

Abstract
There is a need for new adjuvants that will induce immune responses to subunit vaccines. We show that a short peptide, named Hp91, whose sequence corresponds to an area within the endogenous molecule High mobility group box (HMGB1) protein potentiates cellular immune responses to peptide antigen and cellular and humoral immune responses to protein antigen in vivo. Hp91 promoted the in vivo production of the immunomodulatory cytokines, IFN-γ, TNF-α, IL-6, and IL-12 (p70), as well as antigen-specific activation of CD8+ T cells. These results demonstrate the ability of a short immunostimulatory peptide to serve as an adjuvant for subunit vaccines.
Keywords: HMGB-1 peptides, Adjuvants, Vaccine, Immunotherapy, Dendritic cells
1. Introduction
Vaccines traditionally have, and still consist of whole-inactivated or live-attenuated pathogens or toxins [1,2]. The usage of these modified pathogens is however unattractive for several reasons. Live attenuated pathogens can cause disease by reverting to a more virulent phenotype, especially in the non-developed immune system of newborns or immunodeficient patients, and whole inactivated pathogens contain reactogenic components that can cause undesirable vaccine side effects. Therefore, there is growing interest and research to develop a new generation of vaccines containing recombinant protein subunits, synthetic peptides, and plasmid DNA [1]. While these new modalities promise to be less toxic, many are poorly immunogenic when administered without an immune-stimulating adjuvant. As adjuvants are a crucial component of the new generation of vaccines, there is a great need for safer and more potent adjuvants [1-3].
The development of the appropriate type of immune response is essential for successful immunisation. Robust cell-mediated immunity, which is associated with a Th1 type immune response, is thought to be required for the control of intracellular pathogens [4], viruses [5] as well as cancer [6]. Humoral immunity, characterized by a Th2 type response is useful for vaccination against extracellular pathogens, such as bacteria. By choosing an appropriate adjuvant, the immune response can be selectively modulated to initiate a Th1 or Th2-type [7]. Aluminum salts (alum), which are the only vaccine adjuvants currently approved by the US Food and Drug Administration for use in humans [8,9] are not ideal adjuvants for certain pathogens, since they favor a Th2 response with weak or absent Th1 responses [10-14]. Although neutralizing antibodies from a Th2 response can be protective against many pathogens, the generation of Th1 and cytotoxic T lymphocyte (CTL) responses are important, playing crucial roles in the protection and recovery from viruses, intracellular bacteria, and cancer cells.
Pathogen associated molecular patterns (PAMPs) are small molecular sequences commonly associated with pathogens, such as CpG unmethylated bacterial DNA sequences, lipopolysaccharide (LPS), or poly(I:C) [15-18]. While many PAMPs have been investigated for their use as vaccine adjuvants, their development has been slowed for several reasons, including reactogenicity, toxicity, and ability to induce or exacerbate autoimmune diseases[19]. For instance, CpG oligodeoxynucleotides, which signal through TLR9, can activate antigen-presenting cells, induce a wide variety of cytokines, and generate a potent cellular Th1 immune response in mice, initially showed strong clinical promise [20-23]. However, clinical trials in humans utilizing CpG as a cancer immunotherapy adjuvant failed to produce the potent immune responses that were anticipated, and low TLR9 expression in human plasmacytoid DCs may be implicated [24]. Identification of new adjuvants demonstrating low-toxicity and the ability to stimulate a cellular Th1 response in humans would be a great advancement in the development of vaccines for infectious disease and cancer.
In contrast to PAMPs, endogenous molecules and proteins have been proposed and studied as adjuvants. Examples of such endogenous molecules, or danger-associated molecular patterns (DAMPs), include heat stock proteins, cytokines, and high mobility group box 1 (HMGB1) protein [25,26]. Originally identified as a nuclear protein, HMGB1 modulates the innate immune response when released into the extracellular compartment by necrotic and damaged cells [27,28]. HMGB1 is a potent pro-inflammatory cytokine, released by monocytes and macrophages following exposure to LPS, tumor necrosis factor (TNF)-α or IL-1β and as a result of tissue damage [27,29]. Extracellular HMGB1 promotes the maturation of myeloid and plasmacytoid DCs [30-32] and it has been shown to act as immune adjuvant by enhancing immunogenicity of apoptotic lymphoma cells and eliciting antibody responses to soluble ovalbumin protein [33].
We have previously identified a short peptide, named Hp91, within the B box domain of HMGB1 that induces activation of human and mouse DCs [25]. Hp91-activated DCs show increased secretion of pro-inflammatory cytokines and chemokines, including the Th1 cytokine, IL-12. In addition, DCs exposed to HMGB1-derived peptides induced proliferation of antigen-specific syngeneic T cells in vitro [25]. These immunostimulatory properties of Hp91 and the fact that peptides are easy to manufacture make it an attractive candidate as an adjuvant for vaccine development. Here we show that the immunostimulatory peptide (ISP) Hp91 acts as an adjuvant in vivo by enhancing immune responses to peptide and protein antigen.
