

Abstract
Phage holins are small, lethal membrane proteins of two general types: canonical holins, like λ S105, which oligomerizes and forms large membrane holes of unprecedented size; and pinholins, like S2168 of lambdoid phage 21, which forms homo-heptameric channels, or pinholes, with a lumen of < 2 nm. Pinholes depolarize the membrane, leading to activation of secreted endolysins and murein degradation. S2168 has two transmembrane domains, TMD1 and TMD2. TMD2 alone lines the pinhole, making heterotypic interactions involving two surfaces, A and B. Mutational analysis on S2168 suggested that S2168 initially forms inactive dimer, with TMD1 inhibiting TMD2 both in cis and trans. When TMD1 exits the membrane to the periplasm, it liberates TMD2 to participate in the pathway to pinhole formation. In this study, further mutational analysis suggests a refined pinhole formation pathway, with the existence of at least two intermediate states. We propose that the pathway begins in the activated dimer state, with a homotypic TMD2 interface involving the A surface. Evidence is presented for a further oligomeric state involving a heterotypic A:B interaction. Moreover, the data suggest that a glycine-zipper motif present in the A interface of TMD2 is involved in every stage downstream of the inactive dimer.
Keywords: bacteriophage, holin, lysis, SAR domain, membrane protein
INTRODUCTION
Pinholins and SAR endolysins are recently discovered classes of phage lysis proteins, including those involved in Shiga-toxin release by E. coli O157:H7 (Sun et al., 2009, Wagner & Waldor, 2002, Wagner et al., 2002). Pinholins differ from canonical holins, which form large non-specific holes in the cytoplasmic membrane that allow release of the phage endolysin, resulting in degradation of the cell wall (Young & White, 2008). Instead, pinholins merely depolarize the membrane by forming small holes (“pinholes”) incapable of allowing protein transit. This is effective in terms of lysis because of the properties of the SAR endolysin, which is secreted and accumulates in the periplasm, tethered to the membrane in an enzymatically inactive form (Xu et al., 2005, Sun et al., 2009). When depolarization occurs, the SAR endolysin is released from the membrane and refolds to an active form, leading immediately to destruction of the murein. For both canonical holins and pinholins, hole-formation occurs suddenly, after a period of harmless accumulation in the membrane, during which time membrane energization, macromolecular synthesis and virion assembly continues undisturbed. Although the molecular basis of the temporal scheduling of this event, called “triggering”, is unknown, it is dependent on the energized state of the membrane. For example, even a 50% depletion of the proton motive force (pmf) will prematurely trigger the λ holin, resulting in release of endolysin and lysis within seconds (Gründling et al., 2001). Thus for both canonical holins and pinholins, triggering determines the timing of lysis, although the molecular strategies are completely different.
The prototype pinholin is the S2168 protein of the lambdoid phage 21. Genetic, biochemical, structural and modeling studies indicate that S2168 triggers to form heptameric channels of approximately 1.5 nm diameter (Pang et al., 2009). S2168 has two transmembrane domains (TMDs) (Fig. 1A), but only TMD2 is required for the pinhole (Park et al., 2006). A structure has been proposed for the pinhole, based on coarse-grained simulated annealing of TMD2 (Fig. 1B) (Pang et al., 2009). In this model, heterotypic TMD2-TMD2 interactions occur through two interfaces, A and B. The A interface contains a motif, G40xxxS44xxxG48 that resembles glycine-zipper motifs important for both homotypic and heterotypic interactions between transmembrane helices (Russ & Engelman, 2000, Kim et al., 2005).
Figure 1. A. Features of S21.

The amino acid sequences encoded by S21 are shown. S2171 and S2168 sequences begin at Met1 and Met4, respectively. Residues in each transmembrane domain are underlined. The residues deleted in S2168ΔTMD1 are shaded in gray. The glycine zipper (Gly40, Ser44 and Gly48) is colored in red. The position and sequence (RYIRS) of the irs epitope in irsS2168 are shown in the box.
B. Side view of the pinhole model.
The modeled helix-helix contact surfaces are shown. Surface A (W36A37xxG40V41xG43S44xxL47G48xL50T51xxT54) is colored in gold and labeled in brown, and contains the glycine zipper (Gly40, Ser44 and Gly48), colored brown. Contact surface B (A38xxV41L42xxL45xxxF49xxY52L53xN55L56xF58) is shown and labeled in blue. Adapted from Pang et al (2009) with permission.
C. Model of S2168 hole formation pathway (top-down view from periplasm).
The two TMDs (green: TMD1; sectored: TMD2) in a single S2168 molecule are boxed. Initially, both TMDs are inserted in the membrane in an inactive dimer (ID) form, with TMD1 inhibits TMD2 both in cis and in trans. Then both TMD1s are released, which converts the inactive dimer into an activated dimer (AD) form. Activated dimers then aggregate into oligomers, and nucleate to form heptameric pinholes. In TMD2, orange and dark blue represent A and B interaction faces (Fig. 1B), with 0, 4, and 8 indicating the helical positions of the G40, S44, and G48 residues, respectively. The red stop arrows indicate the cis and trans inhibition of TMD1 to TMD2. The interaction face(s) of TMD2 in both the activated dimer and oligomer are not shown; TMD2 is depicted as clear circle with roman numeral II in both cases. The hydrophilic and weakly hydrophobic residues of TMD2 that eventually face the lumen of the pinhole are represented by a red arc. Grey, lipid. Modified from Pang et al (2009) and (2010) with permission.
D. The membrane topology of S2168 (left), S2171 (middle), and irsS2168 (right). TMD1: white box; TMD2: black box. The TMD1 of S2168 is initially inserted in the membrane but later released into the periplasm (Park et al., 2006). The externalization of TMD1 is delayed in the context of S2171 and completely blocked in the context of irsS2168. Adapted from Pang et al (2010) with permission.
E. The phage 21 lysis cassette.
The phage 21 lysis genes S, R, Rz, and Rz1 are located downstream of the late gene promoter pR′21. S21 gene has a dual-start motif, which encodes both a holin S2168 (translated from Met4), and a weak antiholin S2171 (translated from Met1). Both start codons are bold underlined. Adapted from Pang et al (2010) with permission.
