

Abstract
Objective
To determine if cannabinoid receptor 2 (CB2) plays a role in atherosclerosis, we investigated the effects of systemic CB2 gene deletion on hyperlipidemia-induced atherogenesis in low density lipoprotein receptor-deficient (Ldlr−/−) mice.
Methods and results
Ldlr−/− and CB2/Ldlr double knockout (CB2−/−Ldlr−/−) mice were fed an atherogenic diet for 8 and 12 weeks. Morphometric analysis revealed no significant difference between the atherosclerotic lesion area in the proximal aortas of Ldlr−/− and CB2−/−Ldlr−/− mice after 8 or 12 weeks on the atherogenic diet. The macrophage and smooth muscle cell (SMC) content, as revealed by immunohistochemical staining, did not differ significantly between Ldlr−/− and CB2−/−Ldlr−/− lesions after 8 weeks. However, after 12 weeks, CB2−/−Ldlr−/− lesions displayed greater macrophage content (86.6 ± 4.1 versus 75.2 ± 7.5%, P < 0.05) and SMC content (11.1 ± 5.1 versus 4.2 ± 2.4%, P < 0.05) compared to controls. Lesional apoptosis, as determined by in situ TUNEL analysis, was reduced ∼50% in CB2−/−Ldlr−/− lesions after 12 weeks. CB2−/−Ldlr−/− lesions displayed significantly reduced collagen content and increased elastin fiber fragmentation after 12 weeks, which was associated with an ∼57% increase in matrix metalloproteinase 9 (MMP) levels. In vitro, CB2−/− macrophages secreted ∼1.8-fold more MMP9 activity than CB2+/+ macrophages.
Conclusions
CB2 receptor deficiency affects atherogenesis in Ldlr-null mice by increasing lesional macrophage and SMC content, reducing lesional apoptosis and altering extracellular matrix components, in part, by upregulating MMP9. These results suggest that pharmacological manipulation of CB2 receptors might exert multiple and complex effects on atherogenesis and plaque stability.
Keywords: Atherosclerosis, Apoptosis, Cannabinoid receptor 2, Macrophages, Smooth muscle cells, Collagen, Elastin, Matrix metalloproteinase 9
1. Introduction
Atherosclerosis is an inflammatory disease characterized by the buildup of lipids and cellular debris within arterial walls and influenced by numerous biochemical factors [1]. During atherogenesis, monocytes adhere to sites of vascular endothelial injury and migrate into the vascular wall where they proliferate and differentiate into macrophages. In the intima, they ingest atherogenic lipoproteins, such as oxidized low density lipoproteins (OxLDL), and transform into foam cells. OxLDL, and some oxysterol constituents of oxLDL such as 7-ketocholesterol, are well-known inducers of macrophage apoptosis [2]. The role of macrophage apoptosis in atherosclerosis is not fully understood and likely differs depending upon the stage of the disease and the apoptotic inducer. Macrophage apoptosis is an anti-atherosclerotic process that retards early lesion formation [3,4]. In contrast, evidence suggests that macrophage apoptosis in more advanced lesions is a pro-atherosclerotic process that contributes to plaque instability and rupture [5].
The biological effects of cannabinoids, and their endogenous counterparts, the endocannabinoids, are transduced by two functionally distinct G-protein coupled receptors known as CB1 [6] and CB2 [7]. The CB1 receptor is best characterized in the central nervous system where it modulates the neurological and psychotropic effects of cannabinoids [8]. The CB2 receptor is expressed primarily in immune cells, including macrophages, where it exerts the immunomodulatory effects of cannabinoids [9,10]. The CB2 receptor is therefore a target for the development of drugs to treat inflammatory diseases [11,12].
In vitro studies have shown that the CB2 receptor modulates several macrophage processes associated with ongoing atherosclerosis, including migration and proliferation [13,14], as well as the susceptibility to OxLDL/oxysterol-induced apoptosis [15]. In vivo evidence supporting a role for CB2 receptor signaling in atherosclerosis has emerged, including the observation of elevated levels of endocannabinoids in blood of patients with coronary artery disease [16] and, CB2 receptor expression in atherosclerotic lesions of humans and mice, but not in normal vascular tissue [14]. Furthermore, cannabinoid administration induces regression of atherosclerotic lesions in apolipoprotein E-null (ApoE−/−) mice, an effect that is attenuated by co-administration of a CB2-selective antagonist [14,17].
