

Abstract
Proteinase 3 (PR3)-specific antineutrophil cytoplasmic antibodies (ANCA) are highly specific for the autoimmune small vessel vasculitis, Wegener’s granulomatosis (WG). PR3-ANCA have proven diagnostic value but their pathogenic potential and utility as a biomarker for disease activity remain unclear. PR3-ANCA recognize conformational epitopes, and epitope-specific PR3-ANCA subsets with variable impact on biological functions of PR3 have been postulated. The aims of this study were to identify specific PR3 surface epitopes recognized by monoclonal antibodies (moAbs) and to determine whether the findings can be used to measure the functional impact of epitope-specific PR3-ANCA and their potential relationship to disease activity. We used a novel flow cytometry assay based on TALON-beads coated with recombinant human (H) and murine (M) PR3 and 10 custom-designed chimeric human/mouse rPR3-variants (Hm1–5/Mh1–5) identifying 5 separate non-conserved PR3 surface epitopes. Anti-PR3 moAbs recognize 4 major surface epitopes, and we identified the specific surface location of 3 of these with the chimeric rPR3-variants. The ability of PR3-ANCA to inhibit the enzymatic activity of PR3 was measured indirectly using a capture-ELISA system based on the different epitopes recognized by capturing moAbs. Epitope-specific PR3-ANCA capture-ELISA results obtained from patient plasma (n=27) correlated with the inhibition of enzymatic activity of PR3 by paired IgG preparations (r=0.7, P<0.01). The capture-ELISA results also seem to reflect disease activity. In conclusion, insights about epitopes recognized by anti-PR3 moAbs can be applied to separate PR3-ANCA subsets with predictable functional qualities. The ability of PR3-ANCA to inhibit the enzymatic activity of PR3, a property linked to disease activity, can now be gauged using a simple epitope-based capture-ELISA system.
Keywords: ANCA, proteinase 3, Wegener’s granulomatosis, vasculitis
1. Introduction
Antineutrophil cytoplasmic autoantibodies (ANCA) targeting proteinase 3 (PR3) occur in most patients with Wegener’s granulomatosis (WG) [1–3]. PR3-ANCA have undisputed diagnostic value, but their association with disease activity and pathogenic potential remain unclear [4–8]. Circulating PR3-ANCA can persist during remission, and relapses even occur in the absence of rising PR3-ANCA titers. [9, 10]. Moreover, PR3-ANCA levels correlate with disease activity in some patients, but not others [1, 10, 11]. In mice, antibodies directed against murine PR3 had significant pro-inflammatory effects, but a disease phenotype with capillaritis or necrotizing granulomatous inflammation did not occur [12, 13]. In view of these clinical and experimental observations, the concept of general PR3-ANCA pathogenicity under in vivo conditions has been disputed.
The pro-inflammatory effects of ANCA on human neutrophils, monocytes and endothelial cells were established in vitro using purified immunoglobulin G (IgG) from PR3-ANCA positive patients [8, 14, 15]. Most of these effects require binding of PR3-ANCA to its antigen expressed on the cell surface. In addition, PR3-ANCA can interfere with functional properties of PR3. These fall into two categories: those mediated by proteolytic activity of PR3, and those that are independent of enzymatic activity and are mediated by additional membrane and protein interacting sites distant from the active site pocke t[16–18]. Consequently, antibodies binding to different surface epitopes on PR3 can be expected to affect these biological properties in a distinct manner.
PR3-ANCA sera inhibiting the enzymatic activity of PR3 to various degrees have been reported and related to disease activity in a small number of patients [19–23]. The basis of this inhibitory effect and the various surface regions targeted by PR3-ANCA subsets, however, remained unclear. Epitope mapping strategies using linear peptides have yielded unreliable results because PR3-ANCA and most monoclonal antibodies (moAbs) bind to non-linear conformational epitopes [24–27].
The opposing views on ANCA-pathogenicity may be reconciled by considering that PR3-ANCA differ in their binding properties and epitope specificity during remission and relapses, and interfere variably with the functions and clearance of the autoantigen by α1-antitrypsin. Thus, binding specificities of PR3-ANCA could account for a different pathogenic potential and may contribute to the variable severity of disease manifestations. To unravel the pathogenic potential of PR3-ANCA in vivo, we therefore need to better understand how the target antigen interacts with its environment during inflammation, and how these interactions are modified by epitope-specific autoantibodies. Monoclonal antibodies with known epitope recognition are invaluable for molecular structure-function analyses, and they are widely used for antigen capture in diagnostic methods to measure autoantibodies. For these reasons the present study had two major aims: first, the identification of the specific conformational surface epitopes recognized by the currently available anti-PR3 moAbs and second, the translation of these findings into a clinically useful separation of PR3-ANCA subsets by epitope-specific functional impact.
2. Materials and Methods
All reagents were purchased from Sigma (St.Louis,MO), unless specified otherwise. The 293 (adenovirus type 5 transformed human embryonic kidney) cell line was obtained from ATCC (Rockville,MD).
2.1. Monoclonal antibodies
Seven of the 14 moAbs used were gifts from the following investigators: J. Wieslander (4A3,4A5,6A6) [21]; E. Csernok (WGM2) [28]; C.G.M. Kallenberg (12.8,PR3G-2, PR3G-4) [29, 30], and purchased from CellSciences (Canton,MA) (WGM2) and Wieslab, Lund, Sweden (6A6). 2E1 and 1B10 were purchased from Abcam (Cambridge,MA).
Five moAbs were developed by us using standard hybridoma technology. MCPR3-1 and MCPR3-2 have been described [31]. To generate MCPR3-3, purified neutrophil PR3 (Athens Research and Technology,Athens,GA) was used as antigen; for MCPR3–7 and MCPR3–11 pro-rPR3 expressed in 293 cells (MCPR3–7 and MCPR3–11) was the antigen. The use of mice for moAb production was approved by the Institutional Animal Care and Use Committee and conducted according to institutional guidelines.
For the competition studies, PR3G-2, WGM2, MCPR3-2, MCPR3-3 and MCPR3–7 were labeled with fluorescein-isothiocyanate (FITC; EZ-label kit, Pierce, Rockford,IL).
2.2. Bead-based flow cytometry assay
For epitope-specific grouping of moAbs (Figures 1&2) a modification of the method described by Warren et al. was used [32]. Variants of rPR3 (Figure 3) carrying a cmyc-poly-His tag [33] were coated to Talon™-beads (Dynal, Oslo, Norway); antibodies bound to coated antigen were detected by fluorescence-activated cell sorting (FACS).
FIGURE 1. Bead-based FACS assay to detect antibody binding to rPR3.

A. Talon®-beads serve as support matrix for rPR3-variants bound by carboxy-terminal cmyc-poly-His-tag. Bound antigen-specific antibodies (anti-PR3 moAb or PR3-ANCA) are detected indirectly by flow cytometry using a secondary FITC-labeled anti-IgG. The method can be modified to measure the binding of FITC-conjugated anti-PR3 moAbs directly; this was applied to measure the competition between unlabeled epitope-specific anti-PR3 moAbs and FITC-conjugated anti-PR3 moAbs. B–D: Binding of primary antibody to PR3 is proportional to the fluorescent signal (X-axis; white histograms). Black reference histograms indicate lack of binding to uncoated beads.
B–D. representative examples of individual competition assays used to generate the antibody competition matrix (Figure 2B). Shown is the binding of MCPR3-2-FITC to PR3 with or without pre-incubation with different unlabeled moAbs. The left-shift in the histogram corresponds to reduced binding of MCPR3-2-FITC due to competition with the unlabeled moAb compared to the binding of MCPR3-2 FITC in the absence of competitor (expressed as % inhibition). Using the same moAb as unlabeled competitor (MCPR3-2), inhibition was 94 % (B). Using MCPR3-1, recognizing a similar epitope, the inhibition was 51% (C). Using 12.8, which recognizes a different epitope, the inhibition of MCPR3-2 FITC binding was only 4% (D).
