

Abstract
OFD I is an X-linked dominant male-lethal ciliopathy characterized by prominent external features including oral clefts, hamartomas or cysts of the tongue, and digital anomalies. Although these external features are easy to recognize and often lead to diagnosis in early childhood, visceral findings in OFD I, especially the fibrocystic liver and pancreas disease, are under-recognized. In addition, while the occurrence of polycystic kidney disease (PKD) in OFD I is well known, few patients are evaluated and monitored for this complication. We report on two adult females diagnosed with OFD I in infancy, but not evaluated for visceral involvement. In adulthood, they were incidentally found to have severe hypertension and chronic renal insufficiency due to undiagnosed PKD. A pancreatic cystic lesion, also discovered incidentally, was thought to be malignant and led to consideration of major surgery. We present NIH evaluations, including documentation of OFD1 mutations, extreme beading of the intrahepatic bile ducts, pancreatic cysts, and tabulate features of reported OFD I cases having hepatic, pancreatic and renal cystic disease. Liver and pancreas are not routinely evaluated in OFD 1 patients. Increased awareness and lifelong monitoring of visceral complications, particularly involving the liver, pancreas, and kidney, are essential for timely and accurate treatment.
Keywords: oral facial digital type I, ciliopathy, pancreatic cysts, polycytsic kidney disease, congenital hepatic fibrosis, Caroli disease
INTRODUCTION
Oral facial digital syndrome type I (OFD I), first described by Papillon-Leage and Psaume in 1954 [Papillon and Psaume 1954], is a multiple congenital anomaly syndrome with X-linked dominant inheritance resulting in lethality in affected males [Gorlin and Psaume, 1962; Scolari et al., 1997; Thauvin-Robinet et al., 2006]. External manifestations include malformations of the face, oral cavity, hands and feet. Craniofacial findings consist of facial asymmetry, hypertelorism, median cleft or pseudocleft of the upper lip, hypoplasia of the alae nasi, oral clefts and abnormal frenulea, tongue anomalies (clefting, cysts, hamartoma), and anomalous dentition involving missing or extra teeth. Asymmetric brachydactyly and/or syndactyly of the fingers and toes occur frequently. Approximately 50% of OFD I females have some degree of intellectual disability. Some patients have structural central nervous system anomalies such as agenesis of the corpus callosum, cerebellar agenesis or a Dandy-Walker malformation. OFD1, the gene mutated in OFD I, encodes a protein that localizes to the centrosome and basal body of primary cilia; hence, OFD I belongs to the group of ciliopathies.
Fibrocystic disease of the liver and kidneys is a common manifestation in various ciliopathies [Fliegauf et al., 2007; Gunay-Aygun, 2009]. Congenital hepatic fibrosis (CHF) is a constant feature of autosomal recessive polycystic kidney disease (ARPKD) and Meckel syndrome, and it occurs in some patients with Joubert syndrome and related disorders (JSRD) and Bardet-Biedl syndrome. Polycystic liver disease (PLD) affects the majority of adults with autosomal dominant polycystic kidney disease (ADPKD)[Gunay-Aygun, 2008]. Another ciliopathy, renal-hepatic-pancreatic dysplasia (RHPD) is characterized by pancreatic fibrocystic disease in addition to hepatorenal involvement [Bernstein et al., 1987].
Although the external features of OFD I are well described and the occurrence of polycystic kidney disease (PKD) in OFD I is well established [Coll et al., 1997; Connacher et al., 1987; Curry et al., 1992; Doege et al., 1964; Donnai et al., 1987; Feather et al., 1997; Harrod et al., 1976; Kennedy et al., 1991; Majewski et al., 1972; McLaughlin et al., 2000; Odent et al., 1998; Saal et al., 2010; Sabato et al., 1998; Salinas et al., 1991; Scolari et al., 1997; Stapleton et al., 1982; Stoll and Sauvage 2002; Toprak et al., 2006], liver and pancreas involvement have not been recognized to the same degree.
Here we present two adult females diagnosed with OFD I in early childhood and found to have hepatobiliary and pancreatic cystic disease and chronic renal insufficiency due to PKD in adulthood. We detail the clinical, biochemical, and molecular characteristics of these patients and present a review of previously reported OFD I patients with hepatic, pancreatic or renal involvement.
METHODS
Patients
The patients were enrolled in the protocol, “Clinical Investigations into the Kidney and Liver Disease in Autosomal Recessive Polycystic Kidney Disease/Congenital Hepatic Fibrosis and other Ciliopathies” (www.clinicaltrials.gov, trial NCT00068224), approved by the NHGRI Institutional Review Board. Patients or their parents gave written informed consent. Evaluations at the NIH Clinical Center included biochemical and imaging studies and sequencing of the OFD1 gene. Parents were evaluated by ultrasound, and parental DNA was analyzed.