2. Materials and methods
2.1 Reagents and cell lines
The OVA-transfected EL4 line, E.G7-OVA (ATCC, Manassas, VA, USA), was cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Omega Scientific, Tarzana, CA, USA), 10 mM HEPES (Invitrogen), and penicillin (100 U ml−1) - streptomycin (100 μg ml−1) - L-glutamine (2 mM) (Invitrogen).
2.2 Peptides and protein
The peptides, including the ISP Hp91 (DPNAPKRPPSAFFLFCSE), Hp121 (SIGDVAKKLGEMWNNTAA), the MHC-Class I (H-2Kb)-restricted peptide epitope of ovalbumin (OVA-I: OVA 257-264 aa, SIINFEKL), and the MHC-Class II (I-Ab)-restricted peptide epitope of ovalbumin (OVA-II: OVA 323-339 aa, ISQAVHAAHAEINEAGR) were all purchased from GenScript Corp (Piscataway, NJ, USA) and CPC Scientific (San Jose, CA, USA). Hp91 and Hp121 peptides were synthesized with an N-terminal biotin. Peptides were routinely synthesized with greater than 95% purity. LPS-free chicken egg white ovalbumin protein was kindly provided by Dr. Thomas Moran (Department of Microbiology, Mount Sinai School of Medicine, New York, NY, USA). Unless otherwise stated, all peptides and proteins were dissolved in PBS in preparation for immunisation.
2.3 Mice and Immunisations
Female C57BL/6 mice 8-12 weeks of age were used for most experiments. All mice were purchased from Charles River Laboratories (Boston, MA, USA) and housed at the Moores UCSD Cancer Center animal facility. All animal studies were approved by the Institutional Animal Care and Use Committee of UCSD and were performed in accordance with the institutional guidelines. For most experiments, mice were immunised s.c. with 50 μg of SIINFEKL (OVA-I) peptide and 50 μg of ISQAVHAAHAEINEAGR (OVA-II) peptide. The OVA peptide was co-administered with either Hp91 (30 to 500 μg) or PBS (negative control). For some experiments, a protein vaccine group was included, wherein 500 μg of Hp91 was co-administered with 100 μg of LPS-free OVA protein. As a positive control, mice were immunised s.c. with OVA peptide(s) or protein in Incomplete Freund's Adjuvant “IFA” (Sigma-Aldrich, St. Louis, MO, USA). If not otherwise indicated, mice were immunised and boosted two weeks later and spleens and blood were collected 10-14 days after the final immunisation.
2.4 Intravenous administration of ISP
C57BL/6 mice were injected i.v. with 0, 10 or 100 μg Hp91 dissolved in PBS into the tail vein. Blood was collected after 2h and 24h by retroorbital puncture. Blood was allowed to clot and serum was isolated after centrifugation. Serum was diluted and analysed for systemic cytokine and chemokine release by ELISA (eBioscience, San Diego, CA, USA).
2.5 Detection of antigen-specific antibody production by ELISA
Serum was obtained by retroorbital puncture or cardiac puncture from mice following immunisation. Blood was allowed to clot and serum was isolated after centrifugation. Microtiter plates were coated overnight with OVA protein (Sigma-Aldrich), blocked with BSA, and dilutions of serum were added to the plates for incubation. Plates were washed, incubated with anti-mouse IgG or IgM peroxidase conjugated antibodies (Roche, Basel, Switzerland), developed using Zymed TMB substrate (Invitrogen), and analysed using a microplate reader at 450 nm.
2.6 Spleen cell preparation
Single cell suspensions of splenocytes were prepared by mechanical disruption and separation through a 70 μm nylon cell strainer (BD Biosciences, Franklin Lakes, NJ, USA). Red blood cells were lysed using ammonium chloride buffer (Roche Diagnostics, Indianapolis, IN, USA) and the splenocytes were subsequently resuspended in complete medium (RPMI 1640 with 10% FBS, L-glutamine, penicillin, streptomycin, and HEPES). In some experiments, CD4+ and CD8+ cells were depleted from bulk splenocyte populations using anti-CD4 or anti-CD8α conjugated microbeads (Miltenyi-Biotec, Auburn, CA, USA) according to the manufacturer's instructions.
2.7 Enzyme-linked immunospot assay
Freshly isolated splenocytes were plated in duplicate to wells of a nitrocellulose bottom enzyme-linked immunospot (ELISPOT) plate (Millipore, Millerica, MA, USA) that had been previously coated overnight with 5 μg ml−1 monoclonal anti-mouse IFN-γ antibody (Mabtech, Stockholm, Sweden). Splenocytes were cultured overnight at 37°C with 2.5 μg ml−1 SIINFEKL (OVA-I) peptide, 2.5 μg ml−1 ISQAVHAAHAEINEAGR (OVA-II) peptide, or left unstimulated (medium only). After 18 h, culture supernatants were collected for cytokine analysis and ELISPOT plates were developed using 1 μg ml−1 biotinylated anti-mouse IFN-γ antibody (Mabtech), Streptavidin-HRP (Mabtech), and TMB Substrate (Mabtech). The plate was scanned and the spots were counted using an automated ELISpot Reader System (CTL ImmunoSpot, Shaker Heights, OH, USA).
