

Abstract
Clostridium difficile toxin A is known to cause actin disaggregation through the enzymatic inactivation of intracellular Rho proteins. Based on the rapid and severe cell rounding of toxin A-exposed cells, we speculated that toxin A may be involved in post-translational modification of tubulin, leading to microtubule instability. In the current study, we observed that toxin A strongly reduced α-tubulin acetylation in human colonocytes and mouse intestine. Fractionation analysis demonstrated that toxin A-induced α-tubulin deacetylation yielded monomeric tubulin, indicating the presence of microtubule depolymerization. Inhibition of the glucosyltransferase activity against Rho proteins of toxin A by UDP-2′,3′-dialdehyde significantly abrogated toxin A-induced α-tubulin deacetylation. In colonocytes treated with trichostatin A (TSA), an inhibitor of the HDAC6 tubulin deacetylase, toxin A-induced α-tubulin deacetylation and loss of tight junction were completely blocked. Administration of TSA also attenuated proinflammatory cytokine production, mucosal damage, and epithelial cell apoptosis in mouse intestine exposed to toxin A. These results suggest that toxin A causes microtubule depolymerization by activation of HDAC6-mediated tubulin deacetylation. Indeed, blockage of HDAC6 by TSA markedly attenuates α-tubulin deacetylation, proinflammatory cytokine production, and mucosal damage in a toxin A-induced mouse enteritis model. Tubulin deacetylation is an important component of the intestinal inflammatory cascade following toxin A-mediated Rho inactivation in vitro and in vivo.
Keywords: Bacterial Toxins, Cytoskeleton, Epithelial Cell, Histone Deacetylase, Inflammation, Intestine, Microtubules, Reactive Oxygen Species (ROS), Signal Transduction, Tight Junction
Introduction
Clostridium difficile is the causative pathogen of antibiotic-associated diarrhea and pseudomembranous colitis in humans and animals with a 10% symptomatic infection rate among hospitalized patients (1). Two toxins, A and B, released from C. difficile, are responsible for the massive fluid secretion, apoptosis of surface colonocytes, and acute enteritis seen during infection. The two exotoxins, which share ∼63% amino acid homology, have glucosyltransferase activity (2–4) that inactivates Rho family proteins, leading to actin disaggregation (5, 6). Monoglucosylation of Rho, Rac, and Cdc42 by toxin A at threonine 37 prevents Rho family proteins from participating in the formation of actin filaments (7). This mechanism is believed to be a main cause for the cell rounding that is characteristic of toxin-exposed cells (6, 8). However, despite the presence of a rapid and severe change in the shape of infected cells, the effect of toxin A on the post-translational modification of tubulin and its subsequent influence on microtubule instability have not received detailed attention.
Microtubule instability is critical to cell shape (9), cell movement (10), intracellular transport of organelles (11), and the separation of chromosomes during mitosis (12). This instability results in the continual and rapid turnover of microtubules, in a process that is crucial for cytoskeletal remodeling (9–11). Because microtubules play a pivotal role in mitosis, drugs that influence microtubule polymerization have been used to study the mechanisms of cytoskeletal organization (13). For example, colchicine (14) is known to inhibit the microtubule polymerization that leads to mitosis, whereas taxol (15) stabilizes microtubules and blocks cell division. The instability of microtubules, which are composed of α-tubulin and β-tubulin (16), has also been associated with post-translational modifications of tubulin, such as tyrosination and acetylation. In particular, acetylation of α-tubulin at Lys-40 has been reported to enhance microtubule polymerization (17). Although the precise acetyltransferase for tubulin is not yet known, HDAC6 has been implicated in the deacetylation of tubulin (18–20).
The nine known isoforms of mammalian HDAC can be classified into two groups based on amino acid sequence length and the degree of sequence homology to a single prototypic yeast HDACs. The smaller class I HDACs (400–500 amino acids) include HDAC1, HDAC2, HDAC3, and HDAC8 (21). The larger class II HDACs (∼1,000 amino acids) include HDAC4, HDAC5 (22), HDAC6, and HDAC7 (23), and HDAC9. Most class I and class II HDACs are located in the nucleus except class II HDAC6, which is localized strictly to the cytosol (20, 24–26). Based on the hypothesis that abnormal microtubule depolymerization would probably result in barrier dysfunction of epithelial cells, we studied the possible effects of HDAC6-mediated tubulin deacetylation on inflammation and epithelial damage induced by C. difficile toxin A. Our results indicate a major effect of toxin A on microtubules in vitro and in vivo mediated by deacetylation of tubulin.
EXPERIMENTAL PROCEDURES
Toxin A Preparation and Cell Culture
Toxin A was purified from C. difficile strain VPI 10463 (American Type Culture Collection, Manassas, VA) as described previously (27). The purity of native toxin A was assessed by gel electrophoresis, which confirmed a single protein at the expected molecular mass of 307 kDa (28). HT29 and CaCo2 cells derived from human colorectal adenocarcinoma were maintained in McCoy's 5A medium (Invitrogen) and DMEM (Invitrogen), respectively. Cells were cultured in a 37 °C humidified incubator with 5% CO2.
Antibodies and Reagents
Polyclonal antibodies against tubulin, acetylated tubulin, and HDAC6 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). UDP-2′3′-dialdehyde, N-acetyl-l-cysteine (NAC),3 hydrogen peroxide (H2O2), sodium formate, trichostatin A, trapoxin, sodium butyrate, colchicine, and the antibody against β-actin were purchased from Sigma. The polyclonal antibody against caspase-3 was obtained from Cell Signaling Technology (Beverly, MA).
Immunoblot Analysis
Human colonocytes were washed with cold phosphate-buffered saline (PBS) and lysed in buffer (150 mm NaCl, 50 mm Tris-HCl, pH 8.0, 5 mm EDTA, 1% Nonidet P-40), and equal amounts of protein were fractionated by SDS-PAGE. The appropriate antibodies were applied, and antigen-antibody complexes were detected with the LumiGlo reagent (New England Biolabs).
Fractionation of Monomeric and Polymeric Tubulin
HT29 human colonocytes were exposed to toxin A for 4 or 8 h and then washed twice with PBS. For fractionation of polymerized (polymeric) and depolymerized (monomeric) tubulins, the cells were incubated with 0.3 ml of reaction buffer containing 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm phenylmethylsulfonyl fluoride, 1 mm MgSO4, and 0.1% Triton X-100 for 20 min at 37 °C. The supernatant contained the monomeric tubulin fraction, whereas polymeric tubulin was extracted from the Triton X-100-insoluble fraction by incubation in an SDS lysis buffer (25 mm Tris, pH 7.4, 0.4 m NaCl, 0.5% SDS) for 10 min at 37 °C. These fractions were subjected to 10% SDS-PAGE, and blotted membranes were probed with antibodies against α-tubulin, acetylated α-tubulin, and β-actin.