In the present study, we directly examined the consequences of CB2 gene deletion on the development of atherosclerotic lesions in Ldlr-null mice. Our data demonstrate that although CB2 receptor deficiency does not affect the size of atherosclerotic lesions, it is associated with increased lesional macrophage and SMC content, reduced lesional apoptosis and evidence of increased lesion remodeling. These results may have important clinical implications as the pharmaceutical use of cannabinoid compounds with activity at the CB2 receptor may produce multiple and complex effects in atherosclerotic lesions.
2. Materials and methods
2.1. Animals and diet-induced atherosclerosis protocol
All mice were housed in a pathogen-free, humidity- and temperature-controlled room in the Animal Research Facility at East Tennessee State University, maintained on standard mouse chow (Ralston Purina, St Louis, MO), and provided water ad libitum. CB2 receptor knockout mice (CB2−/−) [10] in the C57BL/J6 background (a kind gift from Dr. Nancy Buckley, California State Polytechnic University, Pomona, CA) were crossed with low density lipoprotein receptor knockout mice (Ldlr−/−) backcrossed to the C57BL/J6 background for ten generations (The Jackson Laboratories, Bar Harbor, ME) to generate CB2−/−Ldlr−/− and CB2+/+Ldlr−/− (control) mice. Genotyping was by polymerase chain reaction analysis of DNA isolated from tail clips using Ldlr specific primers, as described by Jackson Labs, and CB2 gene specific primers as previously described [15]. Timed matings were used to produce age-matched CB2−/−Ldlr−/− and CB2+/+Ldlr−/− mice for all experiments. At 8 weeks of age, groups of male CB2−/−Ldlr−/− and control mice were placed on an atherogenic diet (21% fat, 0.15% cholesterol; Harlan Teklad, Madison, WI) or maintained on standard chow for 8 or 12 weeks. All animal procedures were approved by and conducted in accordance with the guidelines administered by the Institutional Animal Care and Usage Committee of East Tennessee State University and in conformity with the Public Health Service Policy on Humane Care and Use of Laboratory Animals incorporated in the Institute for Laboratory Animal Research Guide for Care and Use of Laboratory Animals.
2.2. Atherosclerosis analysis
Anesthetized mice were euthanized by cardiac perfusion with 10 ml ice-cold phosphate-buffered saline. The heart and proximal aorta were dissected and fixed in phosphate-buffered formalin overnight at 4 °C. After fixation, the heart containing the aortic root and proximal aorta was placed in OCT embedding medium and frozen. OCT-embedded tissue was mounted in a Leica CM1850 cryostat and 50 8-μm cross-sections were collected as described [18]. A total of 15 alternating cryosections from each mouse were stained with oil-red O for lipid and counterstained with hematoxylin [3]. Images were digitally captured with an Olympus BX41 microscope equipped with a MicroPublisher 5.0 RTV CCD color camera (QImaging). Lesions lumenal to the internal elastic lamina were measured morphometrically using the Image-Pro Plus software (Media Cybernetics, Inc). Measurements were performed in blinded fashion independently by two observers.
2.3. In situ terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) analysis
Apoptosis was quantified using an in situ Cell Death Detection POD kit (Roche Applied Sciences, Indianapolis, IN) as previously described [3] with the following modifications. Frozen cryosections were thawed and air dried for 2 h, rehydrated with ethanol dilutions and pretreated with 3% citric acid. TUNEL enzyme was diluted 1:150 in TUNEL dilution buffer. Quantification of TUNEL-positive nuclei was performed in blinded fashion independently by two observers.
