. 3, E–H). These results indicated that not only the wild type KPNA7 protein was nonexistent but also the predicted mutant KPNA7 protein was actually nonexistent, or its expression level was too low to be detected. These data then demonstrated that the phenotypes discussed below were caused by the loss-of-function of KPNA7 by gene mutation.
FIGURE 3.

Kpna7 gene mutation caused loss of KPNA7 protein expression in the mouse. A–D, KPNA7-specific antibody staining in wild type GV (A), MII (B) oocytes, zygotes (Zy, C), and two-cell embryos (D). E–H, KPNA7-specific antibody staining in Kpna7 mutation GV (E), MII (F) oocytes, zygotes (G), and two-cell embryos (H). Significant and specific positive signal had been observed in oocytes and early embryos that isolated from wild type mice but not Kpna7 mutation mice (bars, 20 μm).
Kpna7 Mutation-induced Reproductivity Reduction and Sex Imbalance
To find whether some phenotypes would be caused by Kpna7 mutation, we analyzed the heterozygous and homozygous Kpna7 mutation mice. We found that reproductive ability of males was unaffected. However, reduction of reproductive ability was induced in both the Kpna7+/mut and Kpna7mut/mut females. These results suggested that fetal lethality might be induced in the Kpna7+/mut and Kpna7mut/mut mice in a female-preferential manner. As expected, we found that many embryos died after embryonic day 8.5 (E8.5) (Fig. 4, A–D, total of 18 litters were analyzed). The average litter size of the control group, the heterozygous mutants, and the homozygous mutants was 11.1 ± 2.2, 7.9 ± 2.7, and 5.8 ± 3.1 mice/litter, respectively (Fig. 5A, 10, 50, and 26 litters of wild type, heterozygous, and homozygous mice were examined, respectively). In addition, we observed that reduction of reproductive ability was more adversely affected in multigravida and older females than primigravida/young females (data not shown). Simultaneously, we found that female mice were less than male mice in number (Fig. 5A). To analyze whether sex imbalance was caused by the Kpna7 mutation, we mated the F1 heterozygous female and male mice and then performed Mendelian genetic analysis. The results showed that the F2 mice derived from F1♀(+/mut) × F1♂(+/mut) mice did not align with Mendel's law in sex ratio (male/female, from 1:1 to about 1.4:1) or in wild type/heterozygous/homozygous ratio (from 1:2:1 to 2.8:2.3:1) (Fig. 5B, total of 46 litters were analyzed). We further determined the sex and found that most of the dead embryos were females (Fig. 5C, total of 6 litters were analyzed).
FIGURE 4.

Mutation of Kpna7 caused reproductivity reduction by inducing partial fetal lethality. A–D, uteri were isolated from Kpna7 mutation mice. A, one uterus isolated from a Kpna7 mutation mouse contained embryonic 7.5 day (E7.5) embryos. All the embryos were alive on E7.5. B, one uterus containing E12.5 embryos. Three embryos were alive but others had died and been absorbed. C and D, typical littermates observed in Kpna7 mutation mice. C shows E13.5 littermates isolated from one female. Six of 13 of the fetuses were alive. The other embryos had died and been absorbed. Arrows indicate one typical absorbed embryo and its placenta. D shows E16.5 littermates isolated from one female. Five fetuses were alive. Two residues were found.
FIGURE 5.
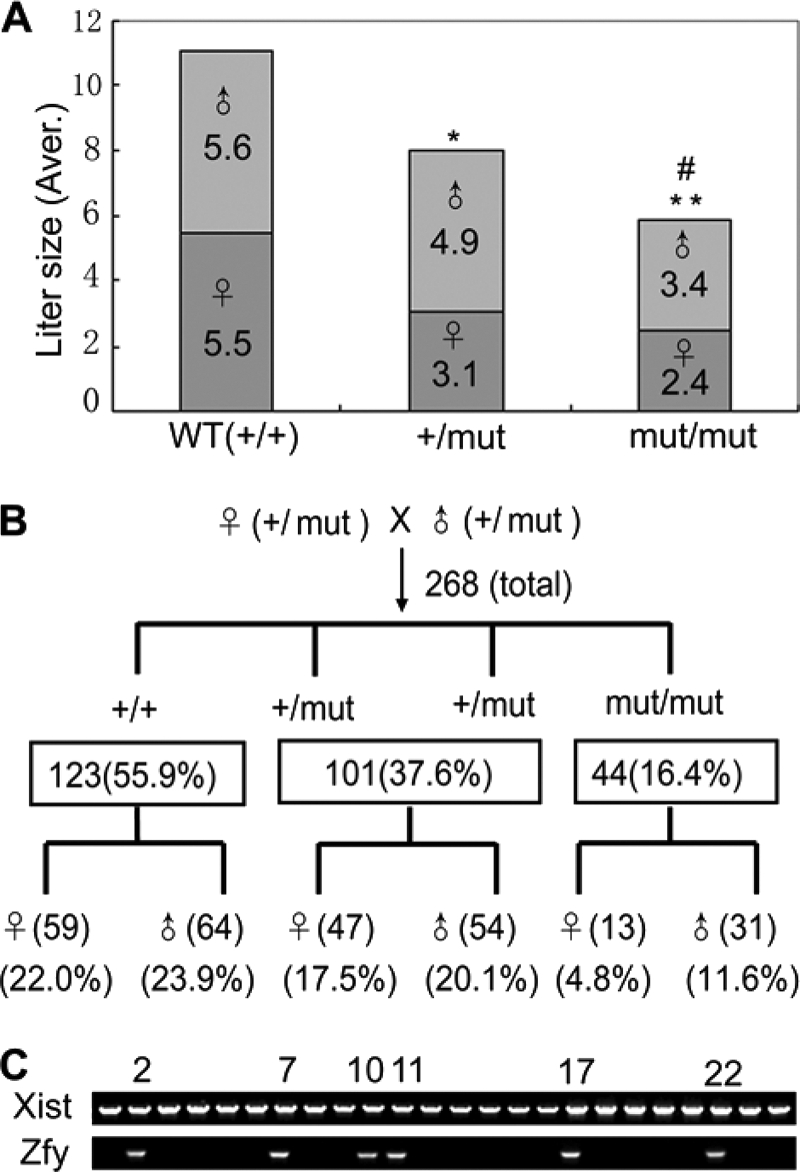