FIGURE 2. Epitope-specific grouping of monoclonal anti-PR3 antibodies revisited.

A. Adapted diagram showing a previously proposed grouping of anti-PR3 moAbs for reference [44].
B. Validation of anti-PR3 moAb grouping and epitope-specific assignment of additional moAbs by bead-based flow cytometry. For clarity, the group numbering shown in A was maintained [44]. PR3-coated beads were incubated with unlabeled competitor-moAbs (y-axis) first; then with the FITC-conjugated moAbs (x-axis). Mature rPR3 was used as antigen for all moAbs; MCPR3-7 was further evaluated with pro-rPR3 because of its preferential binding to pro-PR3. Inhibition of binding of the FITC-conjugated moAbs by the competitor is inversely proportional to the fluorescence intensity and numerically specified in each matrix cell (white: 0–39% inhibition, gray: 40–59% inhibition, black: 60–100% inhibition). The number in each cell represents the mean of 3–5 repeat experiments. Group 5 designates moAbs that did not compete significantly with moAbs from groups 1,3 or 4. We could not confirm the reported overlap of PR3G-4 (proposed group 2) with antibodies from group 1,3 and 4. Instead, there was significant overlap with group 5 moAbs.
C. Revised epitope grouping of anti-PR3 moAbs resulting from our competition studies. Previously untested moAbs (italics) could be assigned to individual groups.
FIGURE 3. Constructs used for the identification of specific PR3-epitopes recognized by monoclonal antibodies.
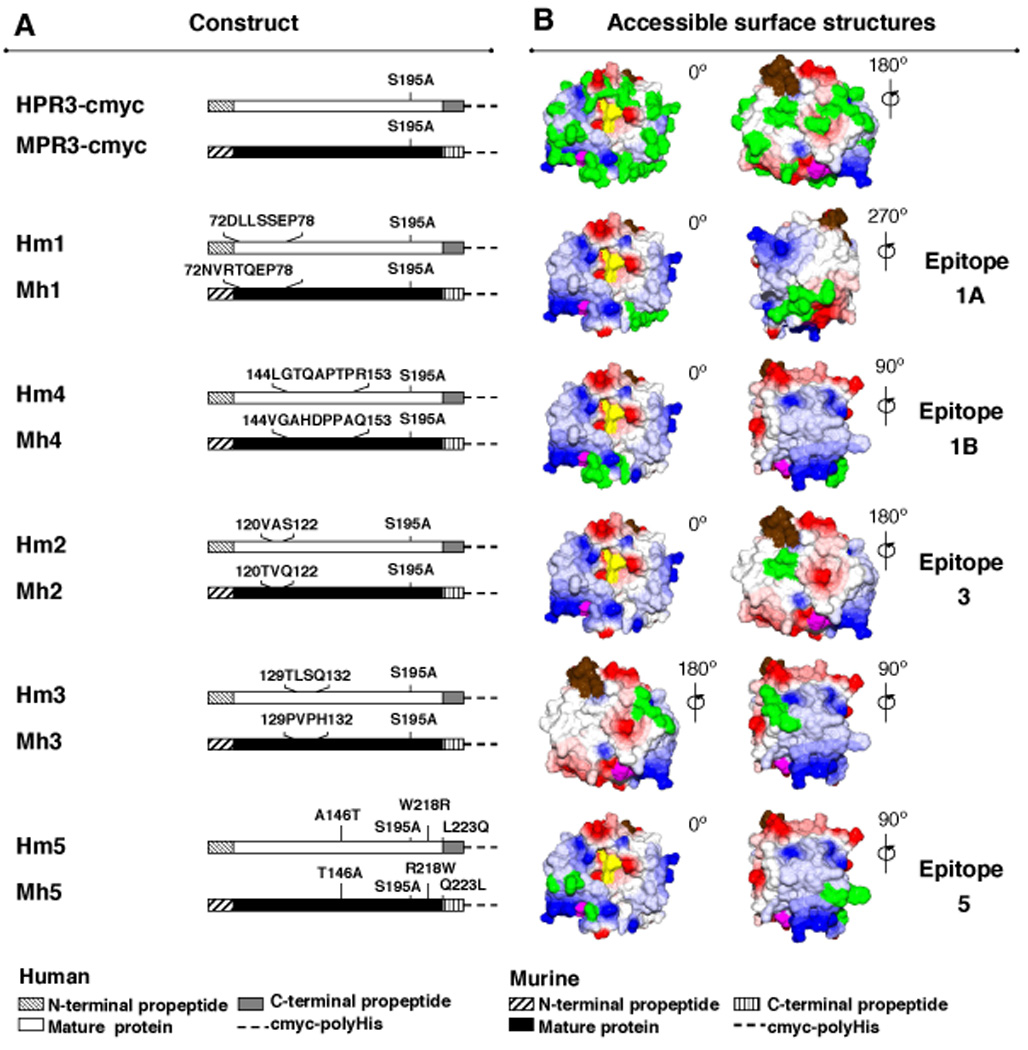
A. Schematic representation of cDNA constructs for human (H) and murine (M) rPR3 (first pair) and human-murine chimeric rPR3-variants (subsequent five pairs) used for the identification of epitopes recognized by moAbs. Each chimeric construct codes for the human to mouse (Hm) and mouse to human (Mh) swap of non-conserved surface amino-acids as indicated.
B. Surface representations of mature human PR3 generated using DeepView/Swiss-Pdb viewer, V4.0, software based on the crystal structure. Colors indicate: red, negative and blue, positive charge; purple, the amino-terminal residues IVGGH of mature PR3; brown, the carboxy-terminus; yellow, active site residues H57, D102, S195; green, residues not conserved between human and mouse PR3 (top pair) and epitope-specific mutations (lower five pairs). PR3 is shown in standard orientation with the active site facing the viewer (0°). Counterclockwise rotations of PR3 are indicated; 180° reveals the back side of PR3.
The polystyrene magnetic beads carry an immobilized cobalt-based chelator on their surface, which binds the imidazole rings of the cmyc-poly-His tag or the rPR3 variants (Figure 3). Briefly, 50µl (2mg, 2.8×109 beads) of stock were washed three times in Talon-buffer (50mM Na phosphate pH 8.0; 300mM NaCl, 0.01% Tween 20) using a Dynal magnet to separate beads from supernatants. After the last wash, 15µg of purified rPR3-variants were added to the bead pellet, re-suspended with 700µl of Talon-buffer and incubated 10 minutes at room temperature (RT). After 4 washes the final pellet was resuspended in 100µl of Talon-buffer and stored at 4°C until use.
Antibodies (moAbs) were incubated with antigen-coated beads in V96 MicroWell™ plates (Nunc™, Roskilde, Denmark). A total of 1µl of stock antigen-labeled beads was diluted in 1ml of PBS, 10µl of diluted beads were put into each V well (equivalent to 2.8×105 beads) and centrifuged at 2000 RPM for 2 minutes, and the supernatant was removed.
To study the binding of antibodies to the immobilized rPR3-variants, moAb (0.25µg/100µl) were incubated with antigen-coated beads for 30 minutes at RT and washed with 100µl of buffer (PBS, 0.1% BSA and 0.01% Tween 20), eliminating supernatant after centrifugation. Then, the bead pellet was incubated 5 minutes with 10µl of FITC-labeled secondary antibodies (goat anti-mouse IgG-FITC) diluted 1:50 in buffer at RT. After washing with 100µl PBS, the coated beads were diluted in 170µl of 1% paraformaldehyde, transferred to 7ml tubes and read immediately by FACScan (BD Bioscience), setting FL=1 at 682, logarithmic scale.