2.4. Histological and immunohistochemical staining
Prior to staining, cryosections were thawed, air dried for 1 h, rehydrated and fixed in 10% formalin. Aortic root collagen deposition was visualized by Mason's Trichrome staining. Elastin was visualized by the Verhoeff-van Gieson method (Elastin kit; Richard-Allan Scientific, Kalamazoo, MI). Elastin breaks were defined by interruption in the elastin fiber together with the reappearance of elastic fiber under high-power field microscopy and expressed as the number of elastin breaks per lesion [19]. For immunohistochemical staining, cryosections were thawed and air dried for 1 h, fixed in acetone for 10 min and then air dried another hour. Sections were pretreated for 0.5 h in 1% H2O2 in PBS, rinsed and blocked in normal goat serum. Vascular smooth muscle cells were detected with anti-smooth muscle α-actin (NeoMarkers, Fremont, CA) (diluted 1:200). Macrophages were detected with MAB1852 (Chemicon International, Temecula, CA) (diluted 1:1000). MMP9 immunoreactivity was detected with anti-MMP9 (1:1000 dilution) (Millipore, Temecula, CA). Sections were developed using a Vector Labs Elite ABC kit (Burlingame, CA) and counterstained with methyl green. Quantification of staining was by computer-assisted image analysis (NIH ImageJ; Bethesda, MD). A threshold for staining that discriminated between positive and negative lesion areas was defined by sampling and the same threshold was then applied to all specimens. The areas positive for stain were computed as the percentage of positively stained area relative to the total intimal lesion area studied. All evaluations were performed by a trained observer unaware of genotype.
2.5. Zymography
Resident peritoneal macrophages were isolated by lavage [20], plated (∼1 × 106 per chamber) in 4-well glass chamber slides (Nalge; Naperville, IL) in DMEM containing 10% FBS and allowed to attach overnight. The cells were rinsed with PBS and refed serum-free growth media (OPTI-MEM) supplemented with and without AM-251 or SR144528. Controls received an equivalent volume of vehicle (DMSO). After 24 h incubation, conditioned media was collected and equal volumes were loaded on to 10% SDS-polyacrylamide gels containing gelatin (1 mg/ml) and electrophoresed under nonreducing conditions. Gel proteins were renatured in 50 mM Tris/100 mM NaCl/2.5% Triton X-100 at room temperature, washed, and then incubated in 50 mM Tris/10 mM CaCl2/0.02% NaN3 at 37 °C for 48–72 h before staining with Coomassie blue. Zones of lysis corresponding to proteolytic activity were quantified using computer-assisted image analysis (NIH ImageJ software).
2.6. Statistical analysis
Data are presented as means ± SD. Differences were analyzed by Student's t-test or one-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant.
3. Results
3.1. CB2 receptor deficiency does not affect the size of atherosclerotic lesions
Ldlr−/− mice lacking the CB2 receptor (CB2−/−Ldlr−/−) were generated by standard mating protocols (Supplemental Fig. 1). At 8 weeks of age, male CB2−/−Ldlr−/− and CB2+/+Ldlr−/− (control) mice were placed on an atherogenic diet for 8 or 12 weeks. Throughout the study, no significant affect of CB2 receptor deficiency on body weight, diet-induced hyperlipidemia or plasma lipoprotein profiles was observed (Supplemental Fig. 2). After 8 and 12 weeks on the atherogenic diet, mice were sacrificed and the extent of atherosclerotic lesion development was quantified in two typical aortic vascular beds; the aortic root using serial cross-sections and on the intima of the thoracic and abdominal aorta by en face. Morphometric analysis revealed no significant difference between the mean area of lesions in the aortic root of control and CB2−/−Ldlr−/− mice after 8 weeks (107,577 ± 33,913 versus 95,366 ± 32,422 μm2, respectively; P = 0.30) or 12 weeks (195,278 ± 46,540 versus 177,093 ± 24,264 μm2, respectively; P = 0.36) (Fig. 1) on an atherogenic diet. En face prepared aortas showed no significant difference between the percentage of the entire aortic surface occupied by lesions in CB2−/−Ldlr−/− mice and control mice (7.5 ± 2.7 versus 5.6 ± 3.2%, respectively; P = 0.27) after 8 weeks of atherogenic diet (Supplemental Fig. 3).