Although TMD1 is not required for the pinhole structure, it has a role in temporal regulation as an inhibitor of the lethal function of TMD2. Genetic and molecular evidence has been presented indicating TMD1 binds to and inhibits TMD2 in both cis and trans in an inactive dimer, which is proposed to be the first kinetically important intermediate in the pinhole formation pathway (Fig. 1C) (Pang et al., 2010). Progress towards hole formation requires a dramatic topological change, with TMD1 exiting the bilayer (Fig. 1C, D). Both TMD1s of the dimer must be externalized before TMD2 can proceed towards pinhole formation.
S2168 is just one of two proteins produced from S21 (Fig. 1E). The other, S2171, a product of a translational start three codons upstream, exhibits retarded externalization of TMD1, presumably because of the extra positively charged residue at its N-terminus (Fig. 1D). Taken together, these observations indicate that S2171, with its retarded topological dynamics, is an antiholin, albeit a weak one. Antiholins are phage-encoded proteins that bind specifically to and inhibit holins, allowing fine-tuning of the holin triggering time. Dual translational starts encoding holin-antiholin pairs are not uncommon in holin genes, and, in several cases (λ, P22, phi29), differences in topological dynamics have been invoked for the inhibitory character of the antiholins (Young & Bläsi, 1995, Bläsi & Young, 1996). A stronger inhibitory phenotype can be obtained by fusing an epitope, irs, containing two positively charged residues, to the N-terminus of S2168 (Fig. 1A). The resultant construct, irsS2168 is completely blocked from the topological change of TMD1, does not support pinhole formation, and exhibits strong dominant negative, or antiholin, character (Fig. 1D).
Despite its intrinsic inhibitory capacity, TMD1 is not essential for lysis timing, as shown by the fact that an S2168ΔTMD1 allele exhibits scheduled triggering and can be prematurely triggered by artificial collapse of the pmf by treatment of induced cells with an uncoupler (Park et al., 2006). This suggests that there is a kinetically distinct pathway downstream of the activated dimer, with only TMD2 in the membrane, ultimately terminating in the sudden formation of the lethal pinholes. Recently, an extensive library of mutant alleles of S2168 was assembled, both by selections for loss of lethal function and by site-directed mutagenesis (Pang et al., 2010). Phenotypic and molecular analysis of this mutant collection, coupled with earlier biochemical and physiological studies, has suggested a model for the orientations of TMD1 and TMD2 in the initial inactive dimer (Fig. 1C). In addition, a multi-step pathway for pinhole formation (Fig. 1C) was suggested. Here we report additional phenotypic and molecular analyses of the non-lethal mutants residing in TMD2 and the early mutant collection. The results are discussed in terms of a scheme for the pinhole formation pathway.
RESULTS
Oligomeric state of non-lethal S2168 TMD2 mutants
In order to verify the existence of the intermediate states towards the pinhole formation after the externalization of TMD1, a collection of the non-lethal S2168 TMD2 mutants was analyzed. In total from a nearly saturated selection for loss of lethality and from site-directed mutagenesis experiments, 15 non-lethal TMD2 mutant alleles of S2168 were obtained, among which 13 encoded S2168 proteins that accumulated to normal levels (Table 1). Alleles which exhibited lower protein accumulation, most of which had missense changes putting charged residues in transmembrane domains, were excluded from analysis, on the grounds that these alleles owed their lysis defect to improper localization or gross misfolding in the membrane (Pang et al., 2010). Lethal pinholes can be detected in vivo by the formation of covalent S2168 oligomers after treatment of induced cells with the membrane-permeant crosslinker, dithiobis[succinimidyl propionate] (DSP). These oligomers can be visualized as a ladder of immuno-reactive bands in non-reducing SDS-PAGE (Pang et al., 2009) (Fig. 2A). In contrast, the irsS2168 protein cannot be cross-linked to species higher than the dimer and can block oligomer formation by S2168, consistent with its negative-dominant, antiholin character. When the non-lethal TMD2 mutants of full-length S2168 were subjected to the same DSP-crosslinking treatment, 8 alleles exhibited patterns indistinguishable from irsS2168, whereas 5 showed ladders resembling the wt (Fig. 2A, Table 1). Although the distinction between the dimer and oligomer cross-linking phenotypes was robust, we sought independent evidence for the existence of these species using Blue-Native (BN) PAGE analysis. For the wt and four of the five oligomer-forming mutants, we were able to detect ~100 kDa complexes that persist in 1% n-dodecyl-β-D-maltopyranoside (DDM) of membrane fractions, consistent with the mass of DDM micelles containing the wt pinholin (Pang et al., 2009) (Fig. 2B). In contrast, these ~100 kDa complexes were not observed in 6 of the 7 mutants showing DSP-cross-linked dimers. The fact that two of the alleles, one in each class, show different behavior in the two methods is interesting but also not surprising, since the DSP-crosslinking is done in the energized membrane whereas BN PAGE has to be done in detergent extracts. Taken together, these results suggest that missense changes can block the pinhole pathway at both dimer and oligomer stages in the membrane.
Table 1.
Properties of non-lethal S2168 TMD2 mutant alleles.
| allele1 | Oligo2 | TMD1 externalization3 | Oligo+wt4 | D/R5 |
|---|---|---|---|---|
| wt | + | 0.6 | ||
| W36C | − | nd | + | D |
| A37V | + | aD | ||
| G40L | + | aD | ||
| G40T | + | aD | ||
| G43L | − | 0.8 | + | D |
| S44L | − | IF | aD | |
| S44C | − | 0.6 | aD | |
| G48L | − | 0.8 | − | D |
| G48T | + | + | D | |
| G48C | − | 0.3 | − | D |
| T51I | − | 0.6 | aD | |
| T54I | − | 0.7 | − | D |
| T54C | + | + | D |
Mutant alleles.
Oligomerization of each mutant as indicated by DSP crosslinking in vivo. + indicates the formation of oligomer. − indicates the formation of dimer.
Effect of each mutation on the externalization of TMD1. The number indicates the ratio of S16C-mediated disulfide-bond linked dimer to the monomer. nd, not determinable. IF, infinite.
