

Abstract
Background
Liver transplantation for type IV glycogen storage disease (branching-enzyme deficiency) results in the resorption of extrahepatic deposits of amylopectin, but the mechanism of resorption is not known.
Methods
We studied two patients with type IV glycogen storage disease 37 and 91 months after liver transplantation and a third patient with lysosomal glucocerebrosidase deficiency (type 1 Gaucher’s disease), in whom tissue glucocerebroside deposition had decreased 26 months after liver replacement, to determine whether the migration of cells from the allograft (microchimerism) could explain the improved metabolism of enzyme-deficient tissues in the recipient. Samples of blood and biopsy specimens of the skin, lymph nodes, heart, bone marrow, or intestine were examined immunocytochemically with the use of donor-specific monoclonal anti-HLA antibodies and the polymerase chain reaction, with preliminary amplification specific to donor alleles of the gene for the beta chain of HLA-DR molecules, followed by hybridization with allele-specific oligonucleotide probes.
Results
Histopathological examination revealed that the cardiac deposits of amylopectin in the patients with glycogen storage disease and the lymph-node deposits of glucocerebroside in the patient with Gaucher’s disease were dramatically reduced after transplantation. Immunocytochemical analysis showed cells containing the HLA phenotypes of the donor in the heart and skin of the patients with glycogen storage disease and in the lymph nodes, but not the skin, of the patient with Gaucher’s disease. Polymerase-chain-reaction analysis demonstrated donor HLA-DR DNA in the heart of both patients with glycogen storage disease, in the skin of one of them, and in the skin, intestine, blood, and bone marrow of the patient with Gaucher’s disease.
Conclusions
Systemic microchimerism occurs after liver allotransplantation and can ameliorate pancellular enzyme deficiencies.
In patients with type IV glycogen storage disease, deficiency of the branching enzyme alpha-1,4-glucan:alpha-1,4-glucan 6-glucosyltransferase is responsible for the accumulation in the liver and elsewhere of an insoluble and irritating amylopectin-like polysaccharide.1 We recently described the absorption of this amylopectin from the extrahepatic tissues after liver transplantation,2 leading Howell to predict that an explanation of the benefit would “clearly teach us a great deal about transplantation.”3 That prediction has been shown to be accurate by our observation in this study that patients with type IV glycogen storage disease in whom liver transplantation was successful became chimeras: the cells of the host organs became mixed with cells of the donor genome that had migrated from the allograft into the tissues of the recipient and apparently served as enzyme carriers. We also found evidence of similar chimerism and consequent metabolic benefits after liver transplantation in a patient who had type 1 Gaucher’s disease, a disorder caused by a deficiency of the lysosomal enzyme beta-glucocerebrosidase.4
Case Reports
Patients 1 and 2 (Type IV Glycogen Storage Disease)
In April 1992, biopsy specimens of liver, endomyocardium, and skin were obtained from Patients 1 and 2, two brothers with type IV glycogen storage disease who had undergone liver replacement for hepatic failure 91 and 37 months earlier.2 The patients were 31 and 20 months old at the time of transplantation and were subsequently treated with cyclosporine and prednisone. Their physical and intellectual development has been steady since the operation.
Patient 3 (Type 1 Gaucher’s Disease)
The diagnosis of type 1 Gaucher’s disease in Patient 3 (who was 20 years old in 1992) was proved by biopsy of a lytic lesion on the femoral neck of his left leg at the age of 4 years, and was confirmed by study of his spleen, which was removed at the age of 6. From 1979 to 1982 he was treated every six to eight weeks with unmodified human placental glucocerebrosidase (8 units per kilogram of body weight intravenously) at the National Institutes of Health. From November 1989 to January 1990 he was treated every two weeks with macrophage-targeted human placental glucocerebrosidase (30 units per kilogram intravenously; Ceredase, Genzyme, Cambridge, Mass.) There was no evidence of benefit during either period of enzyme treatment.
Because of hepatic failure and recurrent bleeding from esophageal varices, the patient underwent orthotopic liver transplantation in April 1990. Thereafter, he was treated with FK 506 and prednisone. His liver function has been normal since then, and his severe preexisting muscle weakness has steadily improved. In July 1990, three months after liver transplantation, enzyme treatment was resumed. In June 1992, 26 months after liver replacement, biopsy specimens were obtained from the allograft, skin, lymph node, jejunum (by endoscopy), and iliac-crest bone marrow.