Fig. 1.
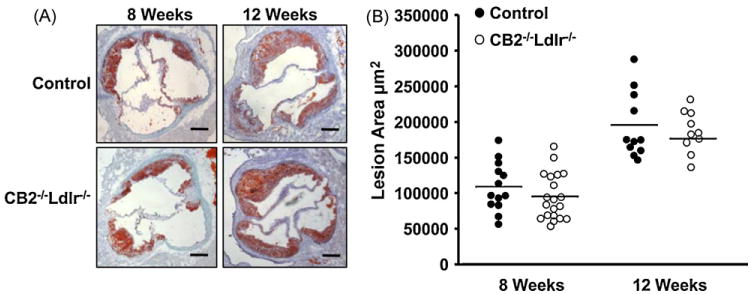
CB2 receptor deficiency does not affect atherosclerotic lesion area in Ldlr−/− mice after 8 and 12 weeks of an atherogenic diet. (A) Representative photomicrographs of oil-red O stained aortic root cross-sections from CB2−/−Ldlr−/− and control mice. Scale bar = 200 μm. (B) Quantification of the aortic root lesion area in CB2−/−Ldlr−/− and control mice. Each data point represents the mean cross-sectional lesion area for one animal; lines represent the mean values of the groups (n ≥ 10).
3.2. CB2 receptor deficiency increases the accumulation of macrophages and smooth muscle cells in atherosclerotic lesions
Immunostaining with a MOMA2 antibody showed extensive macrophage content in the intima of CB2−/−Ldlr−/− and control lesions after 8 and 12 weeks on the atherogenic diet (Fig. 2A). Quantification of the MOMA2-positive areas found no significant difference in the macrophage content of CB2−/−Ldlr−/− and control lesions (79.6 ± 9.0 versus 75.2 ± 5.0%, respectively; P = 0.21) after 8 weeks on the atherogenic diet; however, after 12 weeks, the macrophage content of CB2−/−Ldlr−/− lesions was statistically greater than that of control lesions (86.6 ± 4.1 versus 75.2 ± 7.5%, respectively; P < 0.05) (Fig. 2B). Immunostaining with a smooth muscle α-actin antibody showed equivalent SMC immunoreactivity in the media of lesions from both genotypes and increased SMC immunoreactivity within the intima and the fibrous cap regions of CB2−/−Ldlr−/− lesions (Fig. 2A). Quantification of the smooth muscle α-actin immunoreactive areas revealed comparable SMC content in CB2−/−Ldlr−/− and control lesions (5.0 ± 2.8 versus 5.8 ± 3.7%, respectively; P = 0.6) after 8 weeks and an ∼2.6-fold greater SMC content in CB2−/−Ldlr−/− lesions relative to controls (11.1 ± 5.1 versus 4.2 ± 2.4%, respectively; P < 0.05) after 12 weeks (Fig. 2C).
Fig. 2.
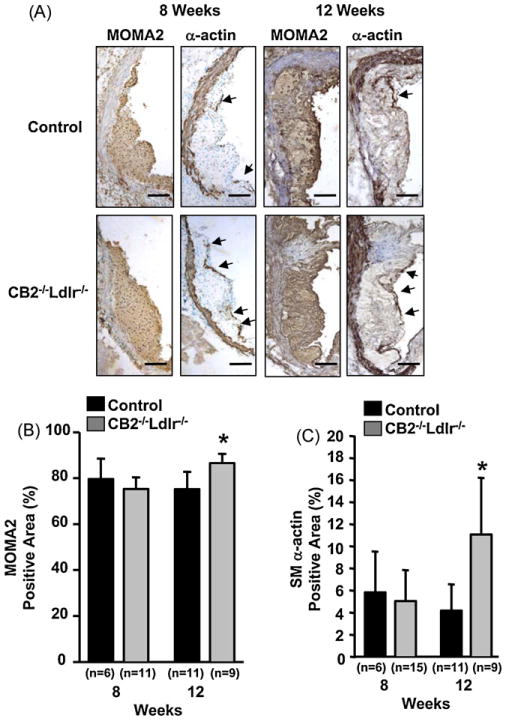
Cellular composition of atherosclerotic lesions in CB2 receptor-deficient Ldlr−/− mice fed an atherogenic diet for 8 and 12 weeks. (A) Representative photomicrographs of serial aortic root cross-sections from CB2−/−Ldlr−/− and control mice fed an atherogenic diet for 8 and 12 weeks immunohistochemically stained for macrophages (MOMA2 antibody) and SMCs (α-actin antibody) as indicated. Arrows indicate regions of SMC immunoreactivity in the lesion cap area. Scale bar = 100 μm. (B and C) Quantification of the macrophage and SMC content of aortic root lesions from CB2−/−Ldlr−/− and control mice. Immunopositive areas were quantified and recorded as the percentage of the total area of the lesion. Values are the means ± SD for each group of mice (n is as indicated in the figure), *P < 0.05.