Sequencing and analysis
All 23 exons of the OFD1 gene and their intronic boundaries were sequenced in two directions using a Beckman CEQ 8000 system (Beckman Coulter, Inc., Fullerton, CA). DNA variant analyses were performed using Sequencher (GeneCodes, Ann Arbor, MI).
Imaging studies
Complete abdominal ultrasound evaluations were performed using standard (4 MHz) and high resolution (7 MHz) ultrasonographic (HR-USG) probes (AVI Sequoia Inc, Mountain View, CA) by a single technologist (K.T.D.). Magnetic resonance imaging (MRI) was performed on a 1.5 Tesla machine (Philips Medical Systems, NA, Bothell, Washington; General Electric Healthcare, Waukesha, WI, USA) without intravenous contrast media. Kidney liver and spleen volumes were calculated from MRI images [Cheong et al., 2007; Grantham et al., 2006] at the NIH Image Processing Center.
Laboratory data
Creatinine clearances were based upon 24-hour urine collections. Serum chemistries were measured using standard methods.
RESULTS
Patient 1
This 38-year-old Caucasian female was diagnosed with OFD I in her first year of life, based upon the presence of a bifid tongue, partial unilateral cleft palate, missing teeth, and digital anomalies including brachydactyly and partial syndactyly of the first and second fingers. In childhood, her general health was good; she tolerated multiple corrective surgeries and dental procedures without complications. The patient underwent menarche at age 15 and had normal secondary sexual development. She reached developmental milestones on time, performed well in school and graduated from college. She was active in sports and received a scholarship in competitive diving. During a routine physical examination at age 29, she was diagnosed with severe hypertension (200/100 mm Hg), and has continued to receive an angiotensin II receptor antagonist in combination with hydrochlorothiazide since then. At age 35, her serum creatinine was 1.5 mg/dL and her AST and ALT were 70, and 201, respectively. Abdominal ultrasound revealed multiple cystic intrahepatic bile duct dilatations, pancreatic cysts and numerous macrocysts in both kidneys. Endoscopic ultrasonography and endoscopic retrograde pancreaticocholangiogram showed multiple cysts within the head of the pancreas; the two largest cysts located in the uncinate process measured 15 × 8 and 5 × 7 mm. (Fig 1A). There was no obvious pancreatic duct connection to the cysts, and the pancreatic duct was not dilated. Because of a concern regarding pancreatic malignancy, a fine needle aspiration of the pancreatic cyst was attempted; the amount of aspirate was not sufficient for complete analysis, but no malignant cells were identified. The patient was told that she might need a pancreatoduodenectomy (Whipple operation) and liver transplantation.
Figure 1.
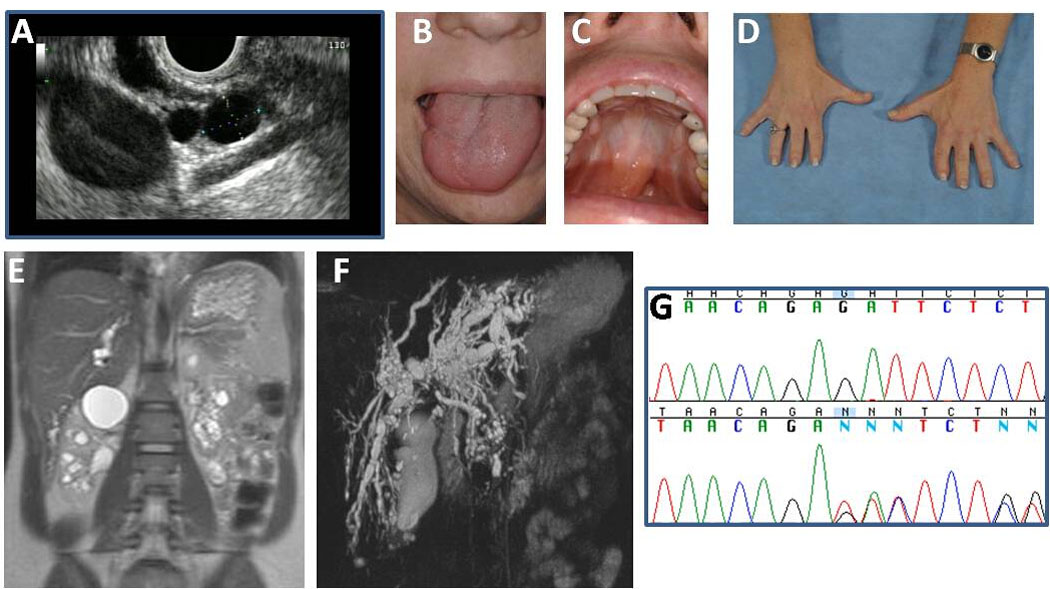
Characteristics of Patient 1. A. Endoscopic ultrasound showing multiple pancreatic cysts. (Performed by Dr. Phuong Nguyen, Hoag Memorial Hospital, Newport Beach, CA). B. Cleft tongue. C. Repaired partial cleft palate. D. Brachydactyly and repaired syndactyly. E. MRCP showing cystic dilatations of the intrahepatic bile ducts. F. MRI showing bilateral multiple renal medullary and cortical macrocysts. G. The patient’s two base pair deletion (g.9966–9967 del GA) in exon 6 of the OFDI gene.