Oligomerization of each mutant in the presence of wt S2168. Labeled same as in 2.
The dominance/recessiveness test. D: dominant-negative. aD: antidominant.
Figure 2. A. Representatives of DSP crosslinking of S2168 varients in vivo.

Whole cells carrying prophage λQ21Δ(SRRzRz1)21 only (lane 1) or with each indicated plasmid were induced and, treated with the cross-linker DSP, and analyzed by SDS-PAGE and western blotting, as described in the Materials and Methods. Plasmid carried in each sample: pS2168 (lane 2), pirsS2168* (lane 3), lane 4–9, derivatives of pS2168, encoding single mutation of S2168 as indicated on top of each lane. Numbers at the right side of each lane indicate the crosslinked oligomers.
B. Blue-native PAGE separation of S2168 and its variants.
Crude membranes from cells carrying prophage λQ21Δ(SRRzRz1)21 only (lane 1) or with derivatives of plasmid pS2168 expressing S2168 (lane 2) or its mutants (lanes 3–6) were analyzed by BN PAGE and immunoblotting, as described in Materials and Methods. The position of oligomeric states of S2168 and its variants were estimated by their apparent molecular weights with detergent micelles. The position of BN PAGE standards are indicated: 242 kDa = B-phycoerythrin; 146 kDa = lactate dehydrogenase; 66 kDa = bovine serum albumin; 20 kDa = soybean trypsin inhibitor.
Mutants blocked at the activated dimer stage
In principle, the eight non-lethal mutations in TMD2 (W36C, G43L, S44L, S44C, G48L, G48C, T51I, and T54I) blocked at the dimer stage might be defective in oligomerization because of an inability to externalize TMD1, like the antiholin irsS2168. Previously, we have shown that TMD1, once externalized to the periplasm, can undergo homotypic interactions detectable by assessing spontaneous disulfide bond formation using the S16C substitution allele (Park et al., 2006). Incorporating this change into each of the non-lethal alleles had no effect on the lytic defect (not shown) or the level of protein accumulation (Fig. 3). Only one TMD2 mutant, G48C, exhibited reduced disulfide bond formation. To verify if this reduction was due to the retention of TMD1 in the bilayer or due to the conformational change of TMD1, cells expressing S2168S16C/G48C were treated with the membrane-impermeant thiol-reagent [2-sulfonatoethyl] methanethiosulfonate (MTSES), followed by denaturation and PEGylation. In such experiments, a positive result for PEGylation indicates Cys residue in question was protected from MTSES treatment, presumably by being embedded in the bilayer. As we showed previously, once cells expressing the functional pinholin S2168S16C triggered at 60 min after induction, Cys16 in TMD1 became accessible to MTSES in the periplasm, from as evidenced by the MTSES-dependent decrease in the PEGylated species and increase in the unmodified one (Fig. 4, lanes 5–6) (Pang et al., 2009). In contrast, in alleles with Cys residues substituted into TMD2, MTSES-accessibility was absolutely correlated with hole formation and lethal function (Pang et al., 2009). Thus in S2168S16C/S44C, the MTSES-dependent reduction in the doubly PEGylated species and concomitant increase in the single and un-PEGylated bands (Fig. 4, lanes 7,8) indicates that Cys16 was in the periplasm, which agrees with the disulfide-bond formation assessment in Fig. 3. In contrast, neither Cys48 alone in the non-lethal S2168G48C nor Cys16 and Cys48 in the non-lethal S2168S16C/G48C were accessible to MTSES, suggesting that the G48C mutation caused retention of TMD1 in the bilayer (Fig. 4, lanes 9–12). Consistent with this notion, G48 is on the A face of TMD2 (Fig. 1B) and, according to the orientation map for the inactive dimer (Fig. 1C), would interact with TMD1 in cis. The other dimer-limited mutants show no defect in the S16C disulfide bond formation assay, and, in fact, one, S44L, appears to be much more efficient than the parental. Ser44 is also on the A face, and, like G48, is part of the G40xxxS44xxxG48 zipper motif, so the altered externalization phenotypes of these two alleles, although opposite, support the idea that the zipper motif is important in the cis interaction with TMD1. Moreover, externalization-proficient, oligomerization-defective phenotypes of G43L, S44L, S44C, G48L, T51I, and T54I, all mutants in the A face of TMD2, suggest that these proteins are blocked at the activated dimer stage (Fig. 1C).
Figure 3. The effect of each mutation on the externalization of S2168 TMD1.

Cultures carrying prophage λQ21Δ(SRRzRz1)21 and derivatives of plasmid pS2168 encoding mutations on the S2168 gene as indicated on top of each lane were induced and precipitated by TCA at the time indicated below each lane. Samples were resuspended in sample loading buffer without reducing agent and analyzed by SDS-PAGE and western blotting. Double and single asterisks indicate the position of dimer and monomer, individually. The dimer to monomer ratio of each mutant is illustrated at the bottom. IF, infinite.
Figure 4. Cysteine-modification analysis of S2168 variants.
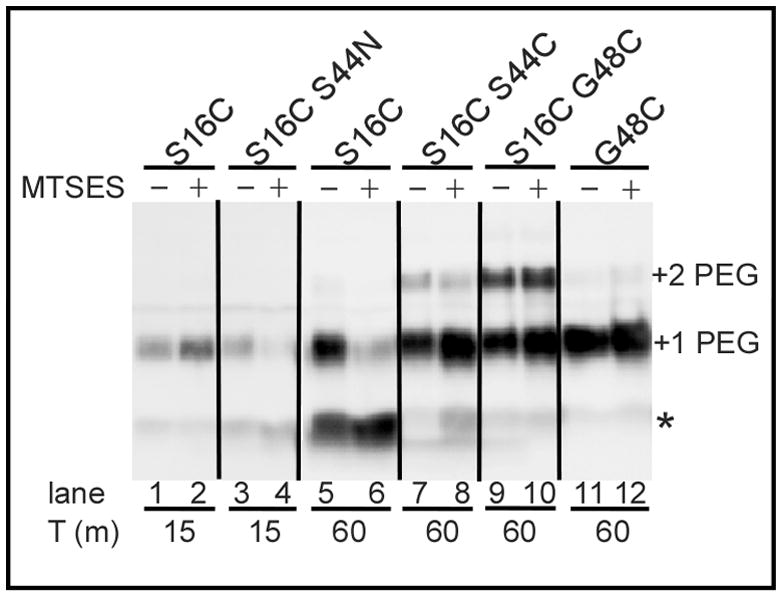
Cells expressing the S2168 mutant alleles (indicated on top) were harvested and treated with the membrane-impermeant thiol reagent MTSES at the time indicated below each lane, subjected to organic denaturation and delipidation, PEGylated with PEG-maleimide, and analyzed by SDS-PAGE and western blotting, as described in Materials and Methods. The asterisk, “+1 PEG”, and “+2 PEG” indicate the position of the unmodified monomer, the singly-PEGylated and the doubly-PEGylated species, respectively.