Mutation of Kpna7 caused reproductivity reduction and sex imbalance. A, statistical analysis of reproductive ability of wild type, Kpna7+/mut, and Kpna7mut/mut mice. The litter size is reduced in Kpna7 heterozygous and homozygous mutation mice than the control group. * and ** representing the average litter size of Kpna7+/mut and Kpna7mut/mut group are smaller than the wild type group (p < 0.01 and p < 0.001, respectively). # representing the litter size of Kpna7mut/mut group is smaller than Kpna7+/mut group (p < 0.05). B, genetic analysis of the heterozygous/homozygous and female/male mice that generated from heterozygous parents. The mice of wild type (+/+), heterozygous (+/mut), and homozygous (mut/mut) were counted. The sex ratio of each gene type was also calculated. C, sex determination of the dead embryos by genomic PCR. Six males (xist and zfy double-positive) and 18 females (zfy-negative) were found in 24 dead embryos collected from six homozygous females of middle or late gestation.
Kpna7 Mutation Induced Preimplantation Development Abnormalities
To characterize whether preimplantation development was affected by the Kpna7 mutation, we performed an in vitro development test. Here and below, the Kpna7 mutation means the homozygous mutation mice. Our results showed that, 144 h after hCG injection, 51.6% of the zygotes (35/68) that were collected from naturally fertilized Kpna7 mutation mice survived through the two-cell stage and developed to the blastocyst stage with a somewhat delayed pattern (Fig. 6, A and C). In contrast, 92.6% of the zygotes of the normal control group were successfully developed into the blastocyst stage (Fig. 6, B and C). Because we had observed the sex imbalance (more Kpna7mut/mut female mice died than male mice), we supposed that there might be abnormalities within the MII oocytes of Kpna7 mutation mice.
FIGURE 6.

Mutation of Kpna7-induced preimplantation developmental abnormalities in vitro. A–C, developmental abnormalities of naturally fertilized zygotes are induced in Kpna7 mutation mice. Many of the zygotes of Kpna7 group failed to develop into blastocyst stage (A). C, statistical analysis of preimplantation development 120 and 144 h after hCG injection. 48.4% of the Kpna7 mutation zygotes fail or are delayed from developing into blastocyst stage. D–F, parthenogenesis developmental abnormalities of naturally fertilized zygotes are induced in Kpna7 mutation mice. D and E, development of parthenogenesis one-cell embryos that collected from Kpna7 mutation and wild type mice. F, statistics analysis of parthenogenesis preimplantation development 120 and 144 h after hCG injection. Most (88.6) of the parthenogenesis one-cell embryos from Kpna7 mutation mice failed to develop into blastocyst stage. G–N, cell cycle is loss of control in parthenogenesis embryos of mutation mice after activation. G and H show embryos 48 h after hCG injection in Kpna7 mutation and wild type group (solid arrow, embryo containing four cells; open arrows, embryos containing two cells). I and J show statistics analysis of two-cell embryos and non-two-cell embryos 48 h after hCG injection in Kpna7 mutation group (I) and wild type group (J), respectively. K–N show four-cell embryos from Kpna7 mice with different morphological styles that observed 22 h after activation (48 h after hCG injection). The embryos are stained with CDC25 (solid arrows, abnormal morphology; open arrows, polar body DNA; bars, A, B, G, and H, 100 μm; D and E, 200 μm; K–N, 20 μm).