To assess competition between moAbs for the recognition of rPR3, unlabeled anti-PR3 moAbs were used as competitors of FITC-labeled anti-PR3 moAbs. The unlabeled moAb (competitor) was incubated with rPR3-coated beads for 30 minutes and washed as indicated, followed by incubation with FITC-labeled moAb for 5 minutes, washing, dilution in 170µl of 1% paraformaldehyde and immediate analysis by FACS.
2.3. Expression of recombinant human-murine chimeric rPR3-variants
Human (H) or murine rPR3 (M) were expressed in 293 cells as described [33–37]. All chimeric rPR3-variants were expressed using this system. The cDNA constructs used are shown in Figure 3A. Amino acid residues are numbered based on the crystal structure of PR3 (pdb entry 1FUJ) and standard alignment with the chymotrypsinogen A sequence [37, 38]. All rPR3 constructs code for the deletion of the two-residue amino-terminal propeptide and the mutation of the Ser-195 to Ala. Consequently, the expressed rPR3 variants are enzymatically inactive but have the mature enzyme surface conformation [34, 35, 39]. Furthermore, all constructs code for a carboxy-terminal cmyc-poly-His tag, used for purification and binding to various solid phases [40, 41]. Neither the Ser-195 to Ala mutation or this tag affect binding of PR3-ANCA or anti-PR3 moAbs [33–35, 39].
Five sets of complementary chimeric rPR3 molecules were generated using either human or murine rPR3 as the backbone (Figure 3A,B). Each pair of chimeric molecules represents one of five distinct patches of accessible surface amino acids, which are not conserved between human and murine PR3 (Figure 3B). The chimeric rPR3-variants, Hm1 to Hm5, were generated using the Quick-Change Site-Directed Mutagenesis Kit (Stratagene, La Jolla,CA) with the tagged human rPR3 construct (H) as template [33]. For the inverse rPR3-variants, Mh1 to Mh5, the tagged murine rPR3 construct (M) served as template onto which the human peptide sequences were grafted [36].
The serum-free culture media supernatants from the 293 cell clones were screened by capture-ELISA using rabbit anti-human PR3 and anti-murine PR3 antibodies for detection of bound rPR3-variants [40]. To purify the rPR3-variants, serum-free media supernatants were concentrated (CentriPlus®, Amicon), imidazole was added to a concentration of 20mM and applied to HiTrap chelating HP columns (GE Healthcare Bio-Sciences AB,NJ). Proteins were eluted with 20mM phosphate, 500mM NaCl and 20mM imidazole. Imidazole was removed with 50mM Na phosphate buffer, 300mM NaCl, 0.01% Tween 20, pH 8.0. Proteins were quantified by Coomassie Plus® (Pierce, Rockford,IL).
2.4. Capture-ELISA
PR3-ANCA were measured by capture-ELISA using MCPR3-2 or MCPR3-3 to capture mature conformation human rPR3 as antigen [31].
2.5. Hydrolysis of N-MeO-Succ-AAPV-pNA
Enzymatic activity of PR3 was determined by measuring the hydrolysis of the substrate N-methoxysuccinyl-Ala-Ala-Pr-Val-p-nitroanilide (MeAAPV) [36]. A total of 4mM of substrate and 133nM of purified PR3 (Athens,GA) were used per microtiter well; each experiment was performed in triplicate. To determine Vmax values, progression curves were plotted based on absorbance at 405 nm measured every 5 minutes over a 4-h period using a VersaMax ™ microplate reader (Molecular Devices,CA).
To determine the inhibitory effect of IgG preparations from PR3-ANCA positive patients on the enzymatic activity of PR3, purified PR3 was pre-incubated with the different moAbs or IgG preparations for 30 minutes prior to the measurement of substrate hydrolysis. PR3 incubated in parallel for 30 minutes with buffer or with isotype-matched IgG not recognizing PR3, served as negative controls.
2.6. Patient samples
Serum and plasma samples used for assay development were left-over samples from patients undergoing routine ANCA testing as part of an evaluation for suspected vasculitis at Mayo Clinic Rochester [6]. Diagnosis, disease activity recorded by the treating physician, and erythrocyte sedimentation rate (ESR) at the time of sampling were determined by review of clinical records. All PR3-ANCA positive patients had WG fulfilling the ACR 1990 criteria and the Chapel Hill Consensus definition for WG [42, 43]. The original PR3-ANCA status was determined by routine clinical testing using standard direct ELISA and indirect immunofluorescence [6]. The sample use was approved by the Institutional Review Board.
2.7. Statistical analysis
Simple descriptive statistics were used for in vitro experiments. Pearson’s correlation coefficient was calculated to analyze correlations between MCPR3-3/MCPR3-2 capture-ELISA ratios and inhibition of proteolytic activity of PR3. Comparisons between MCPR3-3/MCPR3-2 capture-ELISA ratios and disease activity (active disease versus remission) were performed using Wilcoxon rank-sum tests.
3. Results and Discussion
3.1. Rationale for epitope analysis of PR3 and experimental approach
The two primary motivations for investigations of epitope-specific antibodies to PR3 are to understand (i) how the target antigen interacts with its environment during inflammation, and (ii) how - and to what effect - these interactions are modified by epitope-specific autoantibodies.
Mouse moAbs targeting human PR3 recognize surface epitopes that are not conserved on the murine homolog. Depending on the location of their target epitopes, these moAbs have different effects on proteolytic and non-proteolytic biologic functions of PR3. If the epitopes are known, moAbs can be used to study structure-function relationships of PR3 and the role of PR3 in inflammatory conditions including WG. MoAbs are also used as antigen capturing tools for sandwich-ELISAs to measure PR3-ANCA. If the capturing moAbs compete for epitopes recognized by PR3-ANCA, false-negative test results may be the consequence. For these reasons the present study had two major aims: first, the identification of the specific conformational surface epitopes recognized by the currently available anti-PR3 moAbs and second, the translation of these findings into a clinically useful tool to separate PR3-ANCA subsets by epitope-specific functional impact.
To address the first aim, we developed a novel solid-phase assay based on the binding of poly-His-tagged recombinant antigens to Talon-beads with subsequent detection of bound moAbs by FACS. This method was used for moAb-competition studies with human wild-type rRP3-cmyc (H) as coated target antigen ( section 3.2.). For the mapping of specific PR3 surface epitopes we used the custom-designed chimeric rPR3 molecules (section 3.3.). The most significant advantage of this assay is that all rPR3-variants carrying the poly-His tag are coated to the beads via this consistent linker. This minimizes variability of antigen coating efficiency and antigen presentation caused by the mutation-induced changes of the rPR3 structure.
3.2. Monoclonal antibodies against human PR3 recognize four major epitopes
Anti-PR3 moAbs generated by several investigators were reported to recognize different conformational epitopes [21, 31, 44]. Sommarin and colleagues raised three moAbs (6A6, 4A5, and 4A3) using purified human neutrophil PR3 as antigen [21]. According to competition ELISA and biosensor data they recognize three separate epitopes, one of which was shared with 12.8 [21]. We subsequently raised two moAbs using granule rPR3 expressed in HMC-1 cells as antigen (MCPR3-1 and MCPR-2) and showed that they share epitope recognition with 4A3 [31]. Van der Geld and colleagues grouped anti-PR3 moAbs available from several laboratories based on competition in a biosensor assay [30]. For clarity and to promote a consensus epitope terminology for PR3, we based our analyses and subsequent epitope designations on this original grouping (Figure 2A).