2.8 Measurement of cytokines and chemokines
Splenocytes were cultured overnight with 2.5 μg ml−1 OVA-I peptide, 2.5 μg ml−1 OVA-II peptide, 5 μg ml−1 concanavalin A positive control (Sigma), or left unstimulated (media only). After 18 h, cell culture supernatants were collected and analysed for the presence of IL-2 and IL-4 by ELISA (eBioscience).
2.9 LDH Cytotoxicity Assay
Splenocytes were expanded in culture at 3 × 106 cells ml−1 in complete medium with mitomycin-C (Sigma-Aldrich)-treated E.G7-OVA at a 5:1 ratio in 6 well plates. Four days later, live cells were isolated on a lympholyte gradient (Cedarlane Laboratories Limited, Burlington, Ontario, Canada) and cultured in complete medium with 25 U ml−1 IL-2 (R&D Systems, Minneapolis, MN, USA) for two additional days. Cytotoxicity assays were performed using a CytoTox96 Non-Radioactive Cytotoxicity Assay Kit (Promega, Madison, WI, USA). 1 × 104 E.G7-OVA cells per well were plated as target cells. Expanded splenocyte effector cells were incubated with the target cells at effector to target ratios of 1:1, 3:1, 10:1, and 30:1. Cultures were incubated in phenol-red free RPMI (Invitrogen) with 5% FBS (Omega) for 6h at which point the cell culture supernatants were harvested. The lactate dehydrogenase (LDH) released from lysed cells was proportional to the resulting red formazan product, and was quantified using a microplate reader at 490 nm absorbance. The percentage of cytotoxicity was calculated according to the following equation: % Cytotoxicity = [(E – St – Se)/(M – St)] × 100. Abbreviations are as follow; E = LDH release by effector-target coculture, St = spontaneous release by target cells, Se = spontaneous release by effectors, and M = maximal release by target cells.
2.10 Statistical analysis
Data represented are mean ± SEM. Data were analysed for statistical significance using unpaired Student's t-test, 2-way ANOVA, or linear regression. Statistical analyses were done using GraphPad software version 5.01 for Windows (GraphPad Software, San Diego, CA, USA). A p value <0.05 was considered statistically significant for these analyses.
3. Results
3.1 Hp91 induces cytokine release in vivo
We have previously shown that exposure of DCs to an immunostimulatory peptide (ISP) named Hp91 in vitro leads to secretion of inflammatory as well as Th1 skewing cytokines[25]. To examine the adjuvant properties of Hp91 in vivo, serum cytokine responses were measured after intravenous (i.v.) injection of Hp91 into mice. Increased secretion of the Th1 cytokines IFN-γ, IL-12 (p70), as well as TNF-α and IL-6, was observed within 2 h of injection, with levels generally rising further over 24 h (Fig.1).
Fig. 1. Hp91 causes release of cytokines in vivo.

Mice were injected i.v. into the tail vein with Hp91 (10 or 100 μg) or PBS (denoted as 0 μg Hp91). Blood was collected after 2 and 24 h and serum was analysed for IFN-γ, IL-6, IL-12 (p70), and TNF-α by ELISA. Data shown are mean (+/− SEM) from 3-4 mice per group.
3.2 Hp91 enhances CD8 T cell responses to peptide antigen
Since the ISP Hp91 activates DCs and induces antigen-specific T cell responses in vitro[25], we tested whether Hp91 acts as a adjuvant to induce antigen-specific immune responses in vivo. The ISP Hp91 at both doses tested (250 μg and 500 μg) when co-administered with OVA peptides, caused a significant increase in the number of antigen-specific IFN-γ secreting T cells when splenocytes were restimulated with the CD8 epitope SIINFEKL (Fig. 2A), but not when restimulated with the CD4 epitope ISQAVHAAHAEINEAGR (Fig. 2B). Incomplete Freund's Adjuvant (IFA) a known stimulator of cell-mediated immune responses, elicited strong cellular immune responses when splenocytes were restimulated with either the CD8 or CD4 epitope (Fig. 2A,B).
Fig. 2. Cellular immune response in Hp91 immunised mice.