To figure out whether the MII oocytes were normal or not, we performed parthenogenesis test. The results showed that 144 h after hCG injection 88.6% (63/71) of the parthenogenesis one-cell embryos of Kpna7 mutation mice failed to develop into the blastocyst stage. In contrast, only 13.6% of the parthenogenesis one-cell embryos in the wild type group failed to develop into blastocyst stage (Fig. 6, D–F). To find out how the parthenogenesis development was disrupted, we analyzed the preimplantation development of parthenogenesis embryos. Intriguingly, we found that the first and the second cell cycles were greatly ahead of schedule in the Kpna7 mutation mice (Fig. 6, G–N). Through live imaging, we found that parthenogenesis one-cell embryos in the wild type group took place only one time cleavage during 14 h after activation (Fig. 6, H and J; supplemental movie 1). However, most of parthenogenesis one-cell embryos from Kpna7 mutation mice took place two times of cleavage during the same period (Fig. 6, G and I; supplemental movie 2). The first time cleavage took place was 3–4 h after activation, and the second time cleavage took place was 10–12 h after activation (supplemental movie 2). Statistical analysis showed that 22 h after activation (48 h after hCG injection), 81.5% of the embryos from parthenogenesis one-cell embryos of Kpna7 mutation mice had already developed beyond the two-cell stage (Fig. 6I), although only 3.0% of the embryos from wild type group developed beyond the two-cell stage (Fig. 6J). We noticed that many of the embryos beyond the two-cell stage presented atypical morphologies, which seemed being fragmented (Fig. 6G). To analyze the properties of the embryos beyond the two-cell stage, we stained them with CDC25C antibody and DAPI. We did not find significant differences between the wild type group and the Kpna7 mutation group in CDC25C signal. However, DAPI staining showed that most of the embryos contained four nuclei, even in those with “fragmented” morphologies (Fig. 6, K–N).
Kpna7 Mutation Induced Abnormal Expression of Chromatin Modification-associated Genes
Because Kpna7 was predominantly expressed in oocytes and early embryos, we sought to find out whether expression of some genes was changed in mRNA level by Kpna7 mutation. We found that a set of pluripotency- and/or chromatin-associated genes, such as dppa2, dppa4, and piwil2 (Mili), were dramatically down-regulated in mRNA level in late two-cell stage embryos by mutation of Kpna7 (Fig. 7, A and C). The dppa3 gene was not affected (Fig. 7, A and C). However, the hdac3 gene was up-regulated (Fig. 7, A and C). These genes were not affected in MII oocytes as demonstrated by RT-PCR analysis of MII oocytes (Fig. 7B). To confirm the down-regulation of DPPA2 in protein level, we stained the late two-cell embryos with DPPA2 antibody (Fig. 7, D–G). The results showed that DPPA2 protein levels were also down-regulated significantly in 48.4 ± 7.3% (n = 4, totally 34/71) of the late two-cell stage embryos that were collected from Kpna7mut/mut female mice (Fig. 7J). However, only about 1.5% of the late two-cell stage embryos were found to be down-regulated in the wild type group (Fig. 7H).
FIGURE 7.

Mutation of Kpna7 caused down-regulation of a set of chromatin remodeling factors. A and B, qualitative RT-PCR analysis shows that Kpna7 mutation induces down-regulation of dppa2, dppa4, and piwil2 in late two-cell embryos in Kpna7 mutation mice. C, real time RT-PCR analysis also shows that dppa2, dppa4, and piwil2 but not dppa3 are dramatically down-regulated in Kpna7 mutation group. hdac3 mRNA level is up-regulated. D–G, immunostaining analysis of DPPA2 protein levels in late two-cell stage embryos. Arrows indicate the DPPA2 signal (red). D and E show embryos injected with enhanced EGFP and NLS-EGFP, respectively. The DPPA2 signal is high in both wild type (D, solid arrow) and NLS-EGFP-injected late two-cell embryos (E, solid arrow). F and G, typical results of embryos derived from Kpna7 mutation mice. The DPPA2 signal is dramatically down-regulated (F, open arrow) or even hardly detected (G, open arrow) in late two-cell stage embryos. H–J, statistical analysis of DPPA2 staining intensities in late two-cell embryos. H–J show the results of wild type (H), Kpna7 heterozygous (I), and homozygous mutation mice (J), respectively. (Bars, 20 μm.)
Kpna7 Mutation Induced H3K27me3 Down-regulation in Oocytes, Zygotes, and Parthenogenesis Two-cell Embryos
To find out some clues to the phenotypes observed, we performed a series of epigenetic marker staining. We did not find significant changes for H4K8ace, H4K12ace, H3K14ace, H3K4me1, H3K9me3, and HP1B signals in zygotes (Fig. 8, A–F), two-cell embryos (Fig. 8, G–L), and MII oocytes (data not shown). But we did observe down-regulation of the H3K27me3 signal in some MII oocytes (Fig. 8, M and N), zygotes (Fig. 8, O and P), and parthenogenesis two-cell embryos (Fig. 8, Q and R). Statistical analysis demonstrated that H3K27me3 levels were down-regulated in 77.6% of Kpna7 MII oocytes (Fig. 8, S and T), in 42.4% of Kpna7 zygotes (Fig. 8, U and V), and in 63.7% of Kpna7 parthenogenesis late two-cell embryos (Fig. 8, W and X), as compared with the corresponding wild type group.