To verify and extend this proposed grouping of anti-PR3 moAbs, we reexamined currently available moAbs from each group and tested novel moAbs by competition analysis using the bead-based FACS assay. Each unlabeled competitor moAb was tested against representative FITC-conjugated moAbs from the previously identified groups [30]. Figure 2B shows a matrix representing the composite results of these competition experiments.
3.2.1. Epitope 1
The attribution of the moAbs 6A6 and PR3G-2 to group 1 was confirmed. The moAb 12.8 inhibited the binding of PR3G-2-FITC only weakly, but significantly more than that of any other FITC-conjugated moAb. Consequently, the moAbs 6A6, PR3G-2 and 12.8 do indeed recognize a similar epitope. However, in contrast to the previous biosensor data [30], we found no significant overlap with epitopes recognized by other moAbs.
3.2.2. The previously proposed epitope 2 cannot be confirmed as a separate epitope
The moAb PR3G-4 had previously been attributed to a separate group, called group 2, with significant overlap with all other epitopes [30]. In our bead-based FACS assay PR3G-4 did not compete with FITC-conjugated representatives of groups 1, 3 and 4, but it interfered with the binding of FITC-conjugated MCPR3–7 to both mature and pro-PR3 (epitope 5).
3.2.3. Epitope 3
We also found that 4A5 and WGM2 (group 3) recognize a unique epitope. However, in variance to the reported biosensor data, we did observe inhibition of binding of WGM2-FITC by unlabeled MCPR3-2, but not vice versa. This indicates that the surface residues on PR3 targeted by WGM2 and MCPR3-2, respectively, are in close proximity to each other, and that MCPR3-2 has stronger affinity to PR3 than WGM2-FITC. The previously untested moAbs 2E1, 1B10 and MCPR3-3 were attributed to group 3.
3.2.4. Epitope 4
The results from our bead-based FACS assays are consistent with previous reports indicating that 4A3 and MCPR3-2 (group 4) recognize a shared epitope [30, 31]. In addition, MCPR3-1 was attributed to the same group 4.
3.2.5. Epitope 5
The novel moAbs MCPR3–7 and MCPR3–11 did not inhibit binding of FITC-conjugated representatives of groups 1, 3 and 4, indicating that they recognize a separate epitope without overlap with these three epitopes. Therefore, we called it epitope 5. MCPR3–7 and MCPR3–11 are unique moAbs in that they were selected for preferential binding to pro-PR3 over mature PR3. Consequently, separate experiments with FITC-conjugated MCPR3–7 were performed with mature and pro-PR3 as antigen confirming the unique nature of this epitope (Figure 2B).
3.2.6. Conclusions from monoclonal antibody competion studies
Taken together, our competition results confirm the existence of the three independent epitopes designated as 1, 3, and 4. However, in variance to the report by van der Geld and colleagues, we found no overlap between epitope 1 and either epitope 3 or epitope 4, but some overlap between epitopes 3 and 4, suggesting that epitope 1 is topographically distant to epitopes 3 and 4, whereas epitopes 3 and 4 are adjacent to each other. The undescribed moAbs MCPR3-3, 1B10 and 2E1 could be assigned to epitope 3, and MCPR3-1 to epitope 4. Our novel moAbs MCPR3–7 and MCPR3–11 recognize a distinct epitope, which we named epitope 5. These competition assay results do not support the existence of a separate epitope originally proposed as epitope 2 [30]. Based on these data we propose the revised grouping of moAbs as shown in Figure 2C.
3.3. Identification of major PR3 epitopes using human-mouse chimeric rPR3 molecules
3.3.1. Rationale for a selective surface-targeted approach
To date, the specific surface epitopes on PR3 to which the anti-PR3 moAbs bind have remained elusive. Studies performed with overlapping synthetic peptides have not identified the epitopes recognized by moAb, and results about PR3-ANCA binding have been inconclusive [25–27]. This is largely because most PR3-ANCA recognize conformational epitopes [24].
For this reason we had proposed a different approach to map conformation-sensitive PR3-ANCA: the use of human-murine chimeric molecules [37]. This strategy would take advantage of the structural similarities between human and murine PR3 as well as the observed poor cross-reactivity of human PR3-ANCA with murine rPR3[37]. Human and murine PR3 share 69% of amino-acid residues [37]. Surface regions that differ between human and murine PR3 (shown in green in Figure 3B) are also the best candidate regions for moAb-binding. In contrast, immunological tolerance to the highly conserved surface regions most likely prevents antibody formation to these structures in mice.
The initial proof-of-concept experiments were based on human-murine chimeric molecules constructed by en-bloc nucleic sequence domain swap using the two conserved PstI-restriction sites contained in the coding sequence of human and murine PR3 [37, 45]. The recombinant molecules expressed in mammalian systems have preserved serine protease activity and bind PR3-ANCA differentially [45]. Selga and co-workers subsequently used this approach to show a variable PR3-ANCA repertoire in patients with WG and illustrated its limitations [46]. If chimeric constructs are based on segmental sequence swaps dictated by shared restriction sites, the resulting conformational changes are unpredictable; as a result, no distinct binding regions for any moAb could be identified [46]. Therefore, we proceeded with the much more selective surface-oriented approach presented here.
Five small candidate surface epitopes with significant residue differences were identified (Figure 3). For each surface region we generated complementary pairs of human-murine chimeric rPR3-variants, one based on human PR3 with selected residues being replaced by the corresponding murine residues (Hm-variants), and one based on murine PR3 with the corresponding murine amino-acids being replaced by the human counterparts (Mh-variants). All 10 chimeric rPR3-variants were individually coated to Talon-beads and used as antigens for binding by the various moAbs in the FACS assay. To identify the surface epitopes recognized by the moAbs, we looked for loss of binding to Hm chimeric molecules and/or gain of binding to the corresponding Mh chimeric molecule. Representative results obtained with the various moAbs are displayed in Figure 4.
FIGURE 4. Binding of anti-PR3 monoclonal antibodies to human-mouse chimeric rPR3-variants.