Oligomerization proficient non-lethal mutants
Five TMD2 mutants (A37V, G40L, G40T, G48T, T54C) exhibited DSP crosslinking ladders indistinguishable from the lethal parental (Fig. 2A; Table 1). To test whether lethal function could be rescued by artificially depolarizing the membrane, cells induced for these alleles were treated with 2,4-dinitrophenol (DNP). After washing out DNP, these cells resumed growth (Fig. 5A), indicating that the mutant oligomers formed are non-lethal and cannot be triggered to form pinholes. The simplest interpretation is that these mutants are blocked at an oligomeric stage, detectable by DSP cross-linking, before the formation of the pinholes, and the blockage cannot be removed by simply depolarizing the membrane. A similar model has been suggested for the canonical holin S105 of phage lambda, also based on the existence of wt DSP-cross-linking phenotypes in mutants that have lost lethal function, although in the case of λ S, only one of the non-lethal alleles tested exhibited an oligomeric ladder (Gründling et al., 2000).
Figure 5. Growth curves.

Except A, cultures carrying the indicated prophages and plasmids were induced at T=0, and their growth were monitored as A550.
A. The effect of DNP on the triggering of S2168 variants (representatives). Prophage λQ21Δ(SRRzRz1)21 with no plasmid (open circles), or plasmid pS2168 (closed circles), pirsS2168* (open squares), pS2168T54C (closed squares), pS2168G40L (open diamonds) was induced at T=−110. 2mM DNP was added to each culture at T=−70, at which time the S2168 triggers. At T=−60, cultures were taken out and LB media containing DNP was replaced by fresh media as described in Materials and Methods. At T=0, growth of each culture was monitored again.
B, C. The dominance/recessiveness test of S2168 mutants.
B. Representative of the anti-dominant mutants. Closed squares, prophage λQ21Δ(SRRzRz1)21, no plasmid; open circles, prophage λS2168, no plasmid; closed circles, prophage λS2168, plasmid pS2168a; open diamonds, prophage λQ21Δ(SRRzRz1)21, plasmid pS2168aG40L; closed diamonds, prophage λS2168, plasmid pS2168aG40L.
C. Representative of the dominant-negative mutants. Cross, prophage λQ21Δ(SRRzRz1)21, no plasmid; open circles, prophage λQ21Δ(SRRzRz1)21, plasmid pS2168aG48L; closed circles, prophage λS2168, plasmid pS2168aG48L.
Reversed triggering phenotypes in dominance-recessiveness tests
The non-lethal TMD2 alleles were also tested for dominant-recessive character by inducing in the presence, in trans, of the parental S2168 allele borne on a prophage (Table 1; Fig. 5B, C). Under these conditions, all of these non-lethal mutants were sensu stricto dominant, in that their presence in trans to wt resulted in a phenotype different from that observed with the wt allele in both cis and trans positions. Seven of these alleles (W36C, G43L, G48L, G48T, G48C, T54I and T54C) had dominant-negative phenotypes, blocking the triggering of the wt allele in trans (Fig. 5C). Among them, five mutants (W36C, G43L, G48L, G48C, and T54I) were blocked at dimer stage. However, only three (G48L, G48C, T54I) were able to prevent oligomerization of the wt protein (Fig. 6). This was expected for G48C, since it is defective in TMD1 externalization and thus presumably would function with antiholin character, like irsS2168.
Figure 6. DSP crosslinking of S2168 wildtype in the presence of S2168 mutants in vivo.
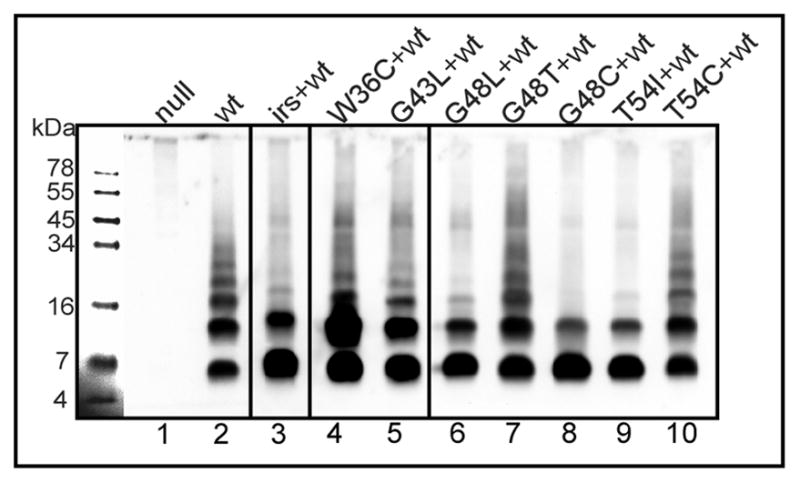
Experiment was performed as in Fig. 2A. Prophage and plasmid carried in each sample: λQ21Δ(SRRzRz1)21, no plasmid (lane 1); prophage λS2168 (lane 2); prophage λS2168, plasmid pirsS2168* (lane 3); lane 4–10, prophage λS2168 with derivatives of pS2168a, encoding single mutation of S2168 as indicated on top of each lane.
The other six non-lethals tested (A37V, G40L, G40T, S44L, S44C, T51I), despite their intrinsic triggering defect, exhibited an early triggering phenotype in trans to the parental, compared to the double-parental control (Fig. 5B). Similar unexpected phenotypic reversal was previously observed in recessiveness-dominance testing of non-lethal mutants of the λ holin (Raab et al., 1988). To distinguish them, these alleles are designated as “anti-dominant” (aD) in Table 1.