Evaluations at the NIH Clinical Center revealed a healthy appearing female who was petite but proportional. Weight was 52.2 kg and height was 155.4 cm. She had a repaired tongue cleft (Fig 1B), a high arched palate with repaired partial cleft (Fig 1C), and repaired gingival and teeth anomalies. Extremity examination revealed brachydactyly of fingers and toes and repaired syndactyly (Fig 1D). The liver was 6 cm below the xiphoid but not palpable at the right costal margin. Spleen and kidneys were not palpable. High-resolution ultrasound and magnetic resonance imaging of the abdomen and magnetic resonance cholangiography revealed cystic dilatations of the intrahepatic biliary ducts throughout the liver, particularly in the left lobe around the porta hepatis (Fig 1E). The common bile duct was not dilated (3.2 mm). Liver echogenicity on ultrasound was moderately increased. Liver, spleen and left and right kidney volumes, calculated based on MRI images, were within normal limits at 784, 172, 73 and 84 ml, respectively. Hepatocellular function was intact; PT, ammonia, direct and total bilirubin, albumin, and total protein levels were normal. Liver enzymes were elevated as follows: ALT 166 (6–41), AST 81 (9–34) AP 256 (37–116), and GGT 556 U/L (5–85). In addition, the kidneys showed multiple macro cysts evenly distributed in the medulla and cortex, along with fine echogenic densities (Fig 1F). The serum creatinine was elevated at 1.64 mg/dl and the creatinine clearance, based on a 24-hour urine collection, was decreased at 64 ml/min/1.73 m2. Urine analysis was unremarkable, and 24-hour protein and calcium excretion were normal. Serum amylase and lipase were normal. Evaluation of cognitive function revealed a full scale IQ of 107 (WAIS III). Sequencing of the OFD1 gene revealed a 2 base pair deletion (g.9966–9967 del GA) in exon 6 (Fig 1G). Maternal and paternal DNA samples were negative for this mutation.
Patient 2
This 37-year-old Caucasian female was diagnosed with OFD I shortly after birth, based upon her cleft tongue, tongue cyst, dental anomalies, high-arched palate, and bilateral short fingers and toes. She also had global developmental delay and learning disability, and was in special education classes at school. She subsequently attended a college geared toward students with special needs, obtaining an associate’s degree in child care training. At age 25 years, she began to experience abdominal pain; an ultrasound showed bilateral cystic kidneys. Blood pressure was normal. Serum creatinine was 1.0 mg/dl. In her early thirties, she developed hypertension and was started on an angiotension II receptor antagonist. The serum creatinine ranged between 1.3 mg/dl and 1.8 mg/dl.
Evaluations at the NIH revealed a well appearing, cooperative female. Her weight was 62.8 kg and height 160.3 cm. She had a cleft tongue (Fig 2A), irregular and missing teeth (Fig 2B), high-arched palate (Fig 2C), patchy alopecia (Fig 2D), and brachydactyly of fingers and toes (Figure 2E and F). The liver was 7 cm at the xiphoid but not palpable at the right costal margin. Spleen and kidneys were not palpable. MRCP showed marked dilatation and beading of the biliary structures around the porta hepatis (Fig 2G and H). The common bile duct was dilated, measuring 6.2 mm. Liver echogenicity on ultrasound was moderately increased, suggesting fibrosis. Spleen size was normal. Measures of hepatocellular synthetic function (PT, total and direct bilirubin and ammonia) and liver enzymes (AP, ALT, AST and GGT) were normal. Kidneys were markedly enlarged and displayed numerous macro cysts (Fig 2G and H). The right kidney measured 21 cm and the left kidney 19.7 cm. Serum creatinine was 2.8 mg/dl and the creatinine clearance, based on a 24-hour urine collection, was 27 ml/min/1.73 m2. The serum parathyroid hormone was elevated at 206 pg/mL (normal 16 – 87), indicating renal osteodystrophy. There was significant proteinuria (2.25 g/day). The full scale IQ was 69 (WAIS-III); the verbal IQ was 69 and the nonverbal performance IQ was 75. The verbal comprehension index was 65, somewhat lower than her perceptual organizational index (78) and working memory index (75). Sequencing of the OFD1 gene revealed a one base-pair deletion (g.18897 del G) in exon 11. Both parental DNA sequences were negative for this mutation.