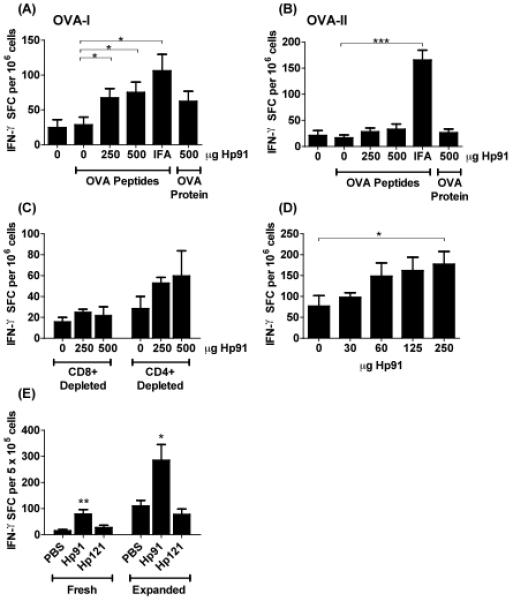
(A-B) Mice were immunised with OVA peptides in PBS (denoted as “0”), Hp91 (250 and 500 μg), or IFA. One group of mice was immunised with OVA protein and Hp91 (500 μg). Freshly-isolated splenocytes from the immunised mice were cultured in the presence of (A) OVA-I (SIINFEKL) peptide (2.5 μg ml−1) or (B) OVA-II (ISQAVHAAHAEINEAGR) (2.5 μg ml−1) in an IFN-γ ELISPOT assay. The number of IFN-γ-secreting cells was determined 18 hours later. The data shown (IFN-γ spot-forming cells per million cells) are means (+/− SEM) for 10 mice/group, except the Hp91-OVA protein group which is n=5. Asterisk, p < 0.05 between groups; Student's t-test. Data are representative of at least 3 independent experiments. (C) Freshly isolated splenocytes from PBS/OVA-peptide and Hp91/OVA-peptide immunised mice were depleted of CD8+ or CD4+ T cells and stimulated overnight in the presence of 2.5 μg ml−1 OVA-I (SIINFEKL) peptide in an IFN-γ ELISPOT assay as above. Data shown are means (+/− SEM) for 5 mice per group. (D) Mice were immunised twice with OVA-I (SIINFEKL) peptide (50ug) co-injected with 0, 30, 60, 125, or 250 μg Hp91 dissolved in PBS. Splenocytes from immunised mice were cultured in an OVA-I IFN-γ ELISPOT assay as above. Spleens were collected 6 days after the boost. Asterisk, p < 0.05 between groups; Student's t-test compared to PBS. (E) Mice were immunized with OVA peptides in PBS, Hp91 or Hp121 (250 μg). Mice received an additional boost s.c. into the contralateral flank one month after the first boost. Freshly-isolated or OVA-I-expanded splenocytes were cultured in an OVA-I IFN-γ ELISPOT assay as above. The number of IFN-γ spot-forming cells are shown as means (+/− SEM) for 4 mice per group. Asterisks, (*<0.05 and **<0.005); 2-way ANOVA.
The OVA-I peptide (SIINFEKL) is recognized in the context of H-2Kb MHC-Class I molecules and is specific for CD8+ T cells. To further confirm that the observed immune response is an OVA-specific CD8+ T cell response, CD4+ or CD8+ cells were depleted from the splenocytes prior to setup up the ELISPOT assay. While greater than 95% of CD4+ cells were depleted, CD8+ depletion was not as complete; 20% of CD8+ cells remained in the cultures as observed by flow cytometry (data not shown). As expected, the number of IFN-γ secreting cells was reduced to near background levels (OVA peptides/PBS) in the CD8+ depleted splenocyte populations (Fig. 2C). In contrast to the CD8+ depletion, the CD4+ depleted splenocytes from the Hp91-OVA immunised groups retained the ability to secrete IFN-γ in response to OVA-I peptide stimulation further supporting the involvement of CD8+ T cells following co-immunisation with the ISP Hp91 and OVA peptide, showing that the ISP Hp91 causes activation and proliferation of antigen-specific CD8+ T lymphocytes in vivo.
To test whether lower doses of Hp91 would suffice as adjuvant for immunization, titrated doses of Hp91 were co-injected with OVA peptide to determine the minimum injection dose required for a significant increase in antigen-specific IFN-γ secreting T cells. As expected, a dose response was observed (Fig. 2D). However, with only 5 mice per group, a significant increase in IFN-γ secreting T cells was observed by ELISPOT only in the group receiving an Hp91 dose of 250 μg, suggesting 250 μg is an optimal dose when immunising small groups of mice. Subsequent experiments showing 500 μg of Hp91 were conducted prior to the titration experiment.
3.3 The ISP adjuvant effect is sequence specific
To test if the in vivo adjuvant effect is related to the sequence of Hp91 or if any peptide will cause similar effects, we used a control peptide named Hp121. Hp121 is also derived from HMGB1 B-box and has the same length, a similar charge, and isoelectric point as Hp91. Hp121 does not cause activation of human DCs [25] or mouse BM-DCs in vitro (data not shown). Mice were immunised s.c. with OVA-I peptide co-injected with either Hp91, Hp121 or PBS control. Mice immunised with Hp91/OVA-I peptide showed a significantly increased number of INF-γ secreting cells as compared to the Hp121/OVA-I peptide and PBS/OVA-I peptide immunised mice using freshly isolated as well as expanded splenocytes (Fig. 2E). No significant increase was observed between the Hp121/OVA peptide and PBS/OVA peptide immunised groups.