Isolation of Nuclear and Cytosolic Proteins
HT29 cells were grown to ∼80% confluence and exposed to toxin A or H2O2 for 3 h. After centrifugation at 500 × g, the cell pellet was resuspended in five volumes of 0.3 m sucrose and 2% Tween 40 in Buffer A (10 mm HEPES-potassium hydroxide (KOH), pH 7.9, containing 10 mm KCl, 1.5 mm MgCl2, 0.1 mm EGTA, 0.5 mm dithiothreitol (DTT), 0.5 mm phenylmethylsulfonyl fluoride (PMSF), 2 μg/ml leupeptin, and 2 μg/ml pepstatin A). After cells were subjected to freezing, thawing, and gentle homogenization, nuclei were isolated by centrifugation at 3,000 rpm in Buffer A. Supernatants were collected as the cytosolic fractions, and the precipitated pellets were lysed in Buffer A for isolation of the nuclear proteins.
Enzymatic Activity Assay for HDAC6
Because HDAC6 is mainly present in the cytosol, whereas the other HDAC family proteins localize to the nucleus, the HDAC activity observed in cytosolic extracts was compared with that in nuclear extracts. Activity was measured from colonocyte nuclear and cytosolic extracts using a fluorescence activity assay kit (Cayman Chemical, Ann Arbor, MI). Using immunoprecipitation assay with an antibody against HDAC6, activity of colonocyte-HDAC6 in the presence and absence of toxin A was confirmed. Briefly, each extract (1 μg of total protein) or each immunoprecipitant was incubated at 37 °C with 100 μm acetylated fluorogenic substrate in HDAC assay buffer. After 30 min, the lysine developer was added, and the mixture was incubated for another 15 min at room temperature. Fluorescence was measured using a Spectra Max M5 fluorescent plate reader (Molecular Devices, Sunnyvale, CA) with excitation at 360 nm and emission at 460 nm.
Animals
This study was approved by the Animal Care and Use Committee of Daejin University (Pocheon, Korea). Male CD1 mice (Daehan Biolink, Daejeon, Korea) weighing 30–35 g were used; they had free access to food and water and were acclimated to these conditions for at least 7 days prior to experiments. Mice were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg). Ileal loops (2 cm) were prepared and injected with control buffer (PBS), toxin A (3 μg), TSA (10 nm), or toxin A (3 μg) plus TSA in a volume of 100 μl of PBS. After 4 h, animals were sacrificed, and ileal loop tissues were collected. Tubulin deacetylation and caspase-3 activation were assessed in tissue extracts by immunoblot analysis with specific antibodies.
Cell Viability
HT29 cells treated with the various agents were incubated with 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide dye for 2 h. The solubilization reagent was added, and the absorbance was determined at 570 nm (model 3550, Bio-Rad).
Measurement of Mouse IL-6 and TNF-α
Toxin A was previously reported to stimulate production of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) from target cells (20, 21). To evaluate whether this occurred in our mouse model of gut inflammation, ileal loops of the above described mice were homogenized (40 s) and centrifuged (11,000 × g, 10 min at 4 °C), and supernatants were collected. Mouse IL-6 and TNF-α were measured by ELISA using appropriate kits obtained from R&D Systems (Minneapolis, MN).
Confocal Scanning Laser Microscopy
For visualization of microtubule structures, colonocytes were exposed to control medium or toxin A for 6 h and then fixed in 4% paraformaldehyde dissolved in PBS. After being washed with PBS, the cells were incubated with a polyclonal anti-acetylated α-tubulin antibody or a polyclonal anti-α-tubulin antibody (diluted 1:500) in PBS containing 0.05% Tween 20 for 6 h at room temperature (29). After three washings, the anti-acetylated tubulin antibody-treated cells were incubated with FITC-labeled anti-mouse IgG (1:500) for 2 h at room temperature. The anti-α-tubulin antibody-treated cells were incubated with Texas Red-labeled anti-rabbit IgG (1:500). The cells were then mounted and analyzed using a Bio-Rad MRC 1024 confocal scanning laser microscope equipped with a krypton/argon mixed gas laser as a light source. Excitation was carried out using the 480- or 594-nm lines from the laser.
Measurement of Transepithelial Resistance (TER)
Colonocytes (CaCo-2 cells) were cultured onto polycarbonate filters (Transwells, Costar, Cambridge, MA) and incubated for 5 days, reaching confluence. After the cells were exposed to toxin A alone, toxin A plus TSA, or colchicine for 8 h, TER was measured with a Millicel ERS Voltohmeter (Millipore, Bedford, MA) (28). The resistance of the supporting membrane and medium was subtracted from all readings.
Statistical Analysis
The results are presented as mean values ± S.E. Data were analyzed using the SIGMA-STAT professional statistics software program (Jandel Scientific Software, San Rafael, CA). Analyses of variance with protected t tests were used for intergroup comparisons.
RESULTS
Toxin A Induces Deacetylation of Tubulin in Human Colonocytes (HT29)
We exposed HT29 cells to toxin A for 2, 4, 6, and 8 h and then measured changes in tubulin acetylation, which is known to influence microtubule polymerization (17). As shown in Fig. 1A, compared with control cells, the basal acetylation level of tubulin in toxin A-treated cells was markedly reduced at 4 and 8 h post-treatment. The total amounts of α-tubulin and β-actin were unchanged in these cells. We then assessed whether toxin A-induced tubulin deacetylation influenced microtubule polymerization. HT29 cells were exposed to toxin A for 0, 4, and 8 h and incubated in cell membrane-permeabilizing buffer for separation of monomeric and polymeric tubulins (30). Toxin A treatment of HT29 cells significantly and time-dependently increased the amount of tubulin in the supernatant (super), indicating that most tubulins were monomeric (Fig. 1B). In contrast, the amount of tubulin in cell pellets (containing polymeric tubulins) was markedly reduced following toxin A exposure. Consistent with these findings, highly acetylated tubulins were detected in control cell pellets but not in pellets from cells exposed to toxin A for 4 h (Fig. 1B). These results suggest that toxin A-induced deacetylation of tubulin causes microtubule depolymerization in human colonocytes. We then assessed whether toxin A increased tubulin deacetylation in the gut epithelium of mice. Ileal loops of CD1 mice were injected with 3 μg of toxin A in 100 μl of PBS (n = 10). Four hours later, ileal mucosal scrapings were collected, protein extracts were subjected to 10% polyacrylamide gel electrophoresis, and blotted membranes were probed with an antibody against acetylated α-tubulin. A marked decrease in α-tubulin acetylation was confirmed in ileal mucosa exposed to toxin A compared with controls (Fig. 1C). Quantitation of the immunoblot results showed that tubulin acetylation was 3-fold lower in toxin A-treated mice than in PBS-treated mice (Fig. 1C, lower panel). Collectively, these results indicate that toxin A causes tubulin deacetylation in both cultured colonocytes and the mouse intestinal epithelium.
FIGURE 1.