Early lysis mutants outside of TMD1
Early lysis phenotypes are common in TMD1; 5 of the 20 residues give rise to 6 mutants (A12L, G14Q, G14C, T15C, A17Q, and G21Q) with triggering times ~35 m earlier than wt (Table 2). These mutants are straightforward to interpret: any mutation that dramatically reduces the dwell-time of TMD1 in the membrane, either by reducing hydrophobicity or disrupting interactions with TMD2, can be expected to advance the triggering time (Fig. 1C) (Pang et al., 2010). In the remaining 48 residues, only one position, S44, gives rise to mutants with dramatically early triggering: S44T and S44N (Table 2, Fig. 7A). S44 is not only the central residue of the GxxxSxxxG homotypic interaction motif but, in the structure proposed for the TMD2 heptamer, it is part of the heterotypic interaction surface A and also exposed to the pinhole lumen (Pang et al., 2009). Interestingly, only one of these two mutants, S44N, exhibits early lysis in the context of ΔTMD1, while the other, S44T, is completely non-functional (Table 2, Fig. 7B). In contrast, three other mutations, S32C in the connecting loop and A37T and A38T in TMD2, exhibit radically early triggering in the deletion allele but are either severely delayed (A37T, A38T) or non-lethal (S32C) in the full length context (Table 2). While these phenotypes cannot yet be interpreted with confidence, the simplest notion is that the S32C, A37T, and A38T changes, while intrinsically more biased towards completion of the TMD2 oligomerization-hole formation pathway, all increase affinity for TMD1 and thus retard its release to the periplasm (Fig. 1C). In contrast, the TMD1 of S44T and S44N are released remarkably early from the membrane, as assessed by the disulfide-bond formation of the S16C mutation in TMD1 (Park et al., 2006) (Fig. 3). The early release of TMD1 by S44N was also verified by the protection of MTSES to Cys16 from PEGylation (Fig. 4). At 15 min after induction of the pinholins, S16C/S44N was protected by MTSES from PEGylation, although S16C alone was not, because it was harvested before triggering.
Table 2.
Triggering time of the early lysis mutants.
| loc1 | allele2 | full-length3 | Δ(TMD1)4 |
|---|---|---|---|
| wt | 0 | 0 | |
| TMD1 | A12L | −35 | |
| G14Q | −35 | ||
| G14C | −35 | ||
| T15C | −35 | ||
| A17Q | −35 | ||
| G21Q | −35 | ||
| loop | S32C | NL | −40 |
| TMD2 | A37T | +30 | −30 |
| A38T | +50 | −20 | |
| S44T | −35 | NL | |
| S44N | −35 | −45 |
Localization of each mutant in the topological domains.
Mutant alleles.
Effect of each mutation on the triggering time (m) of the full length holin S2168 (wt triggers at 50 m). NL, non-lethal.
Effect of each mutation on the triggering time (m) of the truncated holin S2168ΔTMD1 (wt triggers at 50 m). NL, non-lethal.
Adapted from Pang et al (2010) with permission.
Figure 7. Phenotypes of S44T and S44N.
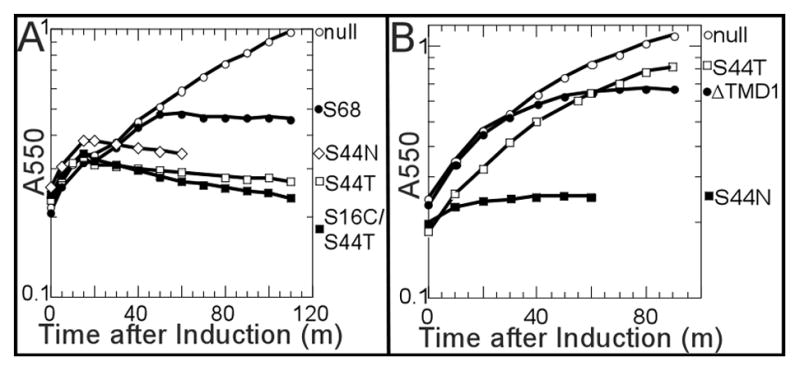
A. Growth curve of S2168 alleles. Lysogen carrying prophage λQ21Δ(SRRzRz1)21 and indicated plasmids were induced at t=0 and monitored for growth as A550. Open circles, no plasmid; closed circles, pS2168; open diamonds, pS2168S44N; open squares pS2168S44T; closed squares, pS2168S16C/S44T.
B. Growth curve of S2168ΔTMD1 alleles. Culture growth was monitored as in A, except that each culture carries plasmids pQ and the indicated plasmids: open circles, plasmid vector pRE (Gründling et al., 2001); closed circles, pS2168ΔTMD1; open squares, pS2168ΔTMD1-S44T; closed squares, pS2168ΔTMD1-S44N.
Discussion
S2168 is the prototype of a recently discovered class of holins, the pinholins. It is a particularly attractive subject for genetic analysis, because of its small size, only 68 residues, and its genetic malleability. Previous studies with the extensive collection of S2168 mutants were able to resolve the orientation of both TMDs in the inactive dimer, the first kinetically important stage in the pinhole formation pathway (Pang et al., 2010). Here we have provided additional analysis of this mutant collection and have suggested a refined pinhole formation model, with a collection of S2168 mutants mapped onto it. In addition, this study suggests a scheme for the orientation of TMD2 in the intermediate stages and at least three roles for the glycine zipper motif in its A surface.
A refined pinhole pathway
Previously, we proposed a model for pinhole formation with S2168 (Pang et al., 2009) (Fig. 1C). At the first kinetically important stage, both TMDs are inserted in the membrane in an inactive dimer (Pang et al., 2010). Once TMD1 is released into the periplasm, S2168 forms an activated dimer form, followed by the aggregation of the dimer into oligomers. Finally, the heptameric pinhole forms, possibly driven by the hydration of the lumenal side chains. Although models for the inactive dimer and the final pinhole with the helical-helical interaction surfaces A and B are suggested by genetic and biochemical methods (Pang et al., 2010, Pang et al., 2009), structures of TMD2s in the intermediate stages were not clear. Here analysis of the oligomeric states of S2168 non-lethal mutants is used to provide evidence for the existence and basic structure of each intermediate.