Methods
The transplantation and biopsy procedures were considered essential for optimal care of all three patients and were performed with their parents’ consent. Tissues were fixed in 10 percent formalin for conventional staining and frozen in Optimum Cold Temperature medium (OCT, Tissue-Tek, Ames Division, Miles Laboratories, Elkhart, Ind.) for immunocytochemical evaluation or in liquid nitrogen for DNA typing. Gaucher’s cells and amylopectin inclusions were examined by light microscopy after staining with periodic acid–Schiff stain (PAS) with and without diastase digestion (PAS-D).
Immunocytochemical Evaluation
The HLA types of the three donors were determined at the time of organ procurement (September 1984 in the donor for Patient I, March 1989 in the donor for Patient 2, and April 1990 in the donor for Patient 3). The HLA types of the recipients were determined just before transplantation. After investigators established which class I and class II antigens were present in the donor but not in the recipient, monoclonal antibodies we re chosen from a library (whose sources were American Tissue and Cell Culture, Genetic Synterus, Seattle, and One Lambda, Canoga Park, Calif.) and used for phenotyping of the biopsy specimens by indirect immunofluorescence and immunoperoxidase (avidin–biotin complex) methods. The liver removed at the time of transplantation was used as a negative control for donor-specific antibodies; as an additional negative-control measure, staining was carried out with irrelevant anti-HLA antibodies and omission of the primary antibody. Livers with known HLA phenotypes stained with matching anti-HLA monoclonal antibodies were used as positive controls.
Polymerase Chain Reaction
Molecular typing of the beta chain of HLA-DR molecules was performed with use of the polymerase chain reaction, according to the protocol of the 11th International Histocompatibility Workshop,3 in which preliminary amplification of the HLA-DR beta-chain gene is performed before allele-specific amplification.
Results
Histologic Studies
Patients with Glycogen Storage Disease
In the patients with glycogen storage disease neither of the liver allografts had evidence of rejection or amylopectin deposition. At the time of our previous report on these patients,2 the area of heart tissue replaced by amylopectin in the younger brother (Patient 2) had decreased from 13 to 6 percent 14 months after transplantation. A further reduction to less than 1 percent occurred during the ensuing 22 months (Fig. 1). The heart of the older brother (Patient 1) was almost free of amylopectin when the first biopsy was performed, more than 5 years after transplantation2; it appeared the same 91 months after transplantation.
Figure 1. Endomyocardial Tissue from Patient 2, with Type IV Glycogen Storage Disease.

Panel A, a photomicrograph of a specimen obtained at biopsy in April 1989, three weeks after liver transplantation, reveals diffuse amylopectin deposition (dark red) in the myocytes (PAS-D with formalin fixation, x250). In Panel B, a specimen obtained in April 1992, there are only traces of amylopectin (arrow) (PAS-D with formalin fixation of frozen tissue, x250).
Patient with Gaucher’s Disease
A hilar lymph node removed with the native liver from the patient with Gaucher’s disease in April 1990 contained numerous glucocerebroside-filled cells that profoundly distorted the nodal architecture. An inguinal lymph node removed 26 months later contained only a few of these cells, located predominantly in the medullary sinuses (Fig. 2). A biopsy of the allograft performed 26 months after transplantation showed that the architecture of the graft was intact, with no evidence of rejection and only a few Gaucher’s cells scattered in the sinusoids. Specimens of the skin and small intestine were normal, and the bone marrow contained only very few Gaucher’s cells.
Figure 2. Sections of Lymph Nodes from Patient 3, with Type 1 Gaucher’s Disease.

Before transplantation (Panel A), a hepatic hilar lymph node resected with the diseased liver in April 1990 shows marked architectural distortion because of massive deposition of glucocerebroside in Gaucher’s cells (PAS, x50). After transplantation (Panel B), an inguinal lymph node resected in 1992 for analysis of chimerism shows restoration of the architecture, with occasional Gaucher’s cells in the sinusoids (PAS, x50).