FIGURE 8.

Epigenetic analysis of oocytes, zygotes, and two-cell embryos isolated from Kpna7 mutation mice. A–F, histone acetylation analysis. H4K8ace (A and B), H4K12ace (C and D), and H3K14ace (E and F) levels in zygotes of Kpna7 mutation mice and wild type mice are similar. G–L, H3K4me1, H3K9me3, and HP1B staining. H3K4me1 (G and H), H3K9me3 (I and J), and HP1B (K and L) staining levels in late two-cell embryos of Kpna7 mutation mice and wild type mice are similar. M–R, H3K27me3 staining in MII oocytes, zygotes, and parthenogenesis two-cell embryos. H3K27me3 levels are down-regulated in MII oocytes (M, N, S, and T) and zygotes (O, P, U, and V) of Kpna7 mutation mice. DAPI and H3K27me3 double staining are shown in top right of M and N to show their colocalization. H3K27me3 levels are down-regulated in parthenogenesis two-cell embryos (Q, R, W, and X) but not in normally fertilized two-cell embryos (data not shown) of Kpna7 mutation mice. (Solid arrows indicate the high levels of staining in wild type group, and open arrows indicate the low levels of staining in Kpna7 mutation group; bars, 20 μm).
Mouse KPNA7 Protein Interacts with KPNB1 (Importin-β1)
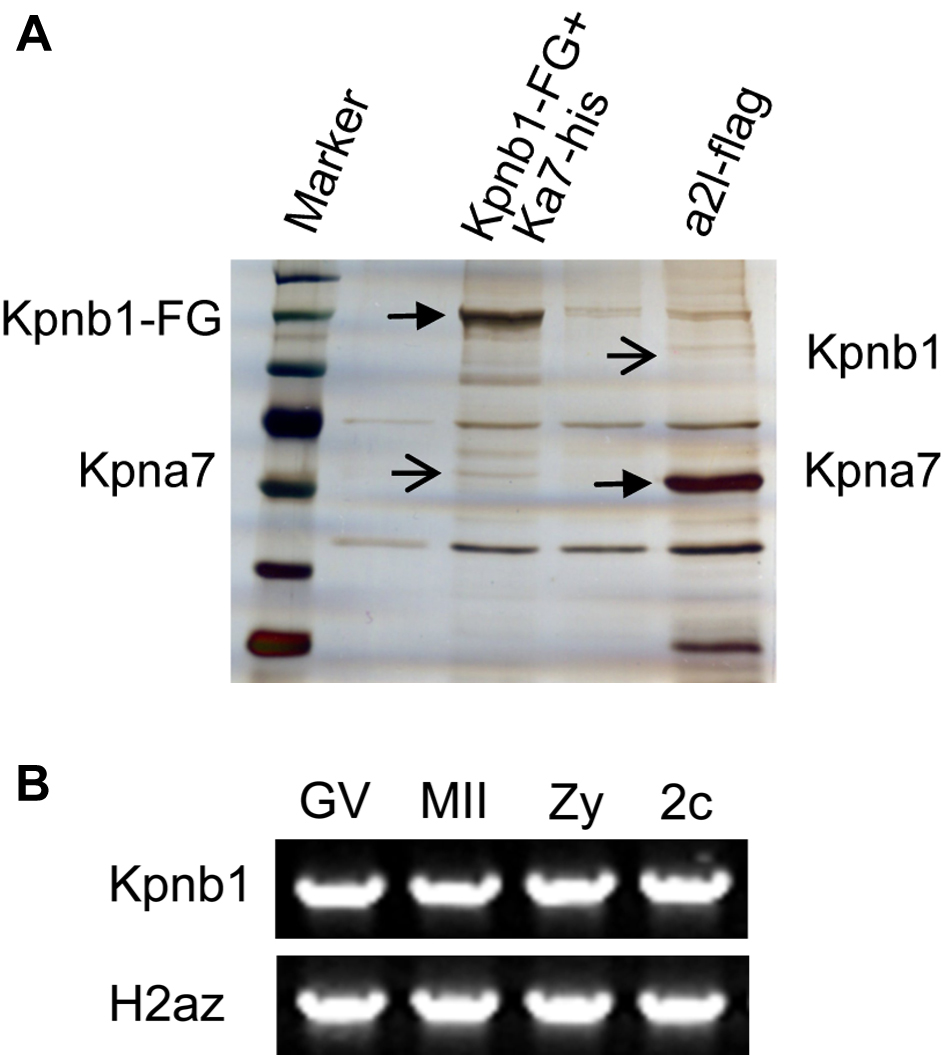
To find out which proteins interact with mouse KPNA7, we performed immunoprecipitation and Western blotting (in somatic cells), silver staining, and mass spectrometry analysis. Immunoprecipitation and Western blotting analysis showed that KPNB1 could immunoprecipitate wild type KPNA7 protein, as well as the known KPNA2 protein (as a positive control), but not the IBB domain-deleted KPNA7 (as a negative control). We also tried to detect whether KPNB1 interacted with the endogenously nonexistent/hardly detectable mutant KPNA7. The results showed that even under overexpression conditions, the mutant KPNA7 protein could only be detected at very low levels (Fig. 9A). Further immunoprecipitation analysis using KPNA7 protein as the bait also pulled down KPNB1 (Fig. 9B). All these results supported the interaction between mouse KPNA7 and KPNB1. Moreover, we confirmed the KPNA7 protein sequence by mass spectrometry analysis of the 60-kDa band that immunoprecipitated with KPNB1-FLAG-EGFP (supplemental Fig. 3A). To test whether KPNB1 was expressed in early embryos, we performed RT-PCR analysis. The results showed that KPNB1 was highly transcribed in oocytes and preimplantation embryos (supplemental Fig. 3B). These data suggested KPNA7 might regulate nuclear importing through interacting with KPNB1.