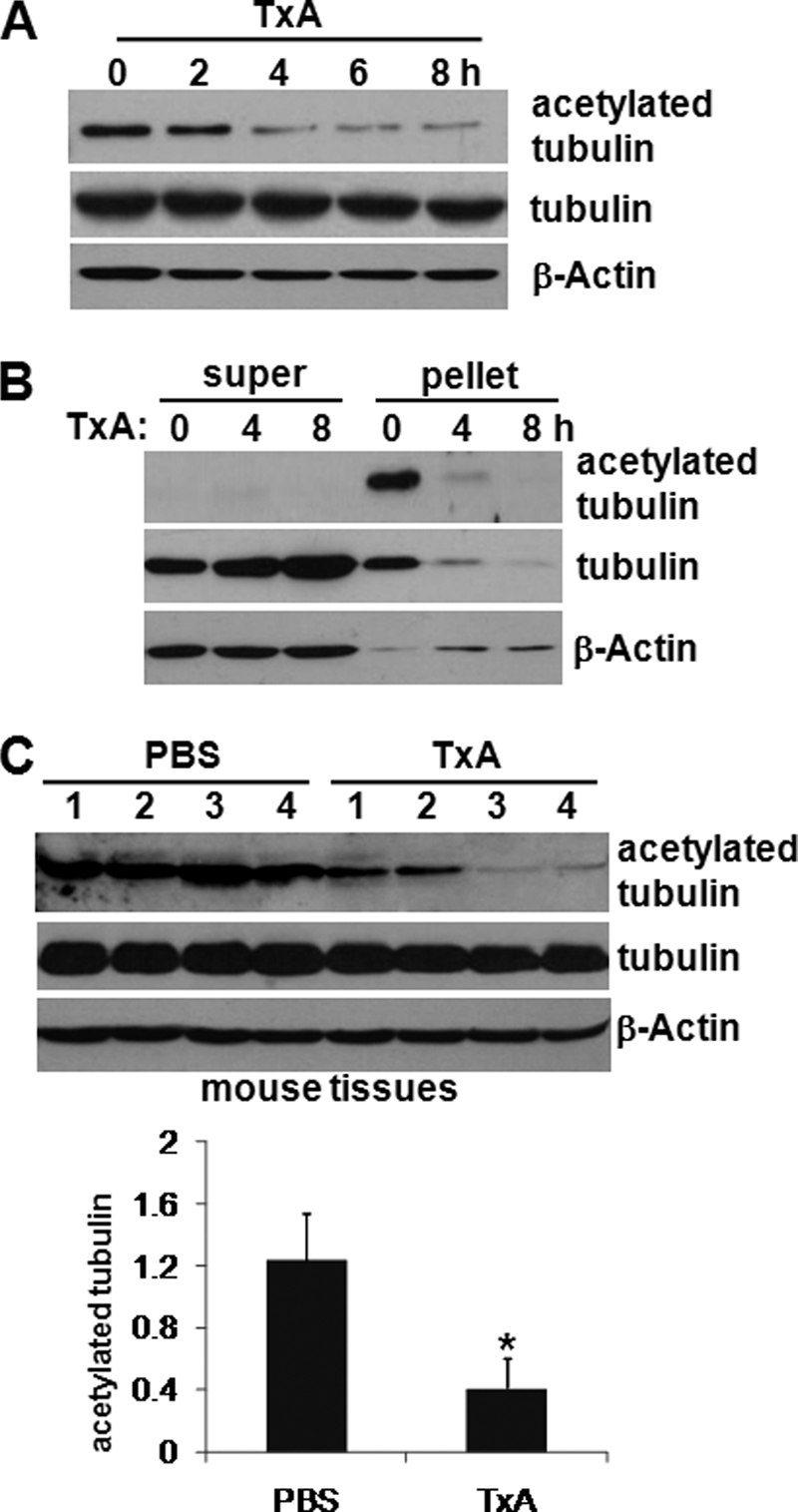
Toxin A inhibits tubulin polymerization through deacetylation in HT29 human colonocytes. A, colonocytes were incubated with toxin A (TxA) (0.5 μg/ml) for the indicated times, cell lysates were subjected to 10% polyacrylamide gel electrophoresis, and blots were probed with antibodies against acetylated α-tubulin, α-tubulin, and β-actin. The presented results are representative of three independent experiments. B, polymerized (polymeric) and depolymerized (monomeric) tubulins were isolated from colonocytes exposed to toxin A for the indicated times (see “Experimental Procedures”). The supernatant (super) was removed as monomeric tubulin, and polymeric tubulin was extracted from the pellet. C, ileal loops of anesthetized CD1 mice (n = 10 mice/group) were closed and injected with toxin A (3 μg of toxin A, 100 μl of PBS) or PBS buffer for 4 h. Ileal loops were removed, and total protein extracts from the tissues were resolved on polyacrylamide gels and probed with an antibody against acetylated α-tubulin (top). Densitometric quantitation was performed (bottom). The results shown are representative of three independent experiments. *, p < 0.05 versus PBS-treated mice. Error bars, S.E.
Depolymerization of Microtubules by Toxin A
We next assessed whether toxin A-induced deacetylation of tubulin resulted in microtubule depolymerization. To do this, we applied immunofluorescence staining with an antibody against acetylated α-tubulin, as described under “Experimental Procedures.” In control human colonocytes, the anti-acetylated α-tubulin antibody clearly revealed typical microtubule shapes in all stained cells (Fig. 2A, arrowheads), whereas the anti-α-tubulin antibody highly detected such shapes in dividing cells that were in the process of forming a mitotic spindle. Exposure of colonocytes to toxin A for 6 h substantially diminished the levels of polymerized microtubules as well as acetylated tubulins compared with that in control cells (Fig. 2B). This finding is consistent with the results in Fig. 1 showing that toxin A treatment caused tubulin deacetylation, leading to increases in monomeric tubulin. This suggests that toxin A, in addition to its role in depolymerizing actin, may also trigger microtubule depolymerization and subsequent cytoskeletal disorganization.
FIGURE 2.

Effects of toxin A-induced tubulin deacetylation on microtubule organization in human colonocytes. A and B, HT29 cells grown on coverslips were fixed and immunostained with anti-acetylated α-tubulin and anti-α-tubulin. Levels of microtubule organization were visualized by confocal microscopy. Microtubules observed in human colonocytes are indicated by arrowheads. The results shown are representative of three independent experiments. B, human colonocytes were exposed to medium (con) or toxin A for 6 h and then immunostained with anti-acetylated α-tubulin or anti-α-tubulin. Marked microtubule disorganization was observed in toxin A (TxA)-treated cells.