3.4 Hp91 induces Th1-type immune response in vivo
Since IL-2 is critical for the activation, survival, and proliferation of T lymphocytes, we tested whether IL-2 secretion is increased in Hp91/OVA peptide immunised mice. Freshly isolated splenocytes were cultured overnight in presence of OVA peptide and culture supernatants were analysed for IL-2 (Figure 3) and IL-4, which may indicate the induction of a Th2 type immune response (data not shown) by ELISA. The highest IL-2 secretion was observed in OVA-I restimulated splenocytes from mice immunised with Hp91/OVA peptides (Fig. 3A), which showed higher IL-2 secretion compared to mice vaccinated with whole OVA protein/Hp91 or OVA peptide/IFA, both of which were also significantly increased as compared to the PBS/OVA control. After exposure to the MHC-Class II specific OVA-II peptide, splenocytes from Hp91/OVA peptide immunised mice showed a low but significant increase in IL-2 secretion as compared to PBS/OVA immunised mice (Figure 3B). IL-4 was not detected in the splenocytes cultures at any of the conditions, though it was detected in the ConA stimulated positive control (data not shown).
Fig. 3. Cytokine secretion in Hp91 immunised mice.
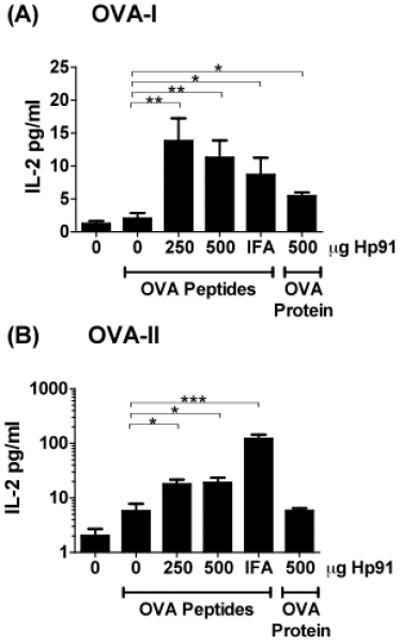
Mice were immunised with OVA peptides (50μg) co-injected with Hp91 (0, 250 or 500 μg dissolved in PBS) or IFA. One group of mice was immunised with OVA protein and Hp91 (500 μg) in PBS. Splenocytes from immunised mice were stimulated overnight with 2.5 μg ml−1 of (A) OVA-I (SIINFEKL) peptide or (B) OVA-II (ISQAVHAAHAEINEAGR). Culture supernatants were collected and analysed for IL-2 secretion by ELISA. Data shown are mean (+/− SEM) for 5-10 mice per group. Asterisk, p < 0.05; Student's t-test.
3.5 Hp91 elicits antibody responses to soluble protein
Immunisation using OVA protein in context of Hp91 promoted an antibody response to OVA which was dominated by the IgG1 isotype (Fig. 4A, 4B). Two-fold serial dilutions of the Hp91/OVA protein group show similar serum titers for the group (Fig. 4C). Minimal increase in IgG2b and IgM OVA specific antibodies was detected (data not shown). As expected, immunisation using OVA peptides (OVA-I and OVA-II) together with Hp91 did not induce antibody responses (Fig. 4A and data not shown).
Fig. 4. Antibody responses in Hp91 immunised mice.

Serum was obtained from immunised mice (5 mice per group) 10 days after the final immunisation and analysed for antibody levels by ELISA. (A) A 1:100 dilution of the serum from immunised mice as indicated, (B) a 1:100 dilution of serum from mice immunised with OVA protein and Hp91 (0 or 250μg in PBS) or (C) a serial dilution of serum from mice immunised with OVA protein and Hp91 (500 μg) in PBS was added to the plates, followed by a peroxidase-conjugated anti-mouse IgG1 antibody. Plates were developed with TMB substrate and absorbance was analysed on a microplate reader. Asterisks, p < 0.001; Student's t-test.
3.6 Co-administration of Hp91 with antigen induces CTL responses
To further investigate whether OVA-specific cytotoxic T lymphocytes (CTL) responses were induced by immunisation with the ISP Hp91, splenocytes from immunised mice were assessed for their ability to lyse OVA-expressing E.G7-OVA cells. The strongest killing was observed using effector splenocytes from mice immunised with Hp91/OVA peptides (Fig. 5), which was even higher than of splenocytes from IFA/OVA peptide immunised mice. When data were analysed by linear regression and slopes were compared, the percent killing of target cells by effectors cells from mice immunized with Hp91/OVA peptides was significantly higher than the PBS control group.
Fig. 5. CTL Induction in immunised mice.