All the non-lethal mutants in TMD2 were localized to interface A, suggesting its importance in the pinhole formation pathway (Fig. 8A). Among all these non-lethal mutants, eight (W36C, G43L, S44L, S44C, G48L, G48C, T51I, and T54I) were blocked in the dimer stage, with only one (G48C) showing a defect in the externalization of TMD1. The six alleles (W36C was not determined) blocked at the dimer stage but proficient at TMD1 externalization support the existence of the activated dimer intermediate (Fig. 8B). Moreover, three of these mutants (S44L, S44C, and T51I) exhibited an anti-dominant phenotype in the presence of wild-type S2168. This suggests that the homotypic interfaces A:A interaction are involved in the activated dimer, since the blockage of these three interface A mutants at the activated dimer stage can be rescued by providing a copy of the wild-type interface A.
Figure 8. A. Helical wheel projection of S2168 TMD2.

Orange and dark blue colored residues are in predicted helical-helical interaction faces A and B, respectively (Pang et al., 2009). The green-colored residue (V41) is in both A and B interfaces. Red asterisks represent the position of non-lethal S2168 mutants, which are all in interface A.
B. Model for the S21 pinhole formation pathway. Same scheme as in Fig. 1C. S21 molecules are first inserted as monomers. ID1, ID2: the inactive dimers 1 and 2; AD, the activated dimer, with the homotypic helical-helical interaction of the glycine zipper containing interface A. In the oligomer state, the A:A interaction transits to A:B heterotypic helical interaction before the heptameric pinhole forms. S2168 mutants are mapped in the pathway with either accelerating the process (blue arrow), or blocking it (blue stop arrow).
Secondly, five other non-lethal mutants in TMD2 (A37V, G40L, G40T, G48T, T54C) were crosslinked into oligomers indistinguishable from the S2168, suggesting the existence of the oligomer state before the pinhole forms (Fig. 8B). The orientation of TMD2s in this stage is not clear, although the simplest notion is that helical interactions between interfaces A and B are involved. We favor this idea because since the A:A interaction is involved in the activated dimer stage, and because mutants in two residues of interface A (G48 and T54) can be blocked in either the dimer (G48L and T54I) or the oligomer (G48T and T54C) stage, suggesting the different interactions involved at these residues. Two of these five mutants (G48T and T54C) are dominant-negative, suggesting that the defect of transition to the pinhole at this stage cannot be rescued by the wild-type S2168, whereas it can be rescued in the anti-dominant mutants (A37V, G40L, and G40T). In addition, among the five dominant-negative mutants blocked at the activated dimer stage, two (W36C and G43L) were crosslinked into oligomers in the presence of wt S2168, indicating that their blockage of the A:A to A:B transition can be accommodated by the wild-type, but not the transition from the oligomer stage to the pinhole. Ultimately, resolution of the intermediate states in the pathway will require a kinetic analysis that shows the population of these states occurs in a sequential manner. Technically, such a study may prove challenging because of the exquisite sensitivity of holins to the proton-motive force, a characteristic that imposes severe restrictions on the experimental manipulation of growing cells.
Taken together, the phenotypic analyses of the mutant collection leads us to further refinement of the pinhole formation pathway (Fig. 8B). The first stage obviously must be the monomer, although no S21 mutants have been identified that are blocked at the monomer stage. It is possible that a dimerization defect might lead to proteolysis and thus low protein accumulation, as has been shown for at least one mutant in the class I λ holin S105 (Gründling et al., 2000). In this model, the first kinetically important stage would be the inactive dimer ID1, with each membrane-inserted TMD1 inhibiting TMD2 both in cis and in trans. At an allele-specific rate, the TMD1 externalizes into the periplasm. For the wild-type S21 allele, a second stage of the inactive dimer (ID2) formed between S2168 and S2171 might exist, with only TMD1 of S2168 externalized, because it has already been demonstrated that TMD1 of the longer form exits more slowly (Park et al., 2006). In both ID stages, the interface A of TMD2, composed of most of the hydrophilic residues, is packed inside the protein clusters and away from lipid. In the activated dimer stage, with both TMD1s released, the interface A is engaged in homotypic intermolecular packing. This state might not be kinetically stable, and may yield to more favored packing, the A:B interaction, which would potentiate transition to an oligomer state. Finally, the hydration of the lumenal-side chains might drive the helices to rearrange at an angle of 34° to the normal, and the heptameric pinholes to form (Pang et al., 2009). The opening of the pinhole would likely lead to a local depolarization of the membrane. If the depolarized state favors any of these transitions, the entire pathway would have a fundamentally “all or nothing” character. As noted previously (Wang et al., 2000), holin function should be all or nothing, to avoid poisoning macromolecular metabolism, and thus virion assembly, before triggering.
Mutations in the GxxxSxxxG motif
The interface A of the S2168 TMD2 contains a G40xxxS44xxxG48 motif, also known as the glycine zipper. The glycine zipper is found to be strongly involved in helical packing against a neighboring helix, as seen in several channel forming proteins (MscL, VacA, KcsA) (Kim et al., 2005). It also contains the GxxxG motif, which plays a significant role in homotypic helices interaction through hydrogen bonding, as in the dimeric protein glycophorin A (Curran & Engelman, 2003). From analysis of the lysis phenotypes of mutants in these residues, we suggested that this motif is involved in both the homotypic TMD2 interaction in the activated dimer form, and the heterotypic TMD2 interaction in the pinhole structure. Moreover, the structure of the inactive dimer predicted by the mutagenesis analysis also suggests that this motif interacts with TMD1 in cis (Pang et al., 2010).