Toxin A-induced Deacetylation of Tubulin Is Dependent on Both the Enzymatic Activity of Toxin A and the Involvement of Downstream Signaling Molecules
Toxin A-induced monoglucosylation of Rho family proteins at threonine 37 inactivates the Rho proteins, resulting in actin disaggregation in various cells (7). Because a recent study showed that Rho-GTPase is involved in the increased microtubule instability observed in HIV virus-infected host cells (31), we assessed whether the enzymatic activity of toxin A was required for toxin A-induced tubulin deacetylation. For this purpose, we used UDP-2′,3′-dialdehyde, which has been shown to inhibit the enzymatic activity of toxin A without inducing structural changes (32). HT29 cells were exposed to UDP-2′,3′-dialdehyde for 1 h prior to toxin A exposure, and changes in tubulin acetylation were measured. As shown in Fig. 3A, toxin A markedly reduced tubulin acetylation in control colonocytes, but this effect was significantly blocked in cells pretreated with UDP-2′,3′-dialdehyde, which by itself did not affect the basal levels of tubulin acetylation. Because toxin A has been shown to rapidly cause reactive oxygen species (ROS) generation as an early downstream signal (33), we assessed whether blockage of ROS production could inhibit toxin A-induced tubulin deacetylation. Human colonocytes were exposed for 1 h to NAC, an H2O2 scavenger, or sodium formate, a hydroxyl radical scavenger, followed by toxin A stimulation for 4 h. Interestingly, NAC but not sodium formate specifically inhibited tubulin deacetylation in cells exposed to toxin A (Fig. 3B). Conversely, direct exposure of HT29 cells to H2O2 increased tubulin deacetylation in a time/dose-dependent manner (Fig. 3C); 500 μm H2O2 completely abrogated acetylation of tubulin within 1 h, and this effect was retained through 2 h post-treatment, indicating that H2O2, one of the main ROS produced in toxin A-treated colonocytes (33), was capable of directly decreasing tubulin acetylation. Together, these findings suggest that toxin A-induced tubulin deacetylation requires the enzymatic activity of toxin A against Rho proteins as well as the generation of ROS, leading to signal transduction.
FIGURE 3.
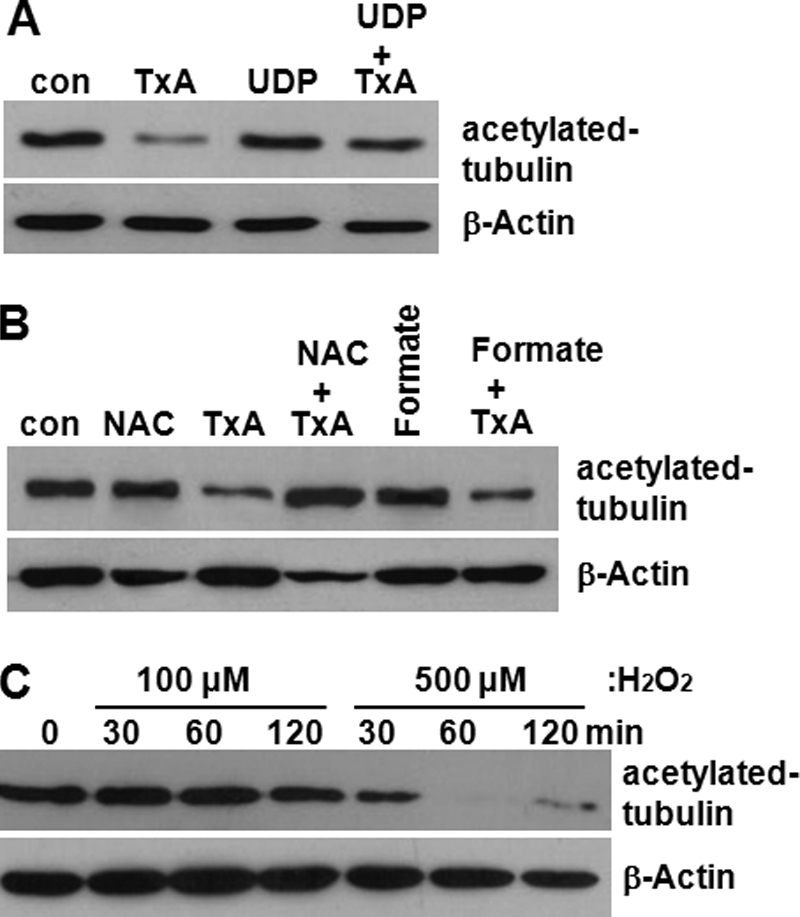
Toxin A enzymatic activity and downstream signaling molecule (H2O2) are involved in toxin A-induced tubulin deacetylation. A, human colonocytes were exposed to medium (con), toxin A alone, UDP-2′,3′-dialdehyde alone (UDP, 1 mm) (32), or UDP-2′,3′-dialdehyde plus toxin A (TxA) for 4 h. The lysates were resolved on 10% polyacrylamide gels and probed with antibodies against acetylated α-tubulin and β-actin. B, colonocytes were exposed to NAC (20 mm) (33), toxin A, toxin A plus NAC, sodium formate (Formate; 1 mm), and sodium formate plus toxin A for 4 h. The results shown are representative of three separate experiments. C, colonocytes were treated with H2O2 (100–500 μm) for the indicated times, and the levels of tubulin deacetylation were measured.