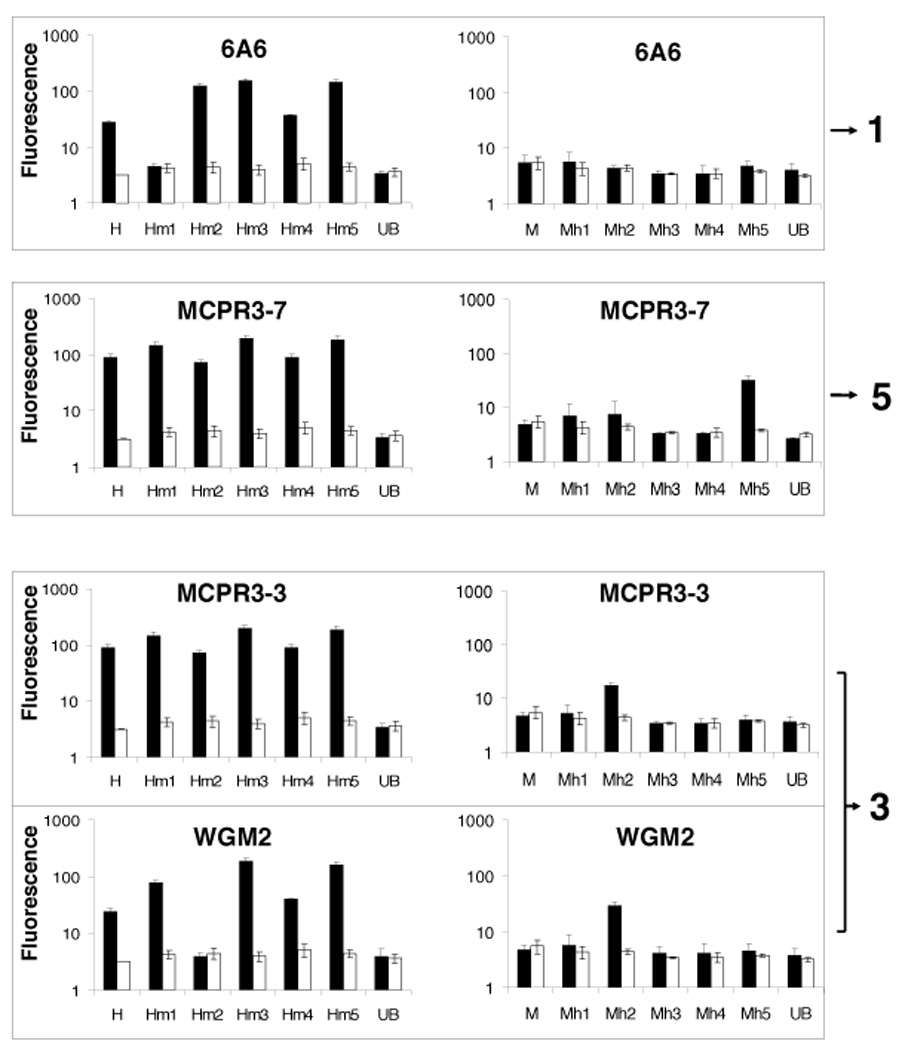
Shown are representative examples of moAb-binding to Talon-bead coated with chimeric rPR3-variants (black bars) and to antigen-free beads (white bars) in parallel experiments measured by FACS. Loss of binding to an Hm-rPR3-variant compared to human rPR3 (H), or gain of binding to a Mh-rPR3-variant compared to murine rPR3 (M) indicate binding of the moAb to the epitope targeted by the mutations (mean±SEM of 3–5 independent experiments). Epitope numbering corresponds to the grouping of moAbs.
3.3.2. Epitope 1
The moAbs 6A6 and PR3G-2 were analyzed as representatives of group 1 moAbs on human and murine rPR3 and on all 10 chimeric molecules. The loss of binding to Hm1 shows that the surface region represented by the Hm1/Mh1 chimeras is the target for these antibodies (epitope 1). It is located “South-East” of the substrate binding pocket of PR3.
3.3.3. Epitope 3
The binding region for the moAbs MCPR3-3, 4A5 and WGM-2 (group 3) was defined by the Hm2/Mh2 chimeras, indicating that epitope 3 is located on the back side of the PR3 molecule. Consequently, moAbs or autoantibodies recognizing this epitope are unlikely to interfere with the enzymatic activity of PR3.
3.3.4 Epitope 4
MCPR3-2 and 4A3 (group 4) were analyzed on all 10 chimeric rPR3 molecules; no loss or gain of binding on any of the Hm or mH variants was detected. This indicates that none of our chimeric molecules represents epitope 4. However, using human-gibbon chimeric rPR3 molecules, Dr. Jenne and coworkers identified epitope 4 as the region located “North-East” of the substrate binding pocket and adjacent to epitope 3 [47] (Figure 5). This is consistent with our finding of mild competition between MPCPR3-2 and WGM-2 (see 3.2.3.).
FIGURE 5. Summary diagram of epitope location and binding regions of different anti-PR3 monoclonal antibodies.

Arabic numbers designate the four major surface epitopes recognized by the anti-PR3 moAbs as derived from the competition studies and binding to chimeric rPR3-variants. Surface representations of mature human PR3 generated using DeepView/Swiss-Pdb viewer, V4.0 software based on the crystal structure. Colors indicate: red, negative and blue, positive surface charge; purple, the amino-terminal residues IVGGH; brown, the carboxy-terminus; yellow, active site residues H57, D102, S195. PR3 is shown in standard orientation with the active site facing the viewer (0°). Counterclockwise vertical rotation by 180° reveals the back side of the molecule. Epitope numbering corresponds to moAb grouping (note that based on our data there is currently no longer a separate epitope designated as epitope 2).
MoAbs recognizing epitope 4 (MCPR3-2 and 4A3) have been used extensively as capturing antibodies in capture-ELISA systems for PR3-ANCA testing in vasculitis because PR3-ANCA from patients with vasculitis rarely compete with these antibodies for binding to epitope 4 [3, 6, 10, 31, 48, 49]. In contrast, about half of the PR3-ANCA occurring in patients with cocaine-induced midline destructive lesions bind to epitope 4 [50], suggesting that epitope-specific PR3-ANCA subsets may be linked to different autoimmune disease phenotypes.
3.3.5. Epitope 5
The constructs Hm5/Mh5 code for mutations of three hydrophobic residues in human PR3 that are not conserved as hydrophobic residues in murine PR3 [37]. This region emerged as the target epitope for the group 5 antibodies, MCPR3–7 and MCPR3–11 (epitope 5). Computational prediction modeling has identified this hydrophobic patch as the most likely binding site of PR3 to plasma membranes, and experiments with human and gibbon rPR3 have confirmed that this region is involved in the binding of human PR3 to the neutrophil membrane receptor NB1 (CD 177) [51, 52].
Against expectations derived from the moAb competition studies (see 3.3.2), PR3G-4 showed no reduction in binding to Hm5 or gain to Mh5 (epitope 5). However, PR3G-4 was the only moAb showing loss of binding to both Hm1 and Hm4 (not shown). The Hm4/Mh4 constructs code for the region located between the Hm1/Mh1 and Hm5/Mh5 regions, and the Hm4/Mh4 constructs share residue-146 mutations with Hm5/Mh5. Together, this indicates that PR3G-4 binds in a region immediately adjacent to, but not including the cluster of hydrophobic residues represented by the Hm5/Mh5 constructs. Yet it is sufficiently close for PR3G-4 to sterically hinder the binding of MCPR3–7 or MCPR3–11 to epitope 5 in the competition studies.
Finally, none of the tested moAbs displayed loss of binding to Hm3 or gain of binding to Mh3. Thus, despite the lack of sequence homology, this region of human PR3 is not very antigenic for mice.
3.4. Estimating the functional effect of epitope-specific PR3-ANCA by capture-ELISA
The second major aim of our study was to translate the identification of PR3 surface epitopes into a clinically useful tool that for the first time allows the separation of PR3-ANCA subsets by epitope-specific functional impact. Our understanding of the moAb binding regions derived from the combination of moAb competition studies and the recognition of the chimeric rPR3-variants (summarized in Figure 5) was used to develop a simple capture ELISA system to measure the occurrence of functionally relevant PR3-ANCA subsets in clinical samples.
3.4.1. Estimating the effect of PR3-ANCA on enzyme activity by capture ELISA
Small studies have suggested that PR3-ANCA that inhibit the enzymatic activity of PR3 or its complexation with the physiologic plasma inhibitor, α1-PI, are associated with disease activity, whereas other PR3-ANCA are not [19, 20, 22]. The cumbersome nature of IgG preparation and subsequent enzyme activity measurements have precluded a formal testing of this hypothesis in large well characterized patient populations with prospectively obtained serial serum samples. Therefore, we evaluated whether our identification of the surface epitopes recognized by the various moAbs summarized in Figure 5 can be translated into a simple capture-ELISA system that provides an approximation of the ability of PR3-ANCA to inhibit the enzymatic activity of PR3.