Phenotypic analysis of the zipper motif mutants is challenging, since it is likely the motif is involved in three interactions in the pathway. However, the completely opposite phenotypes of S2168 mutants on S44 deserve attention. The two non-lethal mutants, S44L and S44C are both TMD1-externalization proficient, dimerization-proficient, oligomerization-deficient, and anti-dominant. Thus replacing S44 with both L and C blocks the transition from the dimer stage (A:A homotypic interaction) to the oligomer (A:B heterotypic interaction) stage. Moreover, the TMD1 of S44L is released much more efficiently than the parental allele or the S44C (Fig. 3). This supports the idea that S44 interacts with TMD1, and suggests that the S44L mutation compromises this interaction and accelerates the release of TMD1, possibly because Leu is bulkier than both Ser and Cys (Fig. 8B). Two other mutants, S44T and S44N trigger extremely early in the context of full length protein. However, they have a remarkably different phenotype in the TMD1-deleted context. S44T completely abolishes the function of S2168ΔTMD1, while S44N accelerates its triggering drastically (Fig. 7A, B). This extreme difference supports our model that, in addition to the cis interaction with TMD1, S44 is also involved in both the homotypic and heterotypic interaction of TMD2 in the pinhole formation pathway. In this model, at some point the activated dimer, which is suggested to be based on the A:A interaction, has to undergo a shift to the A:B interaction that defines the heptamer. In this perspective, S44N and S44T both weaken the cis interaction with TMD1, but have different effects on the homotypic and heterotypic TMD2:TMD2 interactions of the downstream pathway (Fig. 8B). Overall, then, the glycine zipper motif clearly plays a fundamental role in every stage of the pinhole formation pathway downstream of the inactive dimer.
Materials and Methods
Bacterial strains, prophages, plasmids, media and culture growth
E. coli strains, prophages, and plasmids used in this study have been described previously (Pang et al., 2009, Pang et al., 2010). In each case, the phage 21 lysis cassette has been mutated so that only the S2168 gene or its derivatives are expressed. Briefly, lysogen MDS12ΔtonA(λQ21Δ(SRRzRz1)21) carrying the plasmid pS2168 or its derivatives were used to express the wildtype or mutant S2168 alleles. The same lysogen carrying plasmid pirsS2168* was used to express irsS2168, which has codons encoding the irs-epitope (RYIRS) fused to the N-terminus of S2168 gene in the plasmid pS2168a. Plasmid pS2168a has the entire lysis cassette region identical to pS2168, except that it carries AmpR, instead of KanR. E. coli strain MG1655lacIq1tonA::Tn10 (Guyer et al., 1981) carrying plasmids pQ and pS2168ΔTMD1 or its derivatives were used to express the wildtype or mutant S2168ΔTMD1 alleles. For the dominance/recessiveness tests, derivatives of plasmid pS2168a were used. They were induced by the phage 21 late gene activator Q21 expressed from either the lysogen MDS12ΔtonA(λQ21Δ(SRRzRz1)21), or MDS12ΔtonA(λS2168) which provides a chromosomal copy of S2168.
Media, culture growth, and induction of the lysis have been described previously (Pang et al., 2009). Briefly, plasmids carried by lysogens were induced by shifting the culture into 42°C for 15 minutes. Plasmid pQ was induced by 1 mM Isopropyl-β-D-thiogalactopyranoside (IPTG) and 0.2% arabinose. Lysis profiles were obtained by monitoring A550 after induction, as described previously (Smith & Young, 1998, Tran et al., 2005). To assess the effect of DNP to the triggering of holin mutants, at the time when the wildtype holin triggers, 2mM DNP was added into each culture. Culture growth was monitored for additional 10 minutes, then cultures were spun down and the media containing DNP was poured away. Cell pellets were rinsed with LB media twice, and then resuspended in fresh LB media supplemented with corresponding antibiotics. Culture growth was monitored again.
Site-directed mutagenesis, and general DNA manipulations
Procedures for QuikChange (Stratagene) mutagenesis, cloning steps and DNA sequencing have been described (Smith & Young, 1998). Oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). All enzymes were purchased from Promega, except the Pfu polymerase, which was from Stratagene.
Chemical crosslinking in vivo
DSP crosslinking of S2168 and its variants in vivo was described previously (Pang et al., 2009). Briefly, 1 A550 unit of cells were harvested at the time when wildtype S2168 triggered (50 m after induction), washed and resuspended into 640 μl PBS (phosphate buffered saline). The samples were treated with 2mM DSP (Pierce) for 30 m at 25°C and then quenched with 20 mM Tris pH7.5. Proteins were precipitated by trichloroacetic acid (TCA) and analyzed by SDS-PAGE and Western blotting.
Cysteine modification
Chemical modification of cysteines were performed as before (Pang et al., 2009). Briefly, 0.25 A550 unit of cells were harvested immediately after the holin triggered, or in the case of non-lethal S2168 mutants, at the time when S2168 triggered. The samples were washed and resuspended in 0.25 ml of PB (50 mM phosphate buffer, pH7). Each sample was divided into two equal aliquots, with one treated with 10 mM MTSES (Anatrace), and the other with the same amount of water. The reaction was quenched with 50 mM L-cysteine (Sigma-Aldrich), and washed with PB. Proteins from each sample were extracted with chloroform:methanol:water (1:4:1), washed with 95% methanol, and resuspended in 100 μl PEGylation buffer. Fifty μl of each sample was treated with 0.2 mM mPEG-maleimide (Creative Biochem), and precipitated with cold ethanol. The protein pellets were collected by centrifugation, dried, and resuspended in sample loading buffer with 5% β-mercaptoethanol for analysis by SDS-PAGE and western blotting.
TCA precipitation, SDS-PAGE and Western blotting
TCA precipitation, SDS-PAGE and Western blotting were performed as described (Pang et al., 2009). Briefly, 10% TCA was used to immediately stop cell growth and precipitate protein. Precipitates were resuspended in sample loading buffer supplemented with 5% β-mercaptoethanol, unless for the purpose of detecting the disulfide-bond linked dimer of S2168S16C and its variants, or for analyzing the DSP- crosslinked oligomers. Samples were separated on 10% Tris-Tricine gels and transferred to nitrocellulose membrane (0.1 μm; Whatman, NJ). Antisera against the S21 C-terminal peptide KIREDRRKAARGE were used as primary antibody (Barenboim et al., 1999).