HDAC6 (Histone Deacetylase 6) Mediates Toxin A-induced Tubulin Deacetylation in Colonocytes
Because HDAC6, a unique cytosolic deacetylase, is known to be involved in tubulin deacetylation (18, 19, 24), we examined whether toxin A-mediated tubulin deacetylation is associated with HDAC6 activity in human colonocytes, using the selective HDAC6 inhibitor, TSA (34). As expected, TSA-treated human colonocytes showed increased tubulin acetylation compared with control cells exposed to culture medium (Fig. 4A). The maximum TSA-induced increase in tubulin acetylation was observed at 1 h post-treatment, and this level was maintained through 6 h post-treatment (Fig. 4A). In contrast, sodium butyrate (But) and trapoxin (TPX), broad spectrum chemical inhibitors of all HDACs except HDAC6 (34), did not affect the basal level of tubulin acetylation in human colonocytes (Fig. 4A), suggesting that HDAC6 is the deacetylase for tubulin in human colonocytes. Interestingly, preincubation of colonocytes with TSA for 1 h completely inhibited the deacetylation of tubulin in response to toxin A, whereas sodium butyrate and trapoxin did not affect toxin A-induced tubulin deacetylation (Fig. 4B). Immunostaining and confocal microscopy also revealed that the marked tubulin deacetylation induced by toxin A was blocked in TSA-pretreated cells, whereas sodium butyrate and trapoxin did not inhibit toxin A-induced tubulin deacetylation (Fig. 4C). TSA alone stimulated a marked increase in microtubule polymerization compared with medium control. Using a 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide assay to measure changes on cell viability, we next assessed whether the TSA-induced blockade of tubulin deacetylation could inhibit the marked drop in cell viability observed following toxin A treatment. As shown in Fig. 4D, treatment of TSA but not trapoxin significantly blocked the toxin A-induced reduction of cell viability. TSA alone did not appear to have any effect on colonocyte viability (Fig. 4D). Given that HDAC6 appears to be largely responsible for the increased tubulin deacetylation in colonocytes exposed to toxin A, and HDAC6 is unique among the deacetylases in being restricted to the cytosol, we investigated whether toxin A could activate HDAC6 in colonocytes. Cells were incubated with toxin A or H2O2 for 3 h, and cytosolic extracts taken as representing HDAC6 activity and nuclear extracts taken as representing the activity of the other HDACs were isolated, incubated with an acetylated fluorogenic substrate, and developed for visualization of results. As shown in Fig. 4E, the enzymatic activity of cytosolic HDAC6 was significantly increased by toxin A or H2O2 stimulation, compared with the medium-treated control. In contrast, the basal activity of the nuclear HDACs was relatively higher than that in the cytosol, but toxin A and H2O2 exposure decreased nuclear HDAC activity (Fig. 4E). The purity of subcellular fractionation was confirmed by immunoblot analysis with an β-actin antibody (C, medium; T, toxin A H, H2O2). Immunoprecipitation assay with an antibody against HDAC6 also revealed that activities of HDAC6 isolated from colonocytes exposed to toxin A or H2O2 were markedly increased compared with the medium-treated cells. Moreover, these increases were significantly inhibited by pretreatment of TSA, suggesting that exposure of colonocytes to toxin A results in activation of HDAC6, and this is responsible for tubulin deacetylation and subsequent microtubule disassembly. We next assessed whether blockage of HDAC6 by TSA inhibited the toxin A-induced drop of TER. CaCo-2 cells were treated with TSA for 1 h prior to exposure of toxin A for 3, and TER was measured, starting with an initial value of 300 ohms/cm2. As shown in Fig. 4G, the drop of TER by toxin A was significantly inhibited by TSA. The TER in cells treated with TSA alone was relatively higher than that in cells exposed to control medium. These results also suggest that the tubulin assembly regulated by HDAC6 is also strongly associated with barrier function of epithelial cells.
FIGURE 4.

Toxin A causes tubulin deacetylation by activating HDAC6 in human colonocytes. A, colonocytes (HT29) were treated with medium (con), TSA (1 μm), sodium butyrate (But) (1 mm), or trapoxin (TPX) (10 nm) for the indicated times. Cell lysates were resolved on 10% polyacrylamide gels and probed with antibodies against acetylated α-tubulin and β-actin. The results shown are representative of three separate experiments. B, cells were incubated with medium, TSA, sodium butyrate, trapoxin, toxin A alone (TxA), or toxin A plus either TSA, But, or trapoxin for 4 h. C, HT29 cells were grown on coverslips, and levels of microtubule organization were visualized by confocal microscopy and immunostaining with an antibody against acetylated α-tubulin. D, HT29 cells were incubated with medium (con), toxin A, trapoxin or TSA alone, or toxin A plus either trapoxin or TSA. After incubation for 48 h, cell viability was measured by a 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide assay and expressed as a percentage of the results from untreated controls. All reactions were performed in duplicate, and the results shown represent three independent experiments. *, p < 0.001 versus toxin A-stimulated cells. E, toxin A and H2O2 increase HDAC6 activity in colonocytes. HT29 cells were incubated with medium (con), toxin A (0.5 μg/ml), or H2O2 (500 μm) for 3 h, and nuclear and cytosolic extracts were isolated. The purity of subcellular fractionation was confirmed by immunoblot analysis with an β-actin antibody (C, medium; T, toxin A; H, H2O2). Activity of HDAC6 was measured as described under “Experimental Procedures.” All reactions were performed in duplicate, and the results shown represent three independent experiments. *, p < 0.005 versus non-stimulated cells. F, HT29 cells were pretreated with TSA (1 μm) for 1 h and then exposed to medium (con), toxin A, or H2O2 for 3 h. HDAC6 in colonocytes was immunoprecipitated by an HDAC6 antibody, and activity of HDAC6 in the immune complexes was measured. *, p < 0.005 versus toxin A-treated cells; #, p < 0.001 versus H2O2-treated cells. G, colonocytes (CaCo-2) were treated with TSA (1 μm) for 1 h prior to toxin A exposure. TER was determined as described under “Experimental Procedures.” Typical absolute resistance values were 300–350 ohms/cm2. The bars represent the mean ± S.E. of three experiments performed in triplicate; *, p < 0.001 versus toxin A-stimulated cells. Error bars, S.E.
Microtubule Disorganization in Mouse Epithelium Causes Gut Inflammation
Given our observations that toxin A caused tubulin deacetylation and subsequent microtubule depolymerization and toxin A is known to cause gut inflammation, we speculated that microtubule disorganization in epithelial cells might result in an inflammatory response with barrier dysfunction in the gut. To investigate this possibility, we first tested the effect of colchicine, which inhibits microtubule polymerization (14), and taxol, a microtubule hyperstabilizer (15), on microtubule polymerization in human colonocytes. In HT29 cells, treatment of colchicine (5 μm) triggered tubulin deacetylation (Fig. 5A, top) and caused marked depolymerization of microtubules (Fig. 5A, bottom). However, taxol did not increase deacetylation of tubulin and hyperstabilized the microtubules in contrast to colchicine (data not shown).
FIGURE 5.

Disorganization of microtubules in epithelial cells and its association with inflammation. A, HT29 cells were treated with medium (con), DMSO, or colchicine (5 μm) for 2 h, and deacetylation of tubulin was examined (top). Changes in microtubule organization were visualized by immunostaining with an antibody against acetylated α-tubulin, followed by confocal microscopy (bottom). The results shown are representative of three independent experiments. B, colchicine causes a significant drop in the TER of CaCo2 cells. The bars represent the mean ± S.E. (error bars) of three experiments performed in triplicate; *, p < 0.001 versus non-stimulated cells. C–E, ileal loops of anesthetized CD1 mice (n = 8 mice/group) were injected with PBS or colchicine (1 μg of colchicine, 100 μl of PBS) for 4 h. C, tissue extracts obtained from ileal loops were resolved on polyacrylamide gels and probed with an antibody against acetylated α-tubulin. D, mucosal damage was observed in colchicine-treated mice. Treatment with colchicine was associated with abnormally shaped epithelial cells in the ileal tissues of treated mice (arrowheads). E, the concentration of IL-6 was measured from ileal loops exposed to PBS, colchicine, or toxin A (TxA). The bars represent the mean ± S.E. of three independent experiments, each with triplicate determinations. *, p < 0.005 versus PBS-treated mice. Error bars, S.E.
Because changes in microtubule organization triggered loss of epithelial barrier function as well as cell morphology changes (35), we next assessed whether colchicine, an inhibitor of microtubule polymerization, caused barrier dysfunction of colonic epithelial cells leading to inflammatory response in the gut. CaCo-2 cells were grown on semipermeable filters and exposed to colchicine for 8 h, and TER was measured. As shown in Fig. 5B, colchicine treatment resulted in a marked decrease in TER, suggesting that microtubule disassembly is strongly associated with barrier dysfunction of colonocytes.