Figure 2.
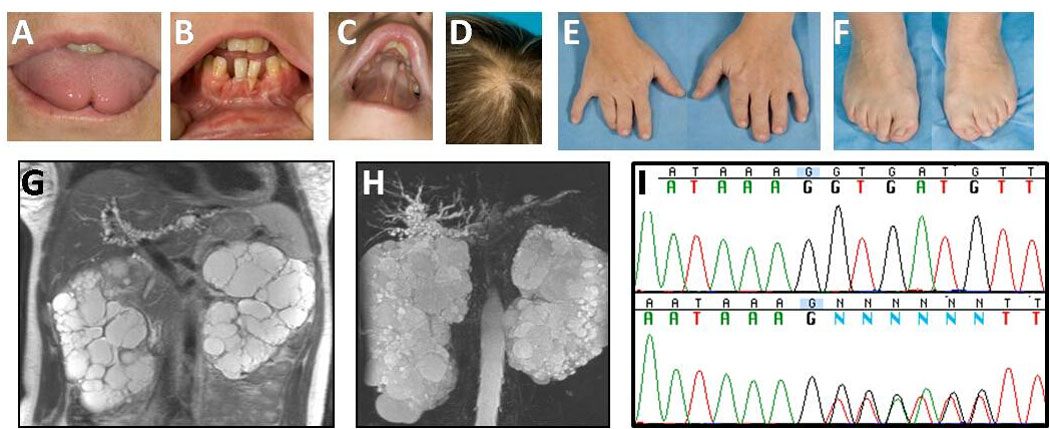
Characteristics of Patient 2. A. Cleft tongue. B. Missing and irregular teeth. C. High-arched palate. D. Patchy alopecia. E. and F. Brachydactyly. G. and H. Enlarged polycystic kidneys and beaded dilatations of the intahepatic bile ducts. I. The patient’s one base-pair deletion (g.18897 del G) in exon 11 of the OFDI gene.
In Table I, we listed the hepatic, pancreatic, renal, central nervous system, and physical findings of 35 females with OFD I and visceral disease, reported in 16 publications between 1964 and 2010; a recent review [Saal et al., 2010], on renal disease in OFD I could not be included in this table because individual patient data were not provided. All index patients were clinically typical for OFD I; all had PKD and oral, facial and digital features. OFD1 sequencing was performed and mutations were reported in all four cases published after the identification of the OFD1 gene [Stoll and Sauvage 2002; Toprak et al., 2006].
Table I.
Craniofacial, digital, renal, hepatic, and pancreatic findings of reported OFD1 patients.