Immunocytochemical Studies
The endothelium and bile ducts of all three patients retained the phenotypes of the donors, whereas most of the Kupffer cells of the liver were replaced by those of the recipient, as previously described.6–8
Cells of the donor phenotype were found in the hearts of both patients with glycogen storage disease (Table 1). These cells constituted a minority of the interstitial cardiac cells and were seen at a density of 8 to 10 cells per square millimeter in ultrathin sections (2 μm) (Fig. 3). The vascular endothelial cells of the heart were exclusively of the phenotype of the recipient. The skin of both patients contained donor cells interspersed among the cells of the basal layer of epidermis and in the periadventitial tissue of dermal blood vessels. All epidermal and vascular endothelial cells had the recipient’s phenotype. Cells with the donor’s phenotype could be found in the lymph nodes but not the skin of the patient with Gaucher’s disease (Table 1).
Table 1.
Features of Chimerism Demonstrated by Immunocytochemical Evaluation in Recipients of Liver Grafts for Type IV Glycogen Storage Disease (Patients 1 and 2) and Type 1 Gaucher’s Disease (Patient 3).
| Feature | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Reactivity for anti–donor HLA antibody* | |||
| No. positive/no. tested | 2/5 | 2/5 | 2/2 |
| Site | |||
| Native liver | − | − | NT |
| Allograft | +++ | +++ | ++ |
| Skin | +/rare | +/rare | − |
| Heart | + | + | NT |
| Lymph node | NT | NT | + |
| HLA specificities | |||
| Recipient | |||
| A | 31, 32 | 2, 31 | 2, − |
| B | 50, 61 | 50, 41 | 44, 50 |
| Bw | 6 | 6 | 4, 6 |
| DR | 7, 11 | 2, 7 | 2, 7 |
| Donor† | |||
| A | 2, 26 | 1, − | 3, 31 |
| B | 51, 27 | 7, 8 | 39, 35 |
| Bw | 4 | 6 | |
| DR‡ | 1, 6(13) | 6(13), 6(14) | 1, 11 |
Reactivity ranged from “rare” to “+++”; a minus sign denotes no reaction. NT denotes not tested.
Alleles in boldface type are those recognized by monoclonal antibodies.
The numbers in parentheses are the antigen subspecificities (splits) of the broader specificities.
Figure 3. Immunocytochemical Analysis of Heart, Transplanted Liver, and Native Liver in Patient 1, with Type IV Glycogen Storage Disease.
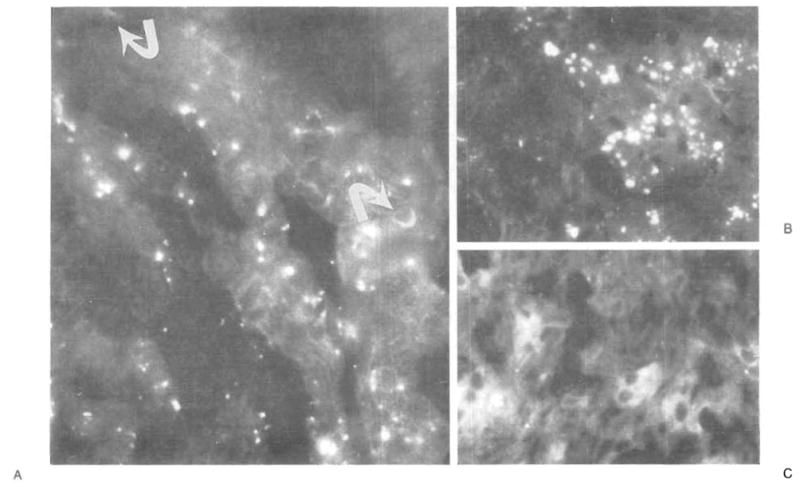
In Panel A, green fluorescence identifies cells with the donor’s phenotype (HLA-DR1,4; arrows) in the interstitium of the heart (x1000). Panel B shows positive green-staining cells (HLA-DR1,4) in the transplanted liver (x400). Panel C (negative control) shows the absence of donor (HLA-DR1,4–positive) cells in the recipient’s native liver (x400). All sections were stained with a single monoclonal antibody specific for HLA-DR1 and DR4. The yellow globules are autofluorescent intracellular pigment.
Polymerase Chain Reaction
Chimerism was observed in the hearts of both patients with glycogen storage disease and in the skin of one of them on polymerase-chain-reaction analysis (Table 2). The HLA type of the patient with Gaucher’s disease was DR2, 7, and that of the donor was DR1,11 (Table 1). When HLA-DR1–specific primers and probes were used (Fig. 4), the donor’s DNA was found in the blood, bone marrow, skin, and small bowel of the recipient (Table 2).