We then tested the effect of colchicine in ileal loops of intact mice. CD1 mice were anesthetized, and PBS or colchicine (5 μm) was injected into the lumen of ileal loops. After 4 h, mucosal tissue extracts were prepared by scraping the mucosa, and the levels of acetylated tubulin were measured by immunoblot analysis. Consistent with the results observed in cultured colonocytes, colchicine-treated mice experienced significant tubulin deacetylation compared with PBS-treated mice (Fig. 5C). H&E staining of intestinal sections revealed that colchicine caused marked mucosal damage (Fig. 5D), as evidenced by abnormally shaped cells and shrinkage of epithelial cell nuclei in colchicine-treated mice (Fig. 5D, arrowheads). To evaluate inflammation in the gut, we next measured the mucosal concentrations of IL-6, a well characterized proinflammatory cytokine, in toxin A-exposed mouse ileum (36). Colchicine significantly increased IL-6 production compared with the PBS control, and the level was similar to that seen in toxin A-administrated mouse ileal loops (Fig. 5E). These results suggest that microtubule disorganization induced by colchicine like toxin A may trigger gut inflammation and mucosal damage.
TSA-induced Blockage of Tubulin Deacetylation Attenuates Inflammation and Enterocyte Apoptosis in a Toxin A-induced Mouse Enteritis Model
Finally, we investigated the possible inhibitory effect of the HDAC6 inhibitor, TSA, on toxin A-induced mucosal damage and acute gut inflammation in mice. We first determined the minimal dose of TSA capable of having a therapeutic effect on the mouse gut epithelium without evidence of cellular toxicity. Closed ileal loops of CD1 mice were exposed to different doses of TSA for 4 h, and the levels of tubulin deacetylation and TNF-α expression, a marker of acute inflammation in toxin A-induced mouse enteritis (37), were measured from tissue extracts. As shown in Fig. 6A, treatment with 100 or 1,000 nm TSA increased TNF-α and tubulin acetylation in the mouse epithelium. In contrast, 10 nm TSA increased tubulin acetylation but did not increase TNF-α levels. Therefore, 10 nm TSA was used in the subsequent experiments. For those experiments, closed ileal loops of CD1 mice were treated with PBS (control), TSA alone, toxin A alone, or TSA plus toxin A for 4 h, and then the levels of tubulin deacetylation were measured. As expected, treatment with TSA alone increased tubulin acetylation compared with the PBS control, and treatment with toxin A alone decreased tubulin acetylation compared with the PBS control (Fig. 6B). However, the toxin A-induced tubulin deacetylation was significantly blocked by TSA co-treatment (Fig. 6B). Similarly, toxin A-treated mice showed increased TNF-α and mucosal damage, but these effects were abolished by TSA co-treatment (Fig. 6, C and D). Because we previously reported that toxin A-induced colonocyte apoptosis in cultured cells and an animal colitis model was a critical step in toxin A-induced mucosal damage and inflammation (32, 33, 36), we measured caspase-3 activation in tissue extracts. We found that toxin A increased caspase-3 activation, but this effect was significantly decreased by TSA co-treatment (Fig. 6E).
FIGURE 6.

TSA-induced HDAC6 inhibition reduces toxin A (TxA)-induced inflammatory responses and destruction of villi in the mouse ileum. A, to determine a TSA concentration suitable for examination of cellular responses without inflammation or mucosal damage, we prepared closed ileal loops in the distal ilea of CD1 mice (n = 5 mice/group) and injected them with different concentrations of TSA in 100 μl of PBS. Tubulin deacetylation and TNF-α concentrations were measured in the ileal tissues. The bars represent the mean ± S.E. (n = 5 mice/group, each with triplicate determinations); *, p < 0.005 versus PBS-treated mice. B–E, mouse ileal loops were injected with 100 μl of PBS alone or PBS containing TSA (10 nm), toxin A (3 μg of toxin A, 100 μl of PBS), or toxin A plus TSA. After 4 h, animals were sacrificed, and ileal loops were removed and processed. B, tissue lysates were resolved on 10% polyacrylamide gels and probed with antibodies against acetylated α-tubulin and β-actin. The results shown are representative of three separate mouse experiments. C, concentration of TNF-α in the ileal tissues. Data are expressed as mean ± S.E. (n = 5 mice/group, each with triplicate determinations); *, p < 0.005 versus toxin A-injected mice. D, light micrographs of mouse ileum (H&E staining; original magnification, ×200). E, the extracts from ileal tissues were resolved on 15% polyacrylamide gels and probed with antibodies against caspase-3 or β-actin. Data are representative of three independent samples. Error bars, S.E.
DISCUSSION
C. difficile toxin A is known to cause inactivation of Rho family proteins, leading to actin disaggregation in cultured cells (7). Toxin A triggers monoglucosylation of Rho, Rac, and Cdc42 at threonine 37, rendering these factors unable to participate in the formation of actin filaments (7, 38–40). Toxin A-treated cells typically become rounded due to abnormalities in the cytoskeletal components (2, 41–43). Although this has been largely attributed to inactivation of Rho family proteins and subsequent actin disaggregation, we postulated that toxin A could also affect post-translational modification of tubulin and microtubule instability. We report here that toxin A treatment of human colonocytes caused tubulin deacetylation and subsequent microtubule depolymerization. Moreover, we identified HDAC6, the only known cytosolic tubulin deacetylase (19), as a major mediator of toxin A-induced tubulin deacetylation and subsequent microtubule depolymerization in human colonocytes. Post-translational modification of tubulin has been reported to regulate microtubule formation, which is a major component of cytoskeletal maintenance and alteration (9–11). As such, our finding that toxin A induces tubulin deacetylation in colonocytes provides new insight into the molecular mechanism responsible for the rapid and severe cell rounding seen in toxin A-treated cells.
The small Rho-GTPase proteins, Rho (44), Rac (45), and Cdc42 (46), have been reported to regulate microtubule dynamics in addition to their well characterized roles in forming the actin cytoskeleton. For example, Rac1 regulates microtubule dynamics to promote the formation of stable microtubules in migrating cells (45), and RhoA has been reported to stabilize microtubules (44). Given that Rho family proteins are associated with microtubule dynamics, and toxin A has been shown to inactivate Rho family proteins through glucosylation at Thr37, we speculated that toxin A-induced tubulin deacetylation in colonocytes might be associated with inactivation of Rho family proteins. Interestingly, UDP-2′,3′-dialdehyde-induced blockage of toxin A enzymatic activity did not completely inhibit toxin A-induced tubulin deacetylation (Fig. 3A). In addition, preincubation of cells with NAC, which scavenges H2O2, one of the main ROS produced by toxin A treatment of colonocytes (33) significantly blocked tubulin deacetylation in toxin A-treated cells (Fig. 3B), whereas exposure of human colonocytes to H2O2 significantly increased tubulin deacetylation. Considering that toxin A caused production of H2O2 in human colonocytes (33), these results suggest that abnormal tubulin deacetylation in toxin A-treated colonocytes was mainly dependent on the enzymatic activity of toxin A but may also involve toxin A-induced signaling molecules, such as H2O2. Based on the previous report that exposure of hepatocytes to H2O2 activated HDAC family proteins (47), H2O2 may be an essential molecule for activation of HDAC6 in human colonocytes exposed to toxin A.