| Patient No |
Age* | Orofacial Manifestations |
Digital Manifestations |
Polycystic Kidneys at Imaging or Pathology |
Impaired Renal Function (Age* at ESRD) |
Liver Cysts at Imaging or Pathology |
Pancreas Cysts at Imaging or Pathology |
Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | 38 | + | + | + | + | + | + | Present report |
| 2 | 37 | + | + | + | + | + | − | Present report |
| 3 | 72 | + | + | + | NE | NE | NE | Case II-9, Doege et al. [1964] |
| 4 | 38 | + | + | + | + (38) | + | + | Case III-26, Doege et al. [1964] |
| 5 | 48 | + | + | + | + (48) | NE | NE | Harrod et al. [1976] |
| 6 | 11 | + | + | + | + (11) | − | − | Stapleton et al. [1982] |
| 7 | 21 | + | + | + | NE | NE | NE | Case III-1, Donnai et al. [1987] |
| 8 | 21 | + | + | Unilateral | + | NE | NE | Case III-4, Donnai et al. [1987] |
| 9 | 54 | + | + | + | + | NE | NE | Case II-2, Donnai et al. [1987] |
| 10 | 64 | NE | + | + | + (64) | NE | NE | Case II-1, Donnai et al. [1987] |
| 11 | 53 | + | + | + | + (53) | NE | NE | Case I-2, Donnai et al. [1987] |
| 12 | 17 | + | + | + | + (17) | NE | NE | Connacher et al. [1987] |
| 13 | 44 | + | + | + | − | NE | NE | Mother, Connacher et al. [1987] |
| 14 | 15 | + | + | + | + (15) | + | + | Kennedy et al. [1991] |
| 15 | 30 | + | + | + | − | + | − | Case 1, Curry et al. [1992] |
| 16 | 12 | + | NE | + | − | − | − | Case 2, Curry et al. [1992] |
| 17 | 33 | NE | NE | + | − | − | − | Case 3, Curry et al. [1992] |
| 18 | 6 | NE | NE | + | − | − | − | Case 4, Curry et al. [1992] |
| 19 | 5th decade |
+ | NE | + | + | − | − | Case I-2, Feather et al. [1997] |
| 20 | 3rd decade |
+ | NE | Unilateral | − | − | − | Case II-2, Feather et al. [1997] |
| 21 | 3rd decade |
+ | + | + | − | − | − | Case II-4, Feather et al. [1997] |
| 22 | 15 | + | + | + | + (15) | − | + | Case III-1, Feather et al. [1997] |
| 23 | 13 | + | + | + | − | − | − | Case III-2, Feather et al. [1997] |
| 24 | 41 | + | + | + | + (41) | + | NE | Coll et al. [1997] |
| 25 | 26 | + | + | + | + (16) | + | + | Scolari et al. [1997] |
| 26 | 55 | + | + | + | + (55) | − | NE | Mother, Scolari et al. [1997] |
| 27 | 32 | + | + | + | − | + | − | Sabato et al. [1998] |
| 28 | 14 | + | + | + | − | NE | NE | Patient 1, Odent et al. [1998] |
| 29 | 28 | + | + | + | + (22) | NE | NE | Patient 4, Odent et al. [1998] |
| 30 | 35 | + | NE | + | + (35) | NE | NE | Patient 5, Odent et al. [1998] |
| 31 | Unknown | + | NE | + | NE | NE | NE | Mother of patient 5, Odent et al. [1998] |
| 32 | 30 | + | + | + | + | − | − | McLaughlin et al. [2000] |
| 33 | 62 | − | − | + | + | NE | NE | Mother, McLaughlin et al. [2000] |
| 34 | 23 | + | + | + | + | NE | NE | Stoll et al. [2002] |
| 35 | 20 | + | + | + | + (20) | Caroli's Disease | NE | Toprak et al. [2006] |
In years; NE: not evaluated; ESRD:end stage renal disease.
Liver was evaluated by imaging or autopsy in 20 of 35 (51 %) patients (Table I). Multiple liver macrocysts were diagnosed in nine of the 20 (45%) patients evaluated; five of these patients also had dilated intrahepatic bile ducts.
Pancreas was evaluated by imaging or autopsy in 17 of 35 (49%) patients (Table I). Multiple macrocysts in the pancreas were identified in five of the 17 (29%) patients evaluated (Table I). Four of the five patients with pancreatic cysts also had liver cysts. The ages of patients with multiple macrocysts in the liver and/or pancreas ranged from 15 to 38.
All 35 OFD I patients listed in Table I had cystic kidneys. Kidney function was impaired to variable degrees, with creatinine clearances ranging from 65 ml/min/1.73 m2 at age 62 to end stage renal disease (ESRD) at age 11. In 14 patients for whom this information was available, the age at ESRD ranged from 11 to 64 years (mean, 32 ± 18 years), consistent with the median age at onset of ESRD of 34 years reported by Saal et al(2010).
DISCUSSION
Primary cilia are important in development and maintenance of bile ducts [Masyuk et al., 2009] and renal tubules [Masyuk et al., 2009; Yoder 2007]; hence, hepatorenal fibrocystic disease is a common manifestation of various ciliopathies [Gunay-Aygun, 2009; Gunay-Aygun, 2008]. The recent recognition that OFD I is a ciliopathy [Macco and Franco, 2009] makes it easier to understand why this multiple congenital anomaly syndrome is associated with the seemingly unrelated features of fibrocystic disease of kidneys, liver and pancreas.