Table 2.
Distribution of Donor HLA-DR Alleles in the Three Liver Recipients, According to Polymerase-Chain-Reaction Analysis.
| Patient No. | Donor-Specific Allele | Distribution* | |||||
|---|---|---|---|---|---|---|---|
| LIVER | SKIN | HEART | BLOOD | MARROW | SMALL BOWEL | ||
| 1 | DR1 | + | + | + | − | NT | NT |
| 2 | DR6 | + | − | + | − | NT | NT |
| 3 | DR1 | + | + | NT | + | + | + |
The liver tissue was from the donors; all other tissues were from the recipients. A plus sign denotes the presence of a donor-specific allele, and a minus sign its absence. NT denotes not tested.
Figure 4. Demonstration of Donor DNA after Transplantation in Patient 3, with Type 1 Gaucher’s Disease.
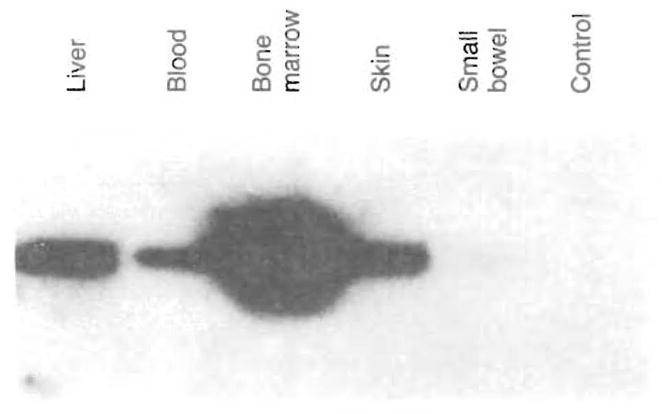
Genomic DNA was extracted from the recipient’s tissues and amplified with HLA-DR beta-chain “generic” oligonucleotide primers to determine the subgroup of the donor’s alleles. “Specific” primers were then used to amplify the alleles selectively. The alleles were identified by hybridizing the amplified DNA to radiolabeled allele-specific probes. After HLA-DR1–specific amplification of DNA from the liver, blood, bone marrow, skin, and small bowel, the DNA was separated by electrophoresis on an agarose gel and then analyzed by Southern blotting. The denatured DNA present on the nylon membrane was hybridized to a labeled HLA-DR1 (donor)–specific oligonucleotide probe. For liver DNA, the quantity analyzed was reduced to 1 percent of the other samples. Although lower in intensity, the signal from donor DNA in the small bowel was clearly positive in the original film, although this can be seen only faintly. The negative control was a reaction run without DNA (last lane).
Discussion
The resorption of amylopectin from the heart previously reported after liver transplantation in patients with glycogen storage disease2 was confirmed by endomyocardial biopsies after additional months of follow-up. Donor cells could be detected in the biopsy specimens and other tissues of the recipients by immunocytochemical and molecular techniques 91 and 37 months, respectively, after liver replacement. Similar donor cells were also found in the skin, lymph nodes, small bowel, bone marrow, and blood of a patient with Gaucher’s disease.
We have proposed elsewhere6,9,10 that the successful engraftment of all whole-organ transplants connotes (and requires) an exchange between the organ and the recipient of cells of lymphocyte–macrophage lineage. Although the same process occurs with all organs, the high density and quality of the migratory cells leaving the liver or coming to it from the recipient are thought to be the basis for the “immunologic privilege” of a hepatic graft,6,9,10 including its so-called tolerogenicity in other organs from the same donor.11
The bone marrow–derived dendritic (antigen presenting) cells delineated by Steinman and Cohn 12 that are normally associated with the induction of T-cell immunity rather than the induction of tolerance 13,14 are thought to be the most prominent of the migratory cells moving from the liver to distant host tissues, with replacement by similar cells of the recipient that home to the graft. The distribution of the donor cells is similar to their distribution after bone marrow transplantation.6,15,16 T he consequent chimerism explains why the liver allograft can cause graft-versus-host disease.17–20
The occurrence of cell migration after liver transplantation explains why patients with lysosomal storage disorders — Wolman’s disease,21 Niemann–Pick disease,22 Gaucher’s disease,23 and the sea-blue histiocyte syndrome (unpublished data) — receive more benefit from liver replacement than simply improved hepatic function 24 We had assumed that the lysosomal storage disorder of our patient with Gaucher’s disease would progress after transplantation, prompting us to resume enzyme therapy; we now suspect that such treatment was unnecessary. For the absorption of the intracellular and extracellular amylopectin deposits of type IV glycogen storage disease, as well as the intracellular glucocerebroside deposits of Gaucher’s disease, it is necessary for the enzyme produced by the donor cells to gain access to the substrate. Evidence from in vitro coculture experiments suggests that once the donor cells have moved to the appropriate location, an enzyme can be transported from normal to abnormal cells.25 The number of allogeneic cells required to produce a metabolic effect appears from our results to be surprisingly low. The presumed transmission of enzymes from a small to a larger cell population raises intriguing questions about the potential cell-to-cell transfer of other molecules, including those involved in immunologic processes.