FIGURE 9.

Biochemical analysis of the interaction between KPNA7 and KPNB1 and proposed models. A and B, mouse KPNA7 and KPNB1 interaction analysis. Western blotting analysis of samples obtained by immunoprecipitation. Expression vectors were cotransfected into 293T cells, respectively. Kb1 and Ka7 represent KPNB1 and KPNA7, respectively. A, KPNB1 protein (Kb1-FG) is used as the bait protein and FLAG antibody is used to perform immunoprecipitation. KPNA2 (Ka2-his) is used as a positive control. KPNA7 protein with deleted IBB domain is used as a negative control. B, KPNA7 and KPNB1 are used as bait proteins, respectively, to further confirm the interaction between KPNA7 and KPNB1. C, proposed model in cell level for how the KPNA7 mutation caused the phenotypes/abnormalities.
DISCUSSION
Transcription Pattern, Alternative Splicing, and Partial Mutation
Importin-α contains a flexible N-terminal IBB domain and a highly structured C-terminal domain containing 10 tandem armadillo (ARM) repeats. The flexible IBB domain controls cNLS binding, assembly, and disassembly of the importin-α/β-cNLS cargo ternary complex (11–16). The long groove created by the entire ARM domain can accommodate either two monopartite cNLS peptides or a single bipartite cNLS peptide (11–16). The two NLS-binding sites on importin-α consist of ARM repeats 2–4 and 7–9, respectively. The mouse Kpna7 gene is predominantly expressed in adult ovary, oocyte, zygote, and two-cell stage embryos. The stage- and tissue-specific expression suggested that it may play important roles in transporting nuclear factors to the nucleus during zygotic gene activation. Two transcript isoforms, the 1500- and 1563-bp-long ORFs, had been cloned from ovary and two-cell stage embryos (supplemental Fig. S1). Deletion of exon 5 and 6 of Kpna7 gene resulted in splicing problems and loss of the short predominant transcript of Kpna7 mRNA. Existence of in-frame splicing in the long transcript generated a 1215-bp-long truncated ORF. However, KPNA7-specific antibody staining indicated that there was not any significant and specific signal detected in the Kpna7 mutation oocytes and early embryos. These data demonstrated that the phenotypes were caused by loss of KPNA7 protein in the Kpna7 mutation mice.
Kpna7 Mutation Caused Reduction in Reproductivity
We had successfully obtained the germ line transmittable chimera mice and finally obtained the heterozygous and homozygous Kpna7 mutation mice. We found the litter size of both heterozygous and homozygous Kpna7 mutation mice was significantly smaller than the control group. These data suggested that gametogenesis and/or embryonic development might have been affected. We further identified that fetal lethality was induced by Kpna7 mutation.
Kpna7 Mutation Caused Sex Imbalance
Another interesting phenomenon we had observed was sex imbalance. We found that most of the live pups were male. We further confirmed that most of the dead fetuses were female. We had analyzed the male mutation mice, and we had not found any abnormality in reproductivity. Statistics analysis suggested that some certain maternal problems might be caused by Kpna7 mutation mice.
Kpna7 Mutation Caused Abnormalities in Preimplantation Development
Preimplantation development of naturally fertilized embryos and parthenogenesis embryos was analyzed. Our data showed that development was moderately affected in naturally fertilized Kpna7 mutation mice. The cell cycles were significantly ahead of the schedule of the wild type. The development was severely affected in parthenogenesis of Kpna7 mutation mice. These data highly suggested that abnormalities of maternal factors and/or zygotic gene activation factors were induced by Kpna7 mutation.