Both of our OFD 1 patients had increased echogenicity of the liver on ultrasound, intact hepatocelluar function with normal or minimally elevated liver enzymes, and an enlarged left lobe of the liver that was palpable under the xiphoid while the right lobe was non-palpable at the costal margin. These finding are typical for CHF caused by various ciliary defects such as ARPKD and some cases with JSRD, especially the COACH syndrome [Callier et al., 2008; Desmet, 1998; Gunay-Aygun, 2009; Gunay-Aygun 2008; Kerr et al., 1978]. CHF in ARPKD and COACH syndrome typically results in portal hypertension manifested with splenomegaly, often starting in childhood and worsening as patients get older [Gunay-Aygun 2009; Kerr et al., 1978]. In contrast, our two OFD1 patients had normal sized spleens and none of the other 33 patients in Table I had splenomegaly, suggesting that CHF in OFD 1 is not typically associated with portal hypertension.
With regard to macroscopic biliary abnormalities, both our patients had strikingly similar beaded cystic dilatations of the intrahepatic biliary structures particularly around the porta hepatis. Whether this type of biliary abnormality is typical for OFD1-associated liver disease or not will be determined as more OFD I patients undergo MRCP.
Pancreatic cystic disease in Patient 1 was asymptomatic without any pain. Amylase and lipase were normal and she had no evidence for malabsorption or diabetes to suggest overt exocrine or endocrine pancreas dysfunction. Similarly, none of the reported cases with pancreas cysts had symptoms to suggest pancreas dysfunction. RHPD, another ciliopathy associated with pancreas involvement, results in pancreatic dysplasia in addition to macrocysts. Pancreas microscopy, reported in 3 OFD I cases (Table I, Patient no 4, 14 and 19) did not reveal dysplasia.
Our patients were diagnosed with OFD I in the first year of life based on their characteristic facial and digital anomalies. However, the possibility of hepatic, pancreatic or renal involvement in OFD I was not pursued. For Patient 1, the consideration of a potential malignant pancreatic lesion may have led to unnecessary invasive evaluations and caused anxiety. In addition, she probably had hypertension for several years prior to its diagnosis at age 29, so she likely suffered systemic vascular damage. Similarly, the PKD and associated renal insufficiency of Patient 2 were not diagnosed until she approached end-stage kidney disease, preventing her from receiving appropriate supportive treatment.
OFD I patients are not aware of, or evaluated for, their risk of developing hepatic, renal and pancreatic fibrocystic disease. A significant proportion of patients are not even evaluated and monitored for PKD, the better recognized visceral involvement in OFD I [Prattichizzo et al., 2008; Saal et al., 2010; Szeto et al., 1977; Thauvin-Robinet et al., 2006]. In their report of OFD1 mutation analysis in 100 unrelated OFD I patients, Prattichizzo et al [Prattichizzo et al., 2008], found that only 40 of 81 patients (49%) had renal imaging; 24 of these 40 (60%) had PKD. In the series reported by Thauvin-Robinet et al., the kidneys were not assessed in 36% (9 of 25) of the patients [Thauvin-Robinet et al., 2006]. The proportion of OFD I patients who undergo imaging for liver and pancreas involvement is even smaller. Only 44% of the 34 patients reported by Saal et al. [2010] had their liver and pancreas evaluated. In the series of Prattichizzo et al.[Prattichizzo et al., 2008], four patients were reported as having pancreatic, hepatic, and/or ovarian cysts; the number of patients who had liver and pancreas imaging was not given.
OFD I patients with less obvious craniofacial features can escape the diagnosis of OFD I, and instead present to nephrology clinics in adulthood with late manifestations of PKD; they are often diagnosed with autosomal dominant polycytsic kidney disease (ADPKD). Eight of 33 patients in our literaure review (24%) were diagnosed with OFD I only after presenting with PKD. Two features that are potentially helpful to distinguish X-linked OFD I-related PKD from ADPKD are the presence of affected females only, in multiple generations and preserved renal contour.
Early diagnosis of the visceral manifestations of OFD I is essential for anticipatory care and prevention of hepatic, pancreatic and renal complications. Timely diagnosis and understanding of the fibrocystic nature of the hepatorenal and pancreatic disease in this condition may also prevent unnecessary and invasive diagnostic and therapautic interventions.
ACKNOWLEDGMENTS
We thank OFD I patients and families who generously participated in this investigation. Supported by the Intramural Research Program of the National Human Genome Research Institute and the NIH Clinical Center.