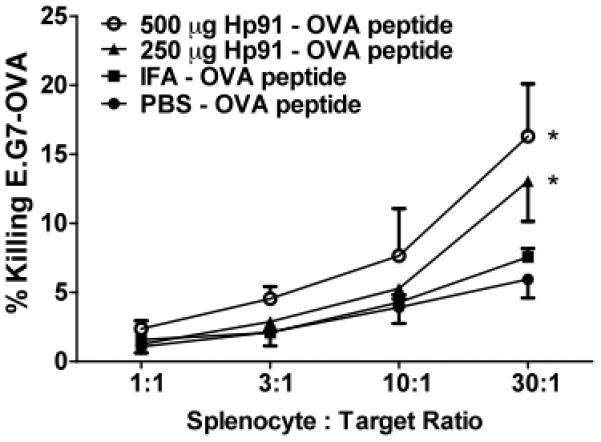
Expanded splenocytes from immunised mice were cultured with E.G7-OVA cells at the indicated effector to target ratios for 6h. Cell culture supernatants were collected and cytotoxicity was quantified in a CytoTox96 non-radioactive cytotoxicity assay. Data from at least 3 mice per group are shown. Data were analysed by linear regression and slopes were compared for significance. Asterisk, p < 0.05; compared to PBS.
4. Discussion
Although subunit vaccines promise to be less toxic, many are poorly immunogenic when administered without adjuvant. Alum, though FDA-approved, generates a weak Th1 response with a questionable safety profile. Thus, there is a great need for safer and more potent adjuvants [1-3].
We have previously shown that the 18 amino acid long ISP Hp91, is a potent stimulus for human DCs with the ability to generate a Th1-type immune response in vitro [25]. Here we demonstrate that Hp91 acts as adjuvant in vivo; inducing cellular immune responses to peptide and both cellular and humoral immune responses to protein antigen. The CD8 immune response was strong, since no in vitro expansion of splenocytes was needed to obtain a significant response as is commonly performed when testing vaccine responses. We show that the ISP Hp91 acts as an immune adjuvant to induce antigen-specific CD8 T cell responses in vivo. In addition, the immunostimulatory effects of Hp91 are related to its sequence, as the HMGB1 control peptide, Hp121, while matching Hp91 in length, isoelectric point, and charge, failed to induce cellular immune response.
The cytokine profile induced by an immune adjuvant plays an important role in the polarization of the immune response. The data show that co-immunisation with the ISP Hp91 and OVA peptides as well as OVA protein results in OVA-specific secretion of IL-2, suggesting that immunised mice are able to mount an adaptive immune response that activates T cells to synthesize and secrete IL-2 for in vivo proliferation of OVA-specific effector T cells. Interestingly, IL-4 levels were undetectable. Intravenous administration of Hp91 resulted in increased secretion of the Th1 cytokines IFN-γ and IL-12 (p70) associated with cell-mediated immunity. This together with the measured IFN-γ secretion by the T cells along with undetectable IL-4 suggests that Hp91 induces a Th1-type of immune response in vivo. We also show that immunisation with Hp91/OVA peptide elicited stronger CTL responses than IFA/OVA.
Although the main objective was to test the potency of Hp91 as adjuvant for peptide vaccines and induction of cellular immune responses, we show that immunisation using OVA protein mixed with Hp91 also induced humoral immune responses. This is very promising for future development of this novel adjuvant, as it could be used for prophylactic vaccination against infectious disease.
We have previously shown that Hp91 can activate DCs, but the exact mechanism of action is still under investigation. The Hp91 peptide sequence corresponds to a region within the B-Box domain of HMGB1 protein [25]. Toll-like receptors (TLRs) 2 and 4 have been shown to be involved in HMGB1 signaling in vitro [34-37] and in vivo data have shown TLR4 to be involved in HMGB1-induced inflammation [38]. We have previously shown that that the HMGB1 immunostimulatory peptides, including Hp91, activate mouse BM-DCs independent of TLRs 2, 4, 9, and MyD88 [25]. HMGB1 has been shown to contribute to LPS-mediated DC maturation via RAGE [39]. The C-terminal motif of HMGB1 (150–183 amino acids) is responsible for RAGE binding [40]. The HMGB1-Bx and the “active” peptides do not contain the C-terminal motif; therefore, it is unlikely that DC maturation induced by HMGB1-Bx or the peptides occurs through RAGE. We have recently shown that utilizing Hp91-loaded nanoparticles for endocytic delivery to DCs demonstrate enhanced potency of the ISP and may suggest that the receptor is not on the cell surface [41], but likely in an intracellular compartment such as an endosome. At this time we cannot exclude that Hp91 signals through a TLR3, MyD88 independent pathway or acts via other receptors. Studies are currently underway to identify the Hp91 receptor.