Kpna7 Mutation Induced Abnormal Expression of a Set of mRNA Levels in Two-cell Embryos
Nuclear reprogramming is one of the key events that happened during zygotic gene activation (1–7, 22), through which the transcriptionally inactive genome changes to an active genome. Here, we analyzed a group of chromatin-related genes that were highly expressed in mature oocytes and early embryos. Among them, we found that dppa2, dppa4, and piwil2 were significantly down-regulated, whereas Hdac3 was up-regulated in late two-cell embryos of Kpna7 mutation mice.
Dppa3/Stella is required in protecting the maternal genome from DNA demethylation in early embryonic development (23). Like DPPA3, DPPA2 and DPPA4 have one DNA-binding SAP domain and one uncharacterized C-terminal domain and are associated with chromatin (24–26). Recent studies indicated that mouse DPPA4 protein associated with transcriptionally active chromatin in ES cells (26). Comprehensive ChIP-on-chip analysis demonstrated that OCT-3/4, SOX-2, and NANOG bind to the dppa4 promoter region in human ES cells (27). However, the roles of DPPA2 and DPPA4 in early embryonic development remain largely unknown. Our results showed that dppa2 and dppa4 but not dppa3 were dramatically down-regulated by the Kpna7 mutation.
PIWI family members play important roles in regulating chromatin structure, mRNA transcription and translation, and mRNA degradation through interactions with piRNAs and associated complexes (28–38). Most recently, piRNAs had been isolated from mature oocytes in mice (39–41). piwil2 (mili) but not piwil1 (miwi) and piwil4 (miwi2) was specifically expressed in mature oocytes (40). Piwi family proteins and piRNAs play important roles in chromatin modification and genome stability (39, 42–44). Our data showed that Kpna7 mutation induced significant down-regulation of piwil2 mRNAs in two-cell embryos. We believe that there must be many other genes whose mRNA levels should be down- or up-regulated by Kpna7 mutation. In the future, the abnormalities that are caused by Kpna7 mutation, including mRNA profiling and proteomic analysis, will be analyzed.
Kpna7 Mutation Induced Down-regulation of H3K27me3 Levels
By knowing the abnormal expression of chromatin modification genes, we further found that epigenetic modifications were induced by Kpna7 mutation. We had analyzed histone acetylation (H4K8ace, H3K12ace, and H3K14ace) (45, 46), methylation (H3K4me1, H3K9me3, and H3K27me3) (45–51), and HP1B (52) intensities in mature oocytes, zygotes, and two-cell embryos. We did not find significant changes of H4K8ace, H3K12ace, H3K14ace, H3K4me1, H3K9me3, and HP1B staining intensities. However, we did find that H3K27me3 levels were significantly down-regulated in many MII oocytes and zygotes and parthenogenesis two-cell embryos of the Kpna7 mutation group. These data indicate that the chromatin state was affected by the Kpna7 mutation. The significant maternal H3K27me3 down-regulation and severe abnormalities of parthenogenesis development may partially provide the reasons for the sex-imbalanced phenotype.
KPNA7 Interacts with KPNB1
Formation of the importin-α/β-cNLS cargo complex is the first step in the transport of many nuclear proteins (11, 12). Our data show that mouse KPNA7 interacts with KPNB1 in somatic cells. Because of the material limitations for obtaining enough samples, we did not perform biochemistry analysis of KPNA7 interactants in oocytes, zygotes, and two-cell embryos. Our RT-PCR results showed that KPNB1 was highly expressed in oocytes, zygotes, and two-cell stage embryos. Our data suggest that KPNA7 may function by binding to KPNB1, thereby regulating nuclear transport and/or chromatin modification (Fig. 9C).
In summary, our results demonstrate that mouse Kpna7 gene is required for normal fertility and fecundity in the mouse. Mutation of Kpna7 induces fetal lethality, accompanied with abnormalities in gene expression and histone/chromatin modifications.
Supplementary Material
Acknowledgments
We thank Dr. Western at Australian Research Council Centre (Australia) for providing the DPPA2 antibody. We thank the staff of the Proteomics Center, National Institute of Biological Sciences, Beijing, for performing mass spectrometry analysis. We thank the laboratory members for helpful comments on the manuscript. We appreciate the anonymous reviewers for their extensive help to improve the manuscript.
This work was supported by the Ministry of Science and Technology of China Grants 2008AA022311, 2010CB944900, and 2008AA1011005.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3, movies 1 and 2, and Tables 1–3.
- H3K9me2/3
- H3K27me3, and H3K4me1, trimethylated and monomethylated histone H3 at lysine 9, 27, and 4, respectively
- ARMs
- armadillo repeats
- NLS
- nuclear localization signal
- cNLS
- classical nuclear localization signal
- GV
- germinal vesicle
- H3K14ace
- H4K8ace, and H4K12ace, acetylated histone H3 and H4 at lysine 14, 8, and 12, respectively
- IBB
- importin-b-binding domain
- hCG
- human chorionic gonadotropin
- MII
- metaphase II
- ES
- embryonic stem.