REFERENCES
- Bernstein J, Chandra M, Creswell J, Kahn E, Malouf NN, McVicar M, Weinberg AG, Wybel RE. Renal-hepatic-pancreatic dysplasia: a syndrome reconsidered. Am J Med Genet. 1987;26:391–403. doi: 10.1002/ajmg.1320260218. [DOI] [PubMed] [Google Scholar]
- Callier P, Faivre L, Thauvin-Robinet C, Marle N, Mosca AL, D'Athis P, Guy J, Masurel-Paulet A, Joly L, Guiraud S, Teyssier JR, Huet F, Mugneret F. Array-CGH in a series of 30 patients with mental retardation, dysmorphic features, and congenital malformations detected an interstitial 1p22.2-p31.1 deletion in a patient with features overlapping the Goldenhar syndrome. Am J Med Genet A. 2008;146A:2109–2115. doi: 10.1002/ajmg.a.32447. [DOI] [PubMed] [Google Scholar]
- Cheong B, Muthupillai R, Rubin MF, Flamm SD. Normal values for renal length and volume as measured by magnetic resonance imaging. Clin J Am Soc Nephrol. 2007;2:38–45. doi: 10.2215/CJN.00930306. [DOI] [PubMed] [Google Scholar]
- Coll E, Torra R, Pascual J, Botey A, Ara J, Perez L, Ballesta F, Darnell A. Sporadic orofaciodigital syndrome type I presenting as end-stage renal disease. Nephrol Dial Transplant. 1997;12:1040–1042. doi: 10.1093/ndt/12.5.1040. [DOI] [PubMed] [Google Scholar]
- Connacher AA, Forsyth CC, Stewart WK. Orofaciodigital syndrome type I associated with polycystic kidneys and agenesis of the corpus callosum. J Med Genet. 1987;24:116–118. doi: 10.1136/jmg.24.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry NS, Milutinovic J, Grossnickle M, Munden M. Renal cystic disease associated with orofaciodigital syndrome. Urol Radiol. 1992;13:153–157. doi: 10.1007/BF02924610. [DOI] [PubMed] [Google Scholar]
- Desmet VJ. Ludwig symposium on biliary disorders--part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–89. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- Doege TC, Thuline HC, Priest JH, Norby DE, Bryant JS. Studies of a Family with the Oral-Facial-Digital Syndrome. N Engl J Med. 1964;271:1073–1078. doi: 10.1056/NEJM196411192712101. [DOI] [PubMed] [Google Scholar]
- Donnai D, Kerzin-Storrar L, Harris R. Familial orofaciodigital syndrome type I presenting as adult polycystic kidney disease. J Med Genet. 1987;24:84–87. doi: 10.1136/jmg.24.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feather SA, Woolf AS, Donnai D, Malcolm S, Winter RM. The oral-facial-digital syndrome type 1 (OFD1), a cause of polycystic kidney disease and associated malformations, maps to Xp22.2-Xp22.3. Hum Mol Genet. 1997;6:1163–1167. doi: 10.1093/hmg/6.7.1163. [DOI] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, Psaume J. Orodigitofacial dysostosis--a new syndrome. A study of 22 cases. J Pediatr. 1962;61:520–530. doi: 10.1016/s0022-3476(62)80143-7. [DOI] [PubMed] [Google Scholar]
- Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF, Jr, Wetzel LH, Baumgarten DA, Kenney PJ, Harris PC, Klahr S, Bennett WM, Hirschman GN, Meyers CM, Zhang X, Zhu F, Miller JP. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:296–306. doi: 10.1002/ajmg.c.30225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunay-Aygun M, Heller Theo, Gahl WA. GeneReviews at GeneTests: Medical Genetics Information Resource (database online) Copyright. Seattle: University of Washington; 2008. Congenital Hepatic Fibrosis Overview. 1997–2008 Available at http://wwwgenetestsorg. [Google Scholar]
- Harrod MJ, Stokes J, Peede LF, Goldstein JL. Polycystic kidney disease in a patient with the oral-facial-digital syndrome - type I. Clin Genet. 1976;9:183–186. doi: 10.1111/j.1399-0004.1976.tb01565.x. [DOI] [PubMed] [Google Scholar]
- Kennedy SM, Hashida Y, Malatack JJ. Polycystic kidneys, pancreatic cysts, and cystadenomatous bile ducts in the oral-facial-digital syndrome type I. Arch Pathol Lab Med. 1991;115:519–523. [PubMed] [Google Scholar]
- Kerr DN, Okonkwo S, Choa RG. Congenital hepatic fibrosis: the long-term prognosis. Gut. 1978;19:514–520. doi: 10.1136/gut.19.6.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski F, Lenz W, Pfeiffer RA, Tunte W, Muller H. The oro-facial-digital syndrome. Symptoms and prognosis. Z Kinderheilkd. 1972;112:89–112. [PubMed] [Google Scholar]