The ability of Hp91 to induce antigen-specific cell mediated, Th1 immune response may make Hp91 suitable as an adjuvant in cancer immunotherapies as well as vaccines against infectious diseases caused by intracellular bacteria [4] or viruses [42,43]. Some of the adjuvants that are being evaluated in clinical and preclinical settings like CD40L [44] and poly(I:C) synthetic double stranded RNA [45], act on myeloid DCs and have shown promising results for tumour immunotherapy [46], emphasizing the importance of activating myeloid DCs. As a peptide adjuvant that acts directly on myeloid DCs [25], Hp91 has several advantages. It can be made synthetically, is inexpensive, can be produced in high quantities at GMP quality, and it can also be genetically engineered to DC targeting molecules like DEC-205 which promotes strong immune responses when linked to a DC stimulatory molecule [47,48]. Since the tested doses of Hp91 have shown no adverse effects in mice to date, this current data suggest this endogenous peptide should be well-tolerated for use in vaccines.
Acknowledgements
We would like to thank D. Futalan, D. Seible, J. Steiner, J. Ahlqvist, and N. Ambren for excellent technical assistance. We would like to thank J.F. Fecteau for critical reading of the manuscript. This work is supported by 5 U54 CA119335 from the National Institutes of Health/NCI (to S.E. and D.M.) and the Swedish Research Council AI52731 and the Swedish International Development Cooperation Agency; SIDA and VINNMER (Vinnova) to (ML).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singh M, O_Hagan D. Advances in vaccine adjuvants. Nature Biotechnology. 1999;17(11):1075–1081. doi: 10.1038/15058. [DOI] [PubMed] [Google Scholar]
- 2.McCluskie MJ, Weeratna RD. Novel adjuvant systems. 2001;1(3):263–271. doi: 10.2174/1568005014605991. [DOI] [PubMed] [Google Scholar]
- 3.Singh M, O'Hagan DT. Recent advances in vaccine adjuvants. Pharm Res. 2002;19(6):715–728. doi: 10.1023/a:1016104910582. [DOI] [PubMed] [Google Scholar]
- 4.Kovarik J, Siegrist CA. The search for novel adjuvants for early life vaccinations: can “danger” motifs show us the way? 2001;49(3):209–215. [PubMed] [Google Scholar]
- 5.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annual Review of Immunology. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 6.Dredge K, Marriott JB, Todryk SM. Dalgleish, A.G. Adjuvants and the promotion of Th1-type cytokines in tumour immunotherapy. Cancer Immunology, Immunotherapy. 2002;51(10):521–531. doi: 10.1007/s00262-002-0309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu-Amano J, Kiyono H, Jackson RJ, et al. Helper T cell subsets for immunoglobulin A responses: oral immunization with tetanus toxoid and cholera toxin as adjuvant selectively induces Th2 cells in mucosa associated tissues. J Exp Med. 1993;178(4):1309–1320. doi: 10.1084/jem.178.4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engers H, Kieny MP, Malhotra P, Pink JR. Third Meeting on novel adjuvants curently in or close to clinical testing world health organization-Organization mondiale de la sante, Fondation Merieux, Annecy France, 7-9 January 2002. Vaccine. 2003;21:3503–3524. doi: 10.1016/s0264-410x(03)00164-6. [DOI] [PubMed] [Google Scholar]
- 9.De Gregorio E, Tritto E, Rappuoli R. Alum adjuvanticity: unraveling a century old mystery. European Journal of Immunology. 2008;38(8):2068–2071. doi: 10.1002/eji.200838648. [DOI] [PubMed] [Google Scholar]
- 10.Gupta RK. Aluminum compounds as vaccine adjuvants. Adv Drug Deliv Rev. 1998;32(3):155–172. doi: 10.1016/s0169-409x(98)00008-8. [DOI] [PubMed] [Google Scholar]
- 11.Audibert FM, Lise LD. Adjuvants: current status, clinical perspectives and future prospects. Immunol Today. 1993;14(6):281–284. doi: 10.1016/0167-5699(93)90046-N. [DOI] [PubMed] [Google Scholar]
- 12.Gupta RK, Siber GR. Adjuvants for human vaccines--current status, problems and future prospects. Vaccine. 1995;13(14):1263–1276. doi: 10.1016/0264-410x(95)00011-o. [DOI] [PubMed] [Google Scholar]
- 13.Bomford R. Will adjuvants be needed for vaccines of the future? Dev Biol Stand. 1998;92:13–17. [PubMed] [Google Scholar]
- 14.Brewer JM, Conacher M, Satoskar A, Bluethmann H, Alexander J. In interleukin-4-deficient mice, alum not only generates T helper 1 responses equivalent to freund's complete adjuvant, but continues to induce T helper 2 cytokine production. European Journal of Immunology. 1996;26(9):2062–2066. doi: 10.1002/eji.1830260915. [DOI] [PubMed] [Google Scholar]
- 15.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 16.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll- like receptor 3. Nature. 2001;413(6857):732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 17.Netea MG, van der Graaf C, Van der Meer JW. Kullberg, B.J. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. Journal of Leukocyte Biology. 2004;75(5):749–755. doi: 10.1189/jlb.1103543. [DOI] [PubMed] [Google Scholar]
- 18.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annual Review of Immunology. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 19.Petrovsky N, Aguilar JC. Vaccine adjuvants: current state and future trends. Immunology and Cell Biology. 2004;82(5):488–496. doi: 10.1111/j.0818-9641.2004.01272.x. [DOI] [PubMed] [Google Scholar]