To interfere with either enzymatic activity or complexation with α1-PI, PR3-ANCA would have to compete for binding of substrates or α1-PI on the front side of PR3. In contrast, PR3-ANCA binding to the back side of PR3 are unlikely to interfere with enzymatic activity. MCPR3-3 binds on the back side of PR3 (epitope 3), and PR3-ANCA yielding low titers in the MCPR3-3 capture-ELISA compete with MCPR3-3 for binding to the back side of PR3. Although epitope 4 is also located on the front side of PR3, the epitope 4-specific moAb MCPR3-2 does not interfere with enzymatic activity, complexation with α1-PI, and binding of PR3-ANCA from patients with WG [3, 6, 31]. Thus, the MCPR3-2 capture-ELISA results approximately reflect the total amount of PR3-ANCA with specificities for epitopes 1, 3 and 5 in the sample. Consequently, we hypothesized that parallel PR3-ANCA determinations by capture-ELISA using either MCPR3-3 or MCPR3-2 for antigen capture can provide an indirect measure for PR3-ANCA binding to epitopes 1 and 5 on the front side of PR3 and for the ability to interfere with enzymatic activity.
To test this hypothesis, we purified IgG from 27 random plasma samples from patients with known WG and determined the ability of these PR3-ANCA containing IgG preparations to inhibit the hydrolysis of MeAAPV by active purified neutrophil PR3. We then tested the paired whole plasma sample for PR3-ANCA by capture-ELISA using MCPR3-3 and MCPR3-2 for antigen capture in parallel. The ratio of the net absorbance on antigen captured by MCPR3-3 over the net absorbance on antigen captured by MCPR3-2 (MCPR3-3/MCPR3-2 ratio) was plotted against the inhibitory capacity of the corresponding IgG preparation (Figure 6A). The MCPR3-3/MCPR3-2 ratio correlated with the ability of the PR3-ANCA to inhibit enzymatic activity of PR3 (r=0.7,p<0.01).
FIGURE 6. Epitope-specific capture-ELISA to gauge the effect of PR3-ANCA on enzymatic activity of PR3 and relationship with disease activity.

A. Plasma samples from 27 patients with WG were tested for PR3-ANCA reactivity by capture-ELISA using MCPR3-2 or MCPR3-3 as antigen-capturing antibodies. The ratio of MCPR3-3/MCPR3-2 net absorbance for each sample is plotted on the X-axis. IgG was purified from the same plasma samples, tested for its ability to inhibit hydrolysis of the substrate MeAAPV by PR3 and plotted on the Y-axis. The scatter plot shows the correlation between the MCPR3-3/MCPR3-2 ratio and the ability of PR3-ANCA to inhibit enzymatic function of PR3.
B. The median MCPR3-3/MCPR3-2 ratio was higher in patients who had active disease compared to patients without detectable clinical disease activity at the time of sampling (0.42 and 0.18, respectively, p=0.038).
C. The median MCPR3-3 / MCPR3-2 ratio of patients with markedly elevated (≥ 45 mm/hr) ESR was also higher (0.45) than that of patients with normal or mildly elevated (< 45 mm/hr) ESR (0.18, p<0.01).
3.4.2. Potential of epitope-specific PR3-ANCA capture ELISA as biomarker
Subsequently, 30 stored serum samples from patients with WG were tested in the same capture-ELISA system. The median MCPR3-3/MCPR3-2 ratio of patients who were in remission at the time of sampling was significantly lower than that of patients with active disease (0.18 versus 0.42,p<0.04) (Figure 6B). Similarly, the median ratio of patients with an ESR < 45 mm/hr was significantly lower than of patients with an ESR ≥ 45 mm/hr (0.18 versus 0.45,p<0.01) (Figure 6C). The correlation between ESR and the MCPR3-3/MCPR3-2 ratio was also significant (r=0.4,p=0.04).
These preliminary results need to be interpreted with caution. First, PR3-ANCA levels cannot be compared between patients. It is well recognized that patients with low disease activity may have high titers and vice versa [1]. This may in part explain the substantial overlap of results found for individual patients in the different clinical categories (Figure 6B&C). Second, single time point assessments may under- or overestimate a significant association between PR3-ANCA titers and clinical activity, because a pathogenic autoantibody may precede clinical disease activity, and therapy may reduce disease activity before antibody levels fall.
For these reasons we also tested serial serum samples from two patients who were first seen with active disease, then went into remission with treatment, and subsequently suffered disease flares (Figure 7). PR3-ANCA testing by standard capture ELISA could not predict the clinical disease flare. In contrast, in both cases the clinical disease flare was preceded by an increase in the MCPR3-3/MCPR3-2 ratio, suggesting that this ratio may represent a better biomarker for impending disease flares than standard PR3-ANCA testing.
FIGURE 7. Epitope-specific capture-ELISA as potential predictor of disease flares.

Two cases with serial clinical follow-up (disease activity score, BVAS/WG) and PR3-ANCA determinations by capture ELISA using MCPR3-2 and MCPR3-3 as capturing antibodies. In both cases standard PR3-ANCA measurements using the MCPR3-2 capture ELISA did not show a useful association with changes in clinical disease activity. In contrast, the MCPR3-3/MCPR3-2 ratio showed an increase preceding the clinical relapse, suggesting potential of this ratio as a better biomarker for disease activity than standard PR3-ANCA testing.
Together, our results indicate that parallel PR3-ANCA measurements using epitope-specific capturing with two different moAbs (MCPR3-3 and MCPR3-2) can be used to estimate the inhibitory capacity of PR3-ANCA sera. In addition, our clinical data suggest that the MCPR3-3/MCPR3-2 ratio is indeed associated with clinical disease activity, similar to what has been reported for the enzyme inhibitory activity of PR3-ANCA IgG [22, 23].
4. Conclusions
As a result of this study, the conformational surface regions recognized by all currently available anti-PR3 moAbs have now been mapped. We have further shown that these insights can be used to separate PR3-ANCA subsets with predictable functional qualities. The ability of PR3-ANCA to inhibit the enzymatic activity of PR3 is a property linked to disease activity that can now be measured with a simple epitope-based capture-ELISA system. This paves the way for large scale analyses of serial samples from large well-characterized longitudinally followed patient cohorts.
Acknowledgements
For gifts of moAbs we thank Drs. E. Csernok (WGM2), J. Wieslander (4A3, 5A6, 6A6) and C. G.M. Kallenberg (PR3G-2,PR3G-4). We are indebted to Dr. David N. Fass for many inspirational discussions and collaboration over the last decade. Furthermore, we thank Ms. Margaret A. Viss for technical assistance with generation of some of the cDNA constructs and Mr. Randy Miller for advise with mouse immunization and FACS analysis. The moAbs MCPR3-7 and MCPR3-11 were generated with the assistance of the Mayo Clinic Monoclonal Antibody Core Facility.