REFERENCES
- 1.Ma J., Svoboda P., Schultz R. M., Stein P. (2001) Biol. Reprod. 64, 1713–1721 [DOI] [PubMed] [Google Scholar]
- 2.Solter D., Hiiragi T., Evsikov A. V., Moyer J., De Vries W. N., Peaston A. E., Knowles B. B. (2004) Cold Spring Harbor Symp. Quant Biol. 69, 11–17 [DOI] [PubMed] [Google Scholar]
- 3.Schultz R. M. (1993) BioEssays 15, 531–538 [DOI] [PubMed] [Google Scholar]
- 4.Nothias J. Y., Majumder S., Kaneko K. J., DePamphilis M. L. (1995) J. Biol. Chem. 270, 22077–22080 [DOI] [PubMed] [Google Scholar]
- 5.Minami N., Suzuki T., Tsukamoto S. (2007) J. Reprod. Dev. 53, 707–715 [DOI] [PubMed] [Google Scholar]
- 6.Santos F., Peters A. H., Otte A. P., Reik W., Dean W. (2005) Dev. Biol. 280, 225–236 [DOI] [PubMed] [Google Scholar]
- 7.Stitzel M. L., Seydoux G. (2007) Science 316, 407–408 [DOI] [PubMed] [Google Scholar]
- 8.Hamatani T., Carter M. G., Sharov A. A., Ko M. S. (2004) Dev. Cell 6, 117–131 [DOI] [PubMed] [Google Scholar]
- 9.Zeng F., Schultz R. M. (2005) Dev. Biol. 283, 40–57 [DOI] [PubMed] [Google Scholar]
- 10.Santos F., Hendrich B., Reik W., Dean W. (2002) Dev. Biol. 241, 172–182 [DOI] [PubMed] [Google Scholar]
- 11.Terry L. J., Shows E. B., Wente S. R. (2007) Science 318, 1412–1416 [DOI] [PubMed] [Google Scholar]
- 12.Goldfarb D. S., Corbett A. H., Mason D. A., Harreman M. T., Adam S. A. (2004) Trends Cell Biol. 14, 505–514 [DOI] [PubMed] [Google Scholar]
- 13.Kutay U., Izaurralde E., Bischoff F. R., Mattaj I. W., Görlich D. (1997) EMBO J. 16, 1153–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobe B. (1999) Nat. Struct. Biol. 6, 388–397 [DOI] [PubMed] [Google Scholar]
- 15.Yasuhara N., Shibazaki N., Tanaka S., Nagai M., Kamikawa Y., Oe S., Asally M., Kamachi Y., Kondoh H., Yoneda Y. (2007) Nat. Cell Biol. 9, 72–79 [DOI] [PubMed] [Google Scholar]
- 16.Conti E., Uy M., Leighton L., Blobel G., Kuriyan J. (1998) Cell 94, 193–204 [DOI] [PubMed] [Google Scholar]
- 17.Kosugi S., Hasebe M., Matsumura N., Takashima H., Miyamoto-Sato E., Tomita M., Yanagawa H. (2009) J. Biol. Chem. 284, 478–485 [DOI] [PubMed] [Google Scholar]
- 18.Nachury M. V., Weis K. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 9622–9627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brinkmann U. (1998) Am. J. Hum. Genet. 62, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S., Hu J., Guo X., Liu J. X., Gao S. (2008) Biol. Reprod. 80, 555–562 [DOI] [PubMed] [Google Scholar]
- 21.Obata Y., Ono Y., Akuzawa H., Kwon O. Y., Yoshizawa M., Kono T. (2000) Hum. Reprod. 15, 874–880 [DOI] [PubMed] [Google Scholar]
- 22.Latham K. E., Garrels J. I., Chang C., Solter D. (1991) Development 112, 921–932 [DOI] [PubMed] [Google Scholar]
- 23.Nakamura T., Arai Y., Umehara H., Masuhara M., Kimura T., Taniguchi H., Sekimoto T., Ikawa M., Yoneda Y., Okabe M., Tanaka S., Shiota K., Nakano T. (2007) Nat. Cell Biol. 9, 64–71 [DOI] [PubMed] [Google Scholar]
- 24.Aravind L., Koonin E. V. (2000) Trends Biochem. Sci. 25, 112–114 [DOI] [PubMed] [Google Scholar]
- 25.Maldonado-Saldivia J., van den Bergen J., Krouskos M., Gilchrist M., Lee C., Li R., Sinclair A. H., Surani M. A., Western P. S. (2007) Stem Cells 25, 19–28 [DOI] [PubMed] [Google Scholar]
- 26.Masaki H., Nishida T., Kitajima S., Asahina K., Teraoka H. (2007) J. Biol. Chem. 282, 33034–33042 [DOI] [PubMed] [Google Scholar]
- 27.Boyer L. A., Lee T. I., Cole M. F., Johnstone S. E., Levine S. S., Zucker J. P., Guenther M. G., Kumar R. M., Murray H. L., Jenner R. G., Gifford D. K., Melton D. A., Jaenisch R., Young R. A. (2005) Cell 122, 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin H. (2007) Science 316, 397. [DOI] [PubMed] [Google Scholar]