- 20.Weiner GJ, Liu HM, Wooldridge JE, Dahle CE, Krieg AM. Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective as immune adjuvants in tumor antigen immunization. Proc Natl Acad Sci U S A. 1997;94(20):10833–10837. doi: 10.1073/pnas.94.20.10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu RS, Targoni OS, Krieg AM, Lehmann PV, Harding CV. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. Journal of Experimental Medicine. 1997;186(10):1623–1631. doi: 10.1084/jem.186.10.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krug A, Towarowski A, Britsch S, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. European Journal of Immunology. 2001;31(10):3026–3037. doi: 10.1002/1521-4141(2001010)31:10<3026::aid-immu3026>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 23.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5(6):471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt C. Clinical setbacks for toll-like receptor 9 agonists in cancer. Nature Biotechnology. 2007;25(8):825–826. doi: 10.1038/nbt0807-825. [DOI] [PubMed] [Google Scholar]
- 25.Telusma G, Datta S, Mihajlov I, et al. Dendritic cell activating peptides induce distinct cytokine profiles. Int Immunol. 2006;18(11):1563–1573. doi: 10.1093/intimm/dxl089. [DOI] [PubMed] [Google Scholar]
- 26.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of Leukocyte Biology. 2007;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 27.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 28.Ulloa L, Messmer D. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 2006;17(3):189–201. doi: 10.1016/j.cytogfr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Vishnubhakat JM, Bloom O, et al. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery. 1999;126(2):389–392. [PubMed] [Google Scholar]
- 30.Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284(5421):1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 31.Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur J Immunol. 2005;35(7):2184–2190. doi: 10.1002/eji.200526066. [DOI] [PubMed] [Google Scholar]
- 32.Messmer D, Yang H, Telusma G, et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol. 2004;173(1):307–313. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- 33.Rovere-Querini P, Capobianco A, Scaffidi P, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5(8):825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park JS, Svetkauskaite D, He Q, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279(9):7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 35.Park JS, Gamboni-Robertson F, He Q, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290(3):C917–924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 36.Tsung A, Klune JR, Zhang X, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204(12):2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26(2):174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 38.van Zoelen MA, Yang H, Florquin S, et al. Role of Toll-Like Receptors 2 and 4, and the Receptor for Advanced Glycation End Products (Rage) in Hmgb1 Induced Inflammation in Vivo. Shock. 2008 doi: 10.1097/SHK.0b013e318186262d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dumitriu IE, Baruah P, Valentinis B, et al. Release of High Mobility Group Box 1 by Dendritic Cells Controls T Cell Activation via the Receptor for Advanced Glycation End Products. J Immunol. 2005;174(12):7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 40.Huttunen HJ, Kuja_Panula J, Rauvala H. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. The Journal of Biological Chemistry. 2002;277(41):38635–38646. doi: 10.1074/jbc.M202515200. [DOI] [PubMed] [Google Scholar]
- 41.Clawson C, Huang CT, Futalan D, et al. Delivery of a peptide via poly(d,l-lactic-co-glycolic) acid nanoparticles enhances its dendritic cell-stimulatory capacity. Nanomedicine. 2010 doi: 10.1016/j.nano.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brazolot Millan CL, Weeratna R, Krieg AM, Siegrist CA, Davis HL. CpG DNA can induce strong Th1 humoral and cell-mediated immune responses against hepatitis B surface antigen in young mice. Proc Natl Acad Sci U S A. 1998;95(26):15553–15558. doi: 10.1073/pnas.95.26.15553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong BR, Josien R, Lee SY, et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med. 1997;186(12):2075–2080. doi: 10.1084/jem.186.12.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loskog A, Totterman TH. CD40L - a multipotent molecule for tumor therapy. Endocr Metab Immune Disord Drug Targets. 2007;7(1):23–28. doi: 10.2174/187153007780059432. [DOI] [PubMed] [Google Scholar]
- 45.Kadowaki N, Ho S, Antonenko S, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194(6):863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jasani B, Navabi H, Adams M. Ampligen: A potential toll-like 3 receptor adjuvant for immunotherapy of cancer. Vaccine. 2009 doi: 10.1016/j.vaccine.2009.01.071. [DOI] [PubMed] [Google Scholar]
- 47.Gurer C, Strowig T, Brilot F, et al. Targeting the nuclear antigen 1 of Epstein-Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood. 2008;112(4):1231–1239. doi: 10.1182/blood-2008-03-148072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trumpfheller C, Caskey M, Nchinda G, et al. The microbial mimic poly IC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. Proc Natl Acad Sci U S A. 2008;105(7):2574–2579. doi: 10.1073/pnas.0711976105. [DOI] [PMC free article] [PubMed] [Google Scholar]