- Masyuk T, Masyuk A, LaRusso N. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Curr Opin Gastroenterol. 2009;25:265–271. doi: 10.1097/MOG.0b013e328328f4ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin K, Neilly JB, Fox JG, Boulton-Jones JM. The hypertensive young lady with renal cysts--it is not always polycystic kidney disease. Nephrol Dial Transplant. 2007;15:1245–1247. doi: 10.1093/ndt/15.8.1245. [DOI] [PubMed] [Google Scholar]
- Odent S, Le Marec B, Toutain A, David A, Vigneron J, Treguier C, Jouan H, Milon J, Fryns JP, Verloes A. Central nervous system malformations and early end-stage renal disease in oro-facio-digital syndrome type I: a review. Am J Med Genet. 1998;75:389–394. [PubMed] [Google Scholar]
- Papillon L, Psaume J. Hereditary abnormality of the buccal mucosa: abnormal bands and frenula. Revue Stomatol. 1954;55:209–227. [PubMed] [Google Scholar]
- Prattichizzo C, Macca M, Novelli V, Giorgio G, Barra A, Franco B. Mutational spectrum of the oral-facial-digital type I syndrome: a study on a large collection of patients. Hum Mutat. 2008;29:1237–1246. doi: 10.1002/humu.20792. [DOI] [PubMed] [Google Scholar]
- Saal S, Faivre L, Aral B, Gigot N, Toutain A, Van Maldergem L, Destree A, Maystadt I, Cosyns JP, Jouk PS, Loeys B, Chauveau D, Bieth E, Layet V, Mathieu M, Lespinasse J, Teebi A, Franco B, Gautier E, Binquet C, Masurel-Paulet A, Mousson C, Gouyon JB, Huet F, Thauvin-Robinet C. Renal insufficiency, a frequent complication with age in oral-facial-digital syndrome type I. Clin Genet. 2010;77:258–265. doi: 10.1111/j.1399-0004.2009.01290.x. [DOI] [PubMed] [Google Scholar]
- Sabato A, Fabris A, Oldrizzi L, Montemezzi S, Maschio G. Evaluation of a patient with hypertension and mild renal failure in whom facial and digital abnormalities are noted. Nephrol Dial Transplant. 1998;13:763–766. doi: 10.1093/ndt/13.3.763. [DOI] [PubMed] [Google Scholar]
- Salinas CF, Pai GS, Vera CL, Milutinovic J, Hagerty R, Cooper JD, Cagna DR. Variability of expression of the orofaciodigital syndrome type I in black females: six cases. Am J Med Genet. 1991;38:574–582. doi: 10.1002/ajmg.1320380416. [DOI] [PubMed] [Google Scholar]
- Scolari F, Valzorio B, Carli O, Vizzardi V, Grazioli L, Bondioni MP, Maiorca R. Oral-facial-digital syndrome type 1 coexisting with polycystic kidney disease. Contrib Nephrol. 1997;122:58–60. doi: 10.1159/000059869. [DOI] [PubMed] [Google Scholar]
- Stapleton FB, Bernstein J, Koh G, Roy S, 3rd, Wilroy RS. Cystic kidneys in a patient with oral-facial-digital syndrome type I. Am J Kidney Dis. 1982;1:288–293. doi: 10.1016/s0272-6386(82)80027-9. [DOI] [PubMed] [Google Scholar]
- Stoll C, Sauvage P. Long-term follow-up of a girl with oro-facio-digital syndrome type I due to a mutation in the OFD 1 gene. Ann Genet. 2002;45:59–62. doi: 10.1016/s0003-3995(02)01116-4. [DOI] [PubMed] [Google Scholar]
- Szeto HH, Inturrisi CE, Houde R, Saal S, Cheigh J, Reidenberg MM. Accumulation of normeperidine, an active metabolite of meperidine, in patients with renal failure of cancer. Ann Intern Med. 1977;86:738–741. doi: 10.7326/0003-4819-86-6-738. [DOI] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Cossee M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, Bieth E, Layet V, Parent P, David A, Goldenberg A, Mortier G, Heron D, Sagot P, Bouvier AM, Huet F, Cusin V, Donzel A, Devys D, Teyssier JR, Faivre L. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43:54–61. doi: 10.1136/jmg.2004.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toprak O, Uzum A, Cirit M, Esi E, Inci A, Ersoy R, Tanrisev M, Ok E, Franco B. Oral-facial-digital syndrome type 1, Caroli's disease and cystic renal disease. Nephrol Dial Transplant. 2006;21:1705–1709. doi: 10.1093/ndt/gfk013. [DOI] [PubMed] [Google Scholar]
- Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–1388. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]