Blue-native PAGE
BN PAGE was performed according to Wittig et al (2006). Twenty A550 units of cells expressing S2168 or its variants were harvested at the time when wildtype S2168 triggered, resuspended in 1 ml sample buffer (50 mM NaCl, 50 mM imidazole, 1mM EDTA, 10% (v/v) glycerol, pH 7), and lysed by passing through a French pressure cell (Spectronic Instruments; Rochester, MY) at 16,000 psi. Whole cells and cell debris were removed by microcentrifugation at 10,000g for 15 minutes at 4°C. Membranes were harvested by ultracentrifugation at 106,000g for 1 hour at 4°C in a Beckman TLA100.3 rotor and extracted overnight at 4°C in 1 ml sample buffer containing 1% (w/v) DDM (Anatrace). After insoluble material was removed by ultracentrifugation at 106,000g for 1 h at 4°C, 20 μl of the soluble supernatant was mixed with 1μl 5% (w/v) Coomassie brilliant blue G-250 (Bethesda Research Laboratories, MD). Samples were loaded on the BN gel (3.5% acrylamide stacking gel with 10% acrylamide separating gel, pH7), with electrophoresis performed at 100 V in the cold until the dye front entered one-third of the gel, when the voltage was increased to 200 V, and the cathode buffer was changed from dark blue (0.02% G-250) to light blue (0.002% G-250), according to Wittig et al (2006). After running, the gel was documented and proteins were transferred to PVDF membrane (0.2 μm; Pall) at 4°C. After destaining with destain buffer (30% (v/v) methanol, 10% (v/v) acetic acid), the membrane was air-dried, documented and completely destained with methanol, and western-blotting was continued. BN PAGE protein standards was purchased from Invitrogen (NativeMark Unstained Protein Standard) and used according to the manufacturer’s instructions.
Acknowledgments
This work was supported by Public Health Service Grant GM27099, by the Robert A. Welch Foundation, and by the Program for Membrane Structure and Function from the Office of the Vice President for Research at Texas A&M University.
References
- Barenboim M, Chang CY, dib Hajj F, Young R. Characterization of the dual start motif of a class II holin gene. Mol Microbiol. 1999;32:715–727. doi: 10.1046/j.1365-2958.1999.01385.x. [DOI] [PubMed] [Google Scholar]
- Bläsi U, Young R. Two beginnings for a single purpose: the dual-start holins in the regulation of phage lysis. Mol Microbiol. 1996;21:675–682. doi: 10.1046/j.1365-2958.1996.331395.x. [DOI] [PubMed] [Google Scholar]
- Curran AR, Engelman DM. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr Opin Struct Biol. 2003;13:412–417. doi: 10.1016/s0959-440x(03)00102-7. [DOI] [PubMed] [Google Scholar]
- Gründling A, Blasi U, Young R. Genetic and biochemical analysis of dimer and oligomer interactions of the lambda S holin. J Bacteriol. 2000;182:6082–6090. doi: 10.1128/jb.182.21.6082-6090.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründling A, Manson MD, Young R. Holins kill without warning. Proc Natl Acad Sci USA. 2001;98:9348–9352. doi: 10.1073/pnas.151247598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer MS, Reed RR, Steitz JA, Low KB. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb Symp Quant Biol. 1981;45(Pt 1):135–140. doi: 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc Natl Acad Sci U S A. 2005;102:14278–14283. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang T, Park T, Young R. Mutational analysis of the S21 pinholin. Molecular Microbiology. 2010;76:68–77. doi: 10.1111/j.1365-2958.2010.07080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang T, Savva CG, Fleming KG, Struck DK, Young R. Structure of the lethal phage pinhole. Proc Natl Acad Sci U S A. 2009;106:18966–18971. doi: 10.1073/pnas.0907941106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T, Struck DK, Deaton JF, Young R. Topological dynamics of holins in programmed bacterial lysis. Proc Natl Acad Sci U S A. 2006;103:19713–19718. doi: 10.1073/pnas.0600943103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab R, Neal G, Sohaskey C, Smith J, Young R. Dominance in lambda S mutations and evidence for translational control. J Mol Biol. 1988;199:95–105. doi: 10.1016/0022-2836(88)90381-6. [DOI] [PubMed] [Google Scholar]
- Russ WP, Engelman DM. The GxxxG motif: A framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- Smith DL, Young R. Oligohistidine tag mutagenesis of the lambda holin gene. J Bacteriol. 1998;180:4199–4211. doi: 10.1128/jb.180.16.4199-4211.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Kuty GF, Arockiasamy A, Xu M, Young R, Sacchettini JC. Regulation of a muralytic enzyme by dynamic membrane topology. Nat Struct Mol Biol. 2009;16:1192–1194. doi: 10.1038/nsmb.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TAT, Struck DK, Young R. Periplasmic domains define holin-antiholin interactions in T4 lysis inhibition. J Bacteriol. 2005;187:6631–6640. doi: 10.1128/JB.187.19.6631-6640.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner PL, Livny J, Neely MN, Acheson DW, Friedman DI, Waldor MK. Bacteriophage control of Shiga toxin 1 production and release by Escherichia coli. Mol Microbiol. 2002;44:957–970. doi: 10.1046/j.1365-2958.2002.02950.x. [DOI] [PubMed] [Google Scholar]
- Wagner PL, Waldor MK. Bacteriophage control of bacterial virulence. Infect Immun. 2002;70:3985–3993. doi: 10.1128/IAI.70.8.3985-3993.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IN, Smith DL, Young R. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000;54:799–825. doi: 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Xu M, Arulandu A, Struck DK, Swanson S, Sacchettini JC, Young R. Disulfide isomerization after membrane release of its SAR domain activates P1 lysozyme. Science. 2005;307:113–117. doi: 10.1126/science.1105143. [DOI] [PubMed] [Google Scholar]
- Young R, Bläsi U. Holins: form and function in bacteriophage lysis. FEMS Microbiology Reviews. 1995;17:191–205. doi: 10.1111/j.1574-6976.1995.tb00202.x. [DOI] [PubMed] [Google Scholar]
- Young R, White RL. Lysis of the host by bacteriophage. In: Mahy BWJ, van Regenmortel MHV, editors. Encyclopedia of Virology. 3. Boston: Academic Press; 2008. pp. 248–258. [Google Scholar]