This study was supported in part by NIH RO1-AR49806 and RC1-AR58303. Dr. Silva was supported in part by a fellowship from the Vasculitis Clinical Research Consortium (U54-RR019497) and by funds from the Mayo Foundation. The funding sources had no involvement in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nölle B, Specks U, Lüdemann J, Rohrbach MS, DeRemee RA, Gross WL. Anticytoplasmic autoantibodies: their immunodiagnostic value. Ann Int Med. 1989;111:28–40. doi: 10.7326/0003-4819-111-1-28. [DOI] [PubMed] [Google Scholar]
- 2.Jenne DE, Tschopp J, Lüdemann J, Utecht B, Gross WL. Wegener's autoantigen decoded. Nature. 1990;346:520. doi: 10.1038/346520a0. [DOI] [PubMed] [Google Scholar]
- 3.Finkielman JD, Lee AS, Hummel AM, Viss MA, Jacob GL, Homburger HA, Peikert T, Hoffman GS, Merkel PA, Spiera R, et al. ANCA are detectable in nearly all patients with active severe Wegener's granulomatosis. Am J Med. 2007;120:643, e9–e14. doi: 10.1016/j.amjmed.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 4.Merkel PA, Polisson RP, Chang Y, Skates SJ, Niles JL. Prevalence of antineutrophil cytoplasmic antibodies in a large inception cohort of patients with connective tissue disease. Ann Intern Med. 1997;126:866–873. doi: 10.7326/0003-4819-126-11-199706010-00003. [DOI] [PubMed] [Google Scholar]
- 5.Hagen EC, Daha MR, Hermans J, Andrassy K, Csernok E, Gaskin G, Lesavre P, Ludemann J, Rasmussen N, Sinico RA, et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization [see comments] Kidney Int. 1998;53:743–753. doi: 10.1046/j.1523-1755.1998.00807.x. [DOI] [PubMed] [Google Scholar]
- 6.Russell KA, Wiegert E, Schroeder DR, Homburger HA, Specks U. Detection of Anti-Neutrophil Cytoplasmic Antibodies under Actual Clinical Testing Conditions. Clin Immunol. 2002;103:196–203. doi: 10.1006/clim.2001.5200. [DOI] [PubMed] [Google Scholar]
- 7.Specks U. Antineutrophil cytoplasmic antibodies: are they pathogenic? Clin Exp Rheumatol. 2004;22:S7–S12. [PubMed] [Google Scholar]
- 8.Kallenberg CG. Pathogenesis of PR3-ANCA associated vasculitis. J Autoimmun. 2008;30:29–36. doi: 10.1016/j.jaut.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Boomsma MM, Stegeman CA, van der Leij MJ, Oost W, Hermans J, Kallenberg CGM, Limburg PC, Cohen Tervaert JW. Prediction of relapses in Wegener's granulomatosis by measurement of antineutrophil cytoplasmic antibody levels. A prospective study. Arthritis Rheum. 2000;43:2025–2033. doi: 10.1002/1529-0131(200009)43:9<2025::AID-ANR13>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 10.Finkielman JD, Merkel PA, Schroeder D, Hoffman GS, Spiera R, St Clair EW, Davis JC, Jr, McCune WJ, Lears AK, Ytterberg SR, et al. Antiproteinase 3 Antineutrophil Cytoplasmic Antibodies and Disease Activity in Wegener Granulomatosis. Ann Intern Med. 2007;147:611–619. doi: 10.7326/0003-4819-147-9-200711060-00005. [DOI] [PubMed] [Google Scholar]
- 11.Kerr GS, Fleisher TA, Hallahan CW, Leavitt RY, Fauci AS, Hoffman GS. Limited prognostic value of changes in antineutrophil cytoplasmic antibody titer in patients with Wegener's granulomatosis. Arthritis Rheum. 1993;36:365–371. doi: 10.1002/art.1780360312. [DOI] [PubMed] [Google Scholar]
- 12.Pfister H, Ollert M, Frohlich LF, Quintanilla-Martinez L, Colby TV, Specks U, Jenne DE. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood. 2004;104:1411–1418. doi: 10.1182/blood-2004-01-0267. [DOI] [PubMed] [Google Scholar]
- 13.van der Geld YM, Hellmark T, Selga D, Heeringa P, Huitema MG, Limburg PC, Kallenberg CG. Rats and mice immunised with chimeric human/mouse proteinase 3 produce autoantibodies to mouse Pr3 and rat granulocytes. Ann Rheum Dis. 2007;66:1679–1682. doi: 10.1136/ard.2006.064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savage CO, Harper L, Holland M. New findings in pathogenesis of antineutrophil cytoplasm antibody- associated vasculitis. Curr Opin Rheumatol. 2002;14:15–22. doi: 10.1097/00002281-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Jennette JC, Xiao H, Falk RJ. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2006;17:1235–1242. doi: 10.1681/ASN.2005101048. [DOI] [PubMed] [Google Scholar]
- 16.Specks U. What you should know about PR3-ANCA: Conformational requirements of proteinase 3 (PR3) for enzymatic activity and recognition by PR3-ANCA. Arthritis Res. 2000;2:263–267. doi: 10.1186/ar99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Geld YM, Limburg PC, Kallenberg CG. Proteinase 3, Wegener's autoantigen: from gene to antigen. J Leukoc Biol. 2001;69:177–190. [PubMed] [Google Scholar]
- 18.Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie. 2008;90:227–242. doi: 10.1016/j.biochi.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 19.van de Wiel BA, Dolman KM, van der Meer-Gerritsen CH, Nack CE, von dem Borne AEGK, Goldschmeding R. Interference of Wegener's granulomatosis autoantibodies with neutrophil Proteinase 3 activity. Clin Exp Immunol. 1992;90:409–414. doi: 10.1111/j.1365-2249.1992.tb05860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolman KM, Stegeman CA, van de Wiel BA, Hack CE, Von dem Borne AEGK, Kallenberg CGM, Goldschmeding R. Relevance of classic anti-neutrophil cytoplasmic autoantibody (C-ANCA)-mediated inhibition of proteinase 3-a1-antitrypsin complexation to disease activity in Wegener's granulomatosis. Clin Exp Immunol. 1993;93:405–410. doi: 10.1111/j.1365-2249.1993.tb08192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sommarin Y, Rasmussen N, Wieslander J. Characterization of monoclonal antibodies to proteinase 3 and application in the study of epitopes for classical anti-neutrophil cytoplasm antibodies. Exp Nephrol. 1995;3:249–256. [PubMed] [Google Scholar]
- 22.Daouk GH, Palsson R, Arnaout MA. Inhibition of proteinase 3 by ANCA and its correlation with disease activity in Wegener's granulomatosis. Kidney Int. 1995;47:1528–1536. doi: 10.1038/ki.1995.216. [DOI] [PubMed] [Google Scholar]
- 23.van der Geld YM, Tool AT, Videler J, de Haas M, Cohen Tervaert JW, Stegeman CA, Limburg PC, Kallenberg CG, Roos D. Interference of PR3-ANCA with the enzymatic activity of PR3: differences in patients during active disease or remission of Wegener's granulomatosis. Clin Exp Immunol. 2002;129:562–570. doi: 10.1046/j.1365-2249.2002.01926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bini P, Gabay JE, Teitel A, Melchior M, Zhou J-L, Elkon KB. Antineutrophil cytoplasmic autoantibodies in Wegener's granulomatosis recognize conformational epitopes on proteinase 3. J Immunol. 1992;149:1409–1415. [PubMed] [Google Scholar]
- 25.Williams RC, Staud R, Malone CC, Payabyab J, Byres L, Underwood D. Epitopes on proteinase-3 recognized by antibodies from patients with Wegener's granulomatosis. J Immunol. 1994;152:4722–4737. [PubMed] [Google Scholar]
- 26.Chang L, Binos S, Savige J. Epitope mapping of anti-proteinase 3 and anti-myeloperoxidase antibodies. Clin Exp Immunol. 1995;102:112–119. doi: 10.1111/j.1365-2249.1995.tb06644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffith ME, Coulthart A, Pemberton S, George AJ, Pusey CD. Anti-neutrophil cytoplasmic antibodies (ANCA) from patients with systemic vasculitis recognize restricted epitopes of proteinase 3 involving the catalytic site. Clin Exp Immunol. 2001;123:170–177. doi: 10.1046/j.1365-2249.2001.01420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csernok E, Ludemann J, Gross WL, Bainton DF. Ultrastructural localization of proteinase 3, the target antigen of anti-cytoplasmic antibodies circulating in Wegener's granulomatosis. Am J Pathol. 1990;137:1113–1120. [PMC free article] [PubMed] [Google Scholar]