With the fuller understanding of what is actually accomplished by liver transplantation, past conclusions about the mechanisms of inborn errors of metabolism that were deduced from studies of liver-transplant recipients will require reexamination. For example, the decline in serum cholesterol concentrations after liver transplantation in patients with familial hypercholesterolemia26 that was ascribed to the low-density lipoprotein receptors of the allograft hepatocytes27 may reflect in part the presence of these receptors on widely disseminated donor cells that are not hepatocytes. Migratory Kupffer cells alone account for 10 to 15 percent of all cells in the liver and one fifth of the total hepatic lysosomal volume.28
If, as we have proposed,6,9,10 microchimerism differing only in degree is an integral feature of the successful engraftment of any organ, all such transplantation procedures could result in a gain in metabolic function. This could explain the report by Desnick et al.29 that native kidneys that had failed as a result of the accumulation of glycosphingolipid in a patient with Fabry’s disease recovered their function after successful renal transplantation.
Acknowledgments
Supported in part by a grant (DK-29961) from the National Institutes of Health.
References
- 1.Brown BI, Brown DH. Lack of an α-1,4-glucan:α-1,4-glucan 6-glycosyl transferase in a case of Type IV glycogenosis. Proc Natl Acad Sci U S A. 1966;56:725–9. doi: 10.1073/pnas.56.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selby R, Starzl TE, Yunis E, Brown BI, Kendall RS, Tzakis A. Liver transplantation for Type IV glycogen storage disease. N Engl J Med. 1991;324:39–42. doi: 10.1056/NEJM199101033240107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howell RR. Continuing lessons from glycogen storage diseases. N Engl J Med. 1991;324:55–6. doi: 10.1056/NEJM199101033240111. [DOI] [PubMed] [Google Scholar]
- 4.Brady RO, Kanfer JN, Bradley RM, Shapiro D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J Clin Invest. 1966;45:1112–5. doi: 10.1172/JCI105417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsia S, Tong JY, Parris GL, et al. Molecular compatibility and renal graft survival: the HLA DRB1 genotyping. Transplantation. 1992 doi: 10.1097/00007890-199302000-00030. (in press) [DOI] [PubMed] [Google Scholar]
- 6.Starzl TE, Demetris AJ, Murase N, Ildstad S, Ricordi C, Trucco M. Cell migration, chimerism, and graft acceptance. Lancet. 1992;339:1579–82. doi: 10.1016/0140-6736(92)91840-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porter KA. Pathology of the orthotopic homograft and heterograft. In: Starzl TE, editor. Experience in hepatic transplantation. Philadelphia: W.B. Saunders; 1969. pp. 464–5. [Google Scholar]
- 8.Gouw AS, Houthoff HJ, Huitema S, Beelen JM, Gips CH, Poppema S. Expression of major histocompatibility complex antigens and replacement of donor cells by recipient ones in human liver grafts. Transplantation. 1987;43:291–6. doi: 10.1097/00007890-198702000-00025. [DOI] [PubMed] [Google Scholar]
- 9.Starzl TE. Cell migration and chimerism: a unifying concept in transplantation: with particular reference to HLA matching and tolerance induction. Transplant Proc. (in press) [PubMed] [Google Scholar]
- 10.Starzl TE, Demetris AJ, Trucco M, et al. Systemic chimerism in human female recipients of male livers. Lancet. 1992;340:876–7. doi: 10.1016/0140-6736(92)93286-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calne RY, Sells RA, Pena JR, et al. Induction of immunological tolerance by porcine liver allografts. Nature. 1969;223:472–6. doi: 10.1038/223472a0. [DOI] [PubMed] [Google Scholar]