Microtubule instability has been shown to increase during viral infection. For example, internalization of the human immunodeficiency virus (HIV) is regulated by HDAC6; overexpression of active HDAC6 prevents HIV infection through tubulin deacetylation (48). Therefore, it is conceivable that microtubule depolymerization and HDAC6 activation in toxin A-treated human colonocytes may mediate internalization of toxin A after receptor binding. However, based on our previous finding that internalization of toxin A in human colonocytes (NCM460 cells) occurred within 5 min and that 1-h exposure of toxin A caused complete glucosylation (inactivation) of Rho family proteins (28), the tubulin deacetylation noted herein after 4 h of toxin A exposure is not likely to be associated with the internalization of toxin A.
In colonic epithelial cells involved in maintaining the barrier function, cytoskeletal disruption may damage tight junctions and cause failure of focal contact formation, possibly leading to large scale exposure of luminal pathogens to immune cells in lamina propria (32, 36). In this context, toxin A-induced microtubule depolymerization may function along with actin disruption to synergistically contribute to cell rounding and mucosal damage. As shown in Fig. 4, less rounding and better viability were observed in colonocytes exposed to toxin A plus TSA compared with cells exposed to toxin A alone, suggesting that blockage of microtubule depolymerization via TSA-mediated inhibition of HDAC6 abolished these responses. In our toxin A-induced mouse enteritis model, blockage of TSA-induced tubulin deacetylation significantly attenuated toxin A-induced mucosal damage and TNF-α production. Moreover, the increased mucosal cell apoptosis observed in toxin A-treated mice was substantially reduced in mice co-treated with toxin A and TSA. Although actin disaggregation has traditionally been considered the most important outcome of toxin A-induced Rho protein inactivation (7), our findings indicate that toxin A-induced tubulin deacetylation is also a critical step for toxin A-induced cytoskeletal disruption, mucosal damage, and inflammation. Our results further suggest three other important concepts. First, toxin A-induced cytoskeletal disruption in epithelial cells may increase the chance of luminal pathogens being constitutively exposed to the immune system through barrier dysfunction. Second, HDAC6-mediated tubulin acetylation and deacetylation appears to be critical for maintenance of dynamic microtubule instability in the gut epithelium. Third, toxin A-induced HDAC6 activation causes abnormal tubulin deacetylation and subsequent microtubule depolymerization in epithelial cells, which in turn is associated with mucosal damage and inflammation in the gut.
We also assessed whether increased instability of microtubules in mouse colonic epithelial cells could cause inflammatory responses and mucosal damage. Colchicine, a well known inhibitor of microtubule polymerization (14), was used to confirm the relationship between cytoskeletal disruption and gut inflammation. In cultured human colonocytes, colchicine caused marked microtubule depolymerization. In the mouse ileal epithelium, colchicine strongly increased tubulin deacetylation, IL-6 secretion, and mucosal damage. These results suggest that in gut epithelial cells, microtubule disorganization in addition to actin disaggregation by toxin A could be a main trigger of proinflammatory responses. We speculate that this may be due to the initiation of barrier dysfunction followed by exposure of immune cells in the lamina propria to luminal bacteria and their products, which subsequently triggers severe inflammation.
In summary, we report that toxin A, in addition to its well described ability to trigger actin disaggregation, also causes tubulin deacetylation and microtubule depolymerization in human colonocytes. This appears to occur through the action of HDAC6, the only known cytosolic histone deacetylase. The HDAC6 inhibitor, TSA, inhibits toxin A-induced tubulin deacetylation in colonocytes and significantly attenuates proinflammatory cytokine production, mucosal damage, and epithelial cell apoptosis in a toxin A-induced mouse enteritis model. These novel findings provide important new insights into toxin A-induced cytoskeletal disorganization. In addition, our results suggest that blocking HDAC6 with TSA may be a potential therapeutic approach for ameliorating toxin A-induced acute inflammation in the gut.
This work was supported by Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea, Grant A080933.
- NAC
- N-acetyl-l-cysteine
- TSA
- trichostatin A
- TER
- transepithelial resistance
- ROS
- reactive oxygen species.
REFERENCES
- 1.Kelly C. P., LaMont J. T. (2008) N. Engl. J. Med. 359, 1932–1940 [DOI] [PubMed] [Google Scholar]
- 2.Pothoulakis C., LaMont J. T. (1993) Gastroenterol. Clin. North Am. 22, 623–637 [PubMed] [Google Scholar]
- 3.Lamont J. T. (2002) Trans. Am Clin. Climatol. Assoc. 113, 167–180 [PMC free article] [PubMed] [Google Scholar]
- 4.Castagliuolo I., Kelly C. P., Qiu B. S., Nikulasson S. T., LaMont J. T., Pothoulakis C. (1997) Am. J. Physiol. 273, G333–G341 [DOI] [PubMed] [Google Scholar]
- 5.von Eichel-Streiber C., Laufenberg-Feldmann R., Sartingen S., Schulze J., Sauerborn M. (1990) Med. Microbiol. Immunol. 179, 271–279 [DOI] [PubMed] [Google Scholar]
- 6.Riegler M., Sedivy R., Pothoulakis C., Hamilton G., Zacherl J., Bischof G., Cosentini E., Feil W., Schiessel R., LaMont J. T. (1995) J. Clin. Invest. 95, 2004–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Just I., Selzer J., Wilm M., von Eichel-Streiber C., Mann M., Aktories K. (1995) Nature 375, 500–503 [DOI] [PubMed] [Google Scholar]
- 8.Hecht G., Pothoulakis C., LaMont J. T., Madara J. L. (1988) J. Clin. Invest. 82, 1516–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popova J. S., Rasenick M. M. (2003) J. Biol. Chem. 278, 34299–34308 [DOI] [PubMed] [Google Scholar]