- 29.Klattenhoff C., Theurkauf W. (2008) Development 135, 3–9 [DOI] [PubMed] [Google Scholar]
- 30.Hartig J. V., Tomari Y., Förstemann K. (2007) Genes Dev. 21, 1707–1713 [DOI] [PubMed] [Google Scholar]
- 31.Houwing S., Kamminga L. M., Berezikov E., Cronembold D., Girard A., van den Elst H., Filippov D. V., Blaser H., Raz E., Moens C. B., Plasterk R. H., Hannon G. J., Draper B. W., Ketting R. F. (2007) Cell 129, 69–82 [DOI] [PubMed] [Google Scholar]
- 32.Lau N. C., Seto A. G., Kim J., Kuramochi-Miyagawa S., Nakano T., Bartel D. P., Kingston R. E. (2006) Science 313, 363–367 [DOI] [PubMed] [Google Scholar]
- 33.Kuramochi-Miyagawa S., Kimura T., Ijiri T. W., Isobe T., Asada N., Fujita Y., Ikawa M., Iwai N., Okabe M., Deng W., Lin H., Matsuda Y., Nakano T. (2004) Development 131, 839–849 [DOI] [PubMed] [Google Scholar]
- 34.Carmell M. A., Girard A., van de Kant H. J., Bourc'his D., Bestor T. H., de Rooij D. G., Hannon G. J. (2007) Dev Cell 12, 503–514 [DOI] [PubMed] [Google Scholar]
- 35.Aravin A. A., Sachidanandam R., Girard A., Fejes-Toth K., Hannon G. J. (2007) Science 316, 744–747 [DOI] [PubMed] [Google Scholar]
- 36.Kavi H. H., Fernandez H. R., Xie W., Birchler J. A. (2006) Trends Biochem. Sci. 31, 485–487 [DOI] [PubMed] [Google Scholar]
- 37.Parker J. S., Roe S. M., Barford D. (2004) EMBO J. 23, 4727–4737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brower-Toland B., Findley S. D., Jiang L., Liu L., Yin H., Dus M., Zhou P., Elgin S. C., Lin H. (2007) Genes Dev. 21, 2300–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brennecke J., Malone C. D., Aravin A. A., Sachidanandam R., Stark A., Hannon G. J. (2008) Science 322, 1387–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe T., Totoki Y., Toyoda A., Kaneda M., Kuramochi-Miyagawa S., Obata Y., Chiba H., Kohara Y., Kono T., Nakano T., Surani M. A., Sakaki Y., Sasaki H. (2008) Nature 453, 539–543 [DOI] [PubMed] [Google Scholar]
- 41.Tam O. H., Aravin A. A., Stein P., Girard A., Murchison E. P., Cheloufi S., Hodges E., Anger M., Sachidanandam R., Schultz R. M., Hannon G. J. (2008) Nature 453, 534–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aravin A. A., Sachidanandam R., Bourc'his D., Schaefer C., Pezic D., Toth K. F., Bestor T., Hannon G. J. (2008) Mol. Cell 31, 785–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aravin A. A., Hannon G. J., Brennecke J. (2007) Science 318, 761–764 [DOI] [PubMed] [Google Scholar]
- 44.O'Donnell K. A., Boeke J. D. (2007) Cell 129, 37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Endo T., Naito K., Aoki F., Kume S., Tojo H. (2005) Mol. Reprod. Dev. 71, 123–128 [DOI] [PubMed] [Google Scholar]
- 46.Wang F., Kou Z., Zhang Y., Gao S. (2007) Biol. Reprod. 77, 1007–1016 [DOI] [PubMed] [Google Scholar]
- 47.Lachner M., Jenuwein T. (2002) Curr. Opin. Cell Biol. 14, 286–298 [DOI] [PubMed] [Google Scholar]
- 48.Horn P. J., Peterson C. L. (2006) Chromosome Res. 14, 83–94 [DOI] [PubMed] [Google Scholar]
- 49.Grewal S. I., Jia S. (2007) Nat. Rev. Genet. 8, 35–46 [DOI] [PubMed] [Google Scholar]
- 50.Liu H., Kim J. M., Aoki F. (2004) Development 131, 2269–2280 [DOI] [PubMed] [Google Scholar]
- 51.Park K. E., Magnani L., Cabot R. A. (2009) Mol. Reprod. Dev. 76, 1033–1042 [DOI] [PubMed] [Google Scholar]
- 52.Kwon S. H., Workman J. L. (2008) Mol. Cells 26, 217–227 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}