- 12.Steinman RM, Cohn ZA. Identification of a novel cell Type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–62. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–96. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 14.Lechler RI, Batchelor JR. Restoration of immunogenicity to passenger cell-depleted kidney allografts by the addition of donor strain dendritic cells. J Exp Med. 1982;155:31–41. doi: 10.1084/jem.155.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demetris AJ, Murase N, Starzl TE. Donor dendritic cells after liver and heart allotransplantation under short-term immunosuppression. Lancet. 1992;339:1610. doi: 10.1016/0140-6736(92)91875-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricordi C, Ildstad ST, Demetris AJ, Abou El-Ezz AY, Murase N, Starzl TE. Donor dendritic cells repopulation in recipients after rat-to-mouse bone-marrow transplantation. Lancet. 1992;339:1610–1. doi: 10.1016/0140-6736(92)91876-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramsey G, Nusbacher J, Starzl TE, Lindsay GD. Isohemagglutinins of graft origin after ABO-unmatched liver transplantation. N Engl J Med. 1984;311:1167–70. doi: 10.1056/NEJM198411013111807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burdick JF, Vogelsang GB, Smith WJ, et al. Severe graft-versus-host disease in a liver-transplant recipient. N Engl J Med. 1988;318:689–91. doi: 10.1056/NEJM198803173181107. [DOI] [PubMed] [Google Scholar]
- 19.Comenzo RL, Malachowski ME, Rohrer RJ, Freeman RB, Rabson A, Berkman EM. Anomalous ABO phenotype in a child after an ABO-incompatible liver transplantation. N Engl J Med. 1992;326:867–70. doi: 10.1056/NEJM199203263261305. [DOI] [PubMed] [Google Scholar]
- 20.Roberts JP, Ascher NL, Lake J, et al. Graft vs. host disease after liver transplantation in humans: a report of four cases. Hepatology. 1991;14:274–81. [PubMed] [Google Scholar]
- 21.Ferry GD, Whisennand HH, Finegold MJ, Alpert E, Glombicki A. Liver transplantation for cholesteryl ester storage disease. J Pediatr Gastroenterol Nutr. 1991;12:376–8. doi: 10.1097/00005176-199104000-00016. [DOI] [PubMed] [Google Scholar]
- 22.Daloze P, Delvin EE, Glorieux FH, Corman JL, Bettez P, Toussi T. Replacement therapy for inherited enzyme deficiency: liver orthotopic transplantation in Niemann-Pick disease type A. Am J Med Genet. 1977;1:229–39. doi: 10.1002/ajmg.1320010209. [DOI] [PubMed] [Google Scholar]
- 23.DuCerf C, Bancel B, Caillon P, et al. Orthotopic liver transplantation for type I Gaucher’s disease. Transplantation. 1992;53:1141–3. [PubMed] [Google Scholar]
- 24.Parkman R. The application of bone marrow transplantation to the treatment of genetic diseases. Science. 1986;232:1373–8. doi: 10.1126/science.3520819. [DOI] [PubMed] [Google Scholar]
- 25.Neufeld EF. Lessons from genetic disorders of lysosomes. Harvey Lect. 1981;75:41–60. [PubMed] [Google Scholar]
- 26.Starzl TE, Bilheimer DW, Bahnson HT, et al. Heart-liver transplantation in a patient with familial hypercholesterolaemia. Lancet. 1984;1:1382–3. doi: 10.1016/s0140-6736(84)91876-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bilheimer DW, Goldstein JL, Grundy SM, Starzl TE, Brown MS. Liver transplantation provides low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia. N Engl J Med. 1984;311:1658–64. doi: 10.1056/NEJM198412273112603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones EA, Summerfield SA. Kupffer cells. In: Arias IM, Popper H, Schachter D, Shafritz DA, editors. The liver: biology and pathobiology. New York: Raven Press; 1982. pp. 507–23. [Google Scholar]
- 29.Desnick RJ, Simmons RL, Allen KY, et al. Correction of enzymatic deficiencies by renal transplantation: Fabry’s disease. Surgery. 1972;72:203–11. [PubMed] [Google Scholar]