- 29.Goldschmeding R, van der Schoot CE, ten Bokkel Huinink D, Hack CE, van den Ende ME, Kallenberg CGM, von dem Borne AEGK. Wegener's granulomatosis autoantibodies identify a novel diisopropylfluorophosphate-binding protein in the lysosomes of normal human neutrophils. J Clin Invest. 1989;84:1577–1587. doi: 10.1172/JCI114335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Geld YM, Limburg PC, Kallenberg CG. Characterization of monoclonal antibodies to proteinase 3 (PR3) as candidate tools for epitope mapping of human anti-PR3 autoantibodies. Clin Exp Immunol. 1999;118:487–496. doi: 10.1046/j.1365-2249.1999.01079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun J, Fass DN, Hudson JA, Viss MA, Wieslander J, Homburger HA, Specks U. Capture-ELISA based on recombinant PR3 is sensitive for PR3-ANCA testing and allows detection of PR3 and PR3-ANCA/PR3 immunecomplexes. J Immunol Methods. 1998;211:111–123. doi: 10.1016/s0022-1759(97)00203-2. [DOI] [PubMed] [Google Scholar]
- 32.Warren HS, Bettadapura J. A novel binding assay to assess specificity of monoclonal antibodies. J Immunol Methods. 2005;305:33–38. doi: 10.1016/j.jim.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Capizzi SA, Viss MA, Hummel AM, Fass DN, Specks U. Effects of carboxy-terminal modifications of proteinase 3 (PR3) on the recognition by PR3-ANCA. Kidney Int. 2003;63:756–760. doi: 10.1046/j.1523-1755.2003.00765.x. [DOI] [PubMed] [Google Scholar]
- 34.Specks U, Fass DN, Fautsch MP, Hummel AM, Viss MA. Recombinant human proteinase 3, the Wegener's autoantigen, expressed in HMC-1 cells is enzymatically active and recognized by c-ANCA. FEBS Lett. 1996;390:265–270. doi: 10.1016/0014-5793(96)00669-2. [DOI] [PubMed] [Google Scholar]
- 35.Sun J, Fass DN, Viss MA, Hummel AM, Tang H, Homburger HA, Specks U. A proportion of proteinase 3-specific anti-neutrophil cytoplasmic antibodies only react with proteinase 3 after cleavage of its N-terminal activation dipeptide. Clin Exp Immunol. 1998;114:320–326. doi: 10.1046/j.1365-2249.1998.00730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiesner O, Litwiller RD, Hummel AM, Viss MA, McDonald CJ, Jenne DE, Fass DN, Specks U. Differences between human proteinase 3 and neutrophil elastase and their murine homologues are relevant for murine model experiments. FEBS Lett. 2005;579:5305–5312. doi: 10.1016/j.febslet.2005.08.056. [DOI] [PubMed] [Google Scholar]
- 37.Jenne DE, Fröhlich L, Hummel AM, Specks U. Cloning and functional expression of the murine homologue of proteinase 3: implications for the design of murine models of vasculitis. FEBS Lett. 1997;408:187–190. doi: 10.1016/s0014-5793(97)00418-3. [DOI] [PubMed] [Google Scholar]
- 38.Fujinaga M, Chernaia MM, Halenbeck R, Koths K, James MN. The crystal structure of PR3, a neutrophil serine proteinase antigen of Wegener's granulomatosis antibodies. J Mol Biol. 1996;261:267–278. doi: 10.1006/jmbi.1996.0458. [DOI] [PubMed] [Google Scholar]
- 39.Specks U, Wiegert EM, Homburger HA. Human mast cells expressing recombinant proteinase 3 (PR3) as substrate for clinical testing for anti-neutrophil cytoplasmic antibodies (ANCA) Clin Exp Immunol. 1997;109:286–295. doi: 10.1046/j.1365-2249.1997.4561353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee AS, Finkielman JD, Peikert T, Hummel AM, Viss MA, Specks U. A novel capture-ELISA for detection of anti-neutrophil cytoplasmic antibodies (ANCA) based on c-myc peptide recognition in carboxy-terminally tagged recombinant neutrophil serine proteases. J Immunol Methods. 2005;307:62–72. doi: 10.1016/j.jim.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Finkielman JD, Merkel PA, Schroeder D, Hoffman GS, Spiera R, St Clair EW, Davis JC, Jr, McCune WJ, Lears A, Ytterberg SR, et al. Glycosylation of proteinase 3 (PR3) is not required for its reactivity with antineutrophil cytoplasmic antibodies (ANCA) in Wegener's granulomatosis. Clin Exp Rheumatol. 2009;27:S45–S52. [PMC free article] [PubMed] [Google Scholar]
- 42.Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, Calabrese LH, Fries JF, Lie JT, Lightfoot RWJ, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990;33:1101–1107. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 43.Jennette JC, Falk RJ, Andrassy K, Bacon BA, Churg J, Gross WL, Hagen EC, Hoffmann GS, Hunder GG, Kallenberg CGM, et al. Nomenclature of systemic vasculitides: The proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–192. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 44.van der Geld YM, Rarok A, Specks U, Limburg PC, Kallenberg CGM. Monoclonal antibodies directed against proteinase 3 as tools for epitope mapping of c-ANCA positive sera (abstract) Clin Exp Immunol. 1998;112 Suppl1 [Google Scholar]
- 45.Specks U, Jenne DE, Fass DN, Hummel AM, C.J. M. Recombinant human-murine chimaeric proteinase 3 (PR3) molecules as tools for PR3-ANCA epitope mapping. FASEB J. 1999;13:A959. [Google Scholar]
- 46.Selga D, Segelmark M, Wieslander J, Gunnarsson L, Hellmark T. Epitope mapping of anti-PR3 antibodies using chimeric human/mouse PR3 recombinant proteins. Clin Exp Immunol. 2004;135:164–172. doi: 10.1111/j.1365-2249.2004.02314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuhl A, Korkmaz B, Utecht B, Kniepert A, Schönermark U, Specks U, Jenne DE. Mapping of conformational epitopes on human proteinase 3, the autoantigen of Wegener's granulomatosis. J Immunol. 2010 doi: 10.4049/jimmunol.0903887. In press. [DOI] [PubMed] [Google Scholar]
- 48.Baslund B, Segelmark M, Wiik A, Szpirt W, Petersen J, Wieslander J. Screening for anti-neutrophil cytoplasmic antibodies (ANCA): is indirect immunofluorescence the method of choice? Clin Exp Immunol. 1995;99:486–492. doi: 10.1111/j.1365-2249.1995.tb05577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee AS, Finkielman JD, Peikert T, Hummel AM, Viss MA, Jacob GL, Homburger HA, Specks U. Agreement of anti-neutrophil cytoplasmic antibody measurements obtained from serum and plasma. Clin Exp Immunol. 2006;146:15–20. doi: 10.1111/j.1365-2249.2006.03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peikert T, Finkielman JD, Hummel AM, McKenney ME, Gregorini G, Trimarchi M, Specks U. Functional characterization of antineutrophil cytoplasmic antibodies in patients with cocaine-induced midline destructive lesions. Arthritis Rheum. 2008;58:1546–1551. doi: 10.1002/art.23469. [DOI] [PubMed] [Google Scholar]
- 51.Hajjar E, Mihajlovic M, Witko-Sarsat V, Lazaridis T, Reuter N. Computational prediction of the binding site of proteinase 3 to the plasma membrane. Proteins. 2008;71:1655–1669. doi: 10.1002/prot.21853. [DOI] [PubMed] [Google Scholar]
- 52.Korkmaz B, Kuhl A, Bayat B, Santoso S, Jenne DE. A hydrophobic patch on proteinase 3, the target of autoantibodies in Wegener granulomatosis, mediates membrane binding via NB1 receptors. J Biol Chem. 2008;283:35976–35982. doi: 10.1074/jbc.M806754200. [DOI] [PubMed] [Google Scholar]