- 10.Gao J., Huo L., Sun X., Liu M., Li D., Dong J. T., Zhou J. (2008) J. Biol. Chem. 283, 8802–8809 [DOI] [PubMed] [Google Scholar]
- 11.Bicek A. D., Tüzel E., Demtchouk A., Uppalapati M., Hancock W. O., Kroll D. M., Odde D. J. (2009) Mol. Biol. Cell 20, 2943–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez N., Chang Q., Buster D. W., Sharp D. J., Ma A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 7846–7851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J. G., Horwitz S. B. (2002) Cancer Res. 62, 1935–1938 [PubMed] [Google Scholar]
- 14.Han Y., Malak H., Chaudhary A. G., Chordia M. D., Kingston D. G., Bane S. (1998) Biochemistry 37, 6636–6644 [DOI] [PubMed] [Google Scholar]
- 15.Verdier-Pinard P., Wang Z., Mohanakrishnan A. K., Cushman M., Hamel E. (2000) Mol. Pharmacol. 57, 568–575 [DOI] [PubMed] [Google Scholar]
- 16.Mejillano M. R., Himes R. H. (1991) J. Biol. Chem. 266, 657–664 [PubMed] [Google Scholar]
- 17.North B. J., Marshall B. L., Borra M. T., Denu J. M., Verdin E. (2003) Mol. Cell 11, 437–444 [DOI] [PubMed] [Google Scholar]
- 18.Haggarty S. J., Koeller K. M., Wong J. C., Grozinger C. M., Schreiber S. L. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 4389–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., Yao T. P. (2002) Nature 417, 455–458 [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y., Li N., Caron C., Matthias G., Hess D., Khochbin S., Matthias P. (2003) EMBO J. 22, 1168–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin R. J., Nagy L., Inoue S., Shao W., Miller W. H., Jr., Evans R. M. (1998) Nature 391, 811–814 [DOI] [PubMed] [Google Scholar]
- 22.McKinsey T. A., Zhang C. L., Lu J., Olson E. N. (2000) Nature 408, 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karvonen U., Jänne O. A., Palvimo J. J. (2006) Exp. Cell Res. 312, 3165–3183 [DOI] [PubMed] [Google Scholar]
- 24.Kwon S., Zhang Y., Matthias P. (2007) Genes. Dev. 21, 3381–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X., Yuan Z., Zhang Y., Yong S., Salas-Burgos A., Koomen J., Olashaw N., Parsons J. T., Yang X. J., Dent S. R., Yao T. P., Lane W. S., Seto E. (2007) Mol. Cell 27, 197–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawaguchi Y., Kovacs J. J., McLaurin A., Vance J. M., Ito A., Yao T. P. (2003) Cell 115, 727–738 [DOI] [PubMed] [Google Scholar]
- 27.Warny M., Keates A. C., Keates S., Castagliuolo I., Zacks J. K., Aboudola S., Qamar A., Pothoulakis C., LaMont J. T., Kelly C. P. (2000) J. Clin. Invest. 105, 1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim H., Rhee S. H., Pothoulakis C., LaMont J. T. (2009) Exp. Cell Res. 315, 3336–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arregui L., Muñoz-Fontela C., Serrano S., Barasoain I., Guinea A. (2002) J. Eukaryot. Microbiol. 49, 312–318 [DOI] [PubMed] [Google Scholar]
- 30.Chang J. S., Kim S. K., Kwon T. K., Bae S. S., Min D. S., Lee Y. H., Kim S. O., Seo J. K., Choi J. H., Suh P. G. (2005) J. Biol. Chem. 280, 6897–6905 [DOI] [PubMed] [Google Scholar]
- 31.Naranatt P. P., Krishnan H. H., Smith M. S., Chandran B. (2005) J. Virol. 79, 1191–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim H., Kokkotou E., Na X., Rhee S. H., Moyer M. P., Pothoulakis C., Lamont J. T. (2005) Gastroenterology 129, 1875–1888 [DOI] [PubMed] [Google Scholar]
- 33.Kim H., Rhee S. H., Kokkotou E., Na X., Savidge T., Moyer M. P., Pothoulakis C., LaMont J. T. (2005) J. Biol. Chem. 280, 21237–21245 [DOI] [PubMed] [Google Scholar]
- 34.Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D., Osada H., Komatsu Y., Nishino N., Khochbin S., Horinouchi S., Yoshida M. (2002) EMBO J. 21, 6820–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eiserich J. P., Estévez A. G., Bamberg T. V., Ye Y. Z., Chumley P. H., Beckman J. S., Freeman B. A. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 6365–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim H., Rhee S. H., Pothoulakis C., Lamont J. T. (2007) Gastroenterology 133, 875–886 [DOI] [PubMed] [Google Scholar]
- 37.Castagliuolo I., Riegler M., Pasha A., Nikulasson S., Lu B., Gerard C., Gerard N. P., Pothoulakis C. (1998) J. Clin. Invest. 101, 1547–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Just I., Fritz G., Aktories K., Giry M., Popoff M. R., Boquet P., Hegenbarth S., von Eichel-Streiber C. (1994) J. Biol. Chem. 269, 10706–10712 [PubMed] [Google Scholar]
- 39.Just I., Selzer J., von Eichel-Streiber C., Aktories K. (1995) J. Clin. Invest. 95, 1026–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Just I., Wilm M., Selzer J., Rex G., von Eichel-Streiber C., Mann M., Aktories K. (1995) J. Biol. Chem. 270, 13932–13936 [DOI] [PubMed] [Google Scholar]
- 41.Pothoulakis C., Sullivan R., Melnick D. A., Triadafilopoulos G., Gadenne A. S., Meshulam T., LaMont J. T. (1988) J. Clin. Invest. 81, 1741–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pothoulakis C., Lamont J. T. (2001) Am. J. Physiol. Gastrointest. Liver Physiol. 280, G178–G183 [DOI] [PubMed] [Google Scholar]
- 43.Pothoulakis C., Gilbert R. J., Cladaras C., Castagliuolo I., Semenza G., Hitti Y., Montcrief J. S., Linevsky J., Kelly C. P., Nikulasson S., Desai H. P., Wilkins T. D., LaMont J. T. (1996) J. Clin. Invest. 98, 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogers S. L., Wiedemann U., Häcker U., Turck C., Vale R. D. (2004) Curr. Biol. 14, 1827–1833 [DOI] [PubMed] [Google Scholar]
- 45.Malorni W., Quaranta M. G., Straface E., Falzano L., Fabbri A., Viora M., Fiorentini C. (2003) J. Immunol. 171, 4195–4202 [DOI] [PubMed] [Google Scholar]
- 46.Banerjee P. P., Pandey R., Zheng R., Suhoski M. M., Monaco-Shawver L., Orange J. S. (2007) J. Exp. Med. 204, 2305–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miura K., Taura K., Kodama Y., Schnabl B., Brenner D. A. (2008) Hepatology 48, 1420–1429 [DOI] [PubMed] [Google Scholar]
- 48.Valenzuela-Fernández A., Alvarez S., Gordon-Alonso M., Barrero M., Ursa A., Cabrero J. R., Fernández G., Naranjo-Suárez S., Yáñez-Mo M., Serrador J. M., Muñoz-Fernández M. A., Sánchez-Madrid F. (2005) Mol. Biol. Cell 16, 5445–5454 [DOI] [PMC free article] [PubMed] [Google Scholar]