3.3. CB2 receptor deficiency reduces lesional apoptosis in Ldlr-null mice
The percentage of TUNEL-positive nuclei detected in atherosclerotic lesions of CB2−/−Ldlr−/− and control mice fed an atherogenic diet for 8 weeks was not significantly different (0.74 ± 0.29 versus 0.67 ± 0.23%, respectively; P > 0.05). In contrast, lesions from CB2−/−Ldlr−/− mice fed an atherogenic diet for 12 weeks displayed an ∼2-fold decrease in the percentage of TUNEL-positive nuclei in comparison to control lesions (0.49 ±0.16 versus 1.05 ± 0.33%, respectively; P < 0.05) (Fig. 3). TUNEL-positive nuclei were localized to macrophage-enriched areas of lesions (Supplemental Fig. 4).
Fig. 3.
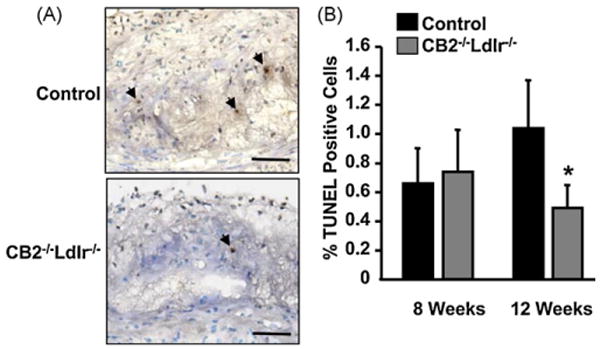
CB2 receptor deficiency decreases lesional apoptosis in Ldlr−/− mice. (A) Representative photomicrographs of in situ TUNEL-POD stained aortic root lesions in cross-sections from CB2−/−Ldlr−/− and control mice after 12 weeks on an atherogenic diet. TUNEL-positive nuclei (brown stain in the nuclei of intimal cells) are indicated by arrows. Scale bar = 50 μm. (B) Quantification of the percentage of TUNEL-positive nuclei in aortic root lesions of CB2−/−Ldlr−/− and control mice after 8 and 12 weeks of an atherogenic diet. Values are means ± SD, n = 6, *P < 0.05.
3.4. CB2 receptor deficiency alters the extracellular matrix composition (ECM) of atherosclerotic lesions
The collagen content of aortic root lesions, as assessed by Mason's trichrome staining, was not discernibly different between CB2−/−Ldlr−/− and control mice after 8 weeks on an atherogenic diet; however, after 12 weeks, CB2−/−Ldlr−/− lesions were consistently observed to have a notably less dense collagen matrix compared to control lesions (Fig. 4A). Quantification confirmed that after 8 weeks of an atherogenic diet, the collagen content of CB2−/−Ldlr−/− lesions was not significantly different than that of control lesions (11.9 ± 6.2 versus 17.8 ± 6.8%, respectively; P = 0.06) and that after 12 weeks, CB2−/−Ldlr−/− lesions had significantly lower collagen content compared to control lesions (13.7 ± 5.5 versus 32.8 ± 9.7%, P < 0.05) (Fig. 4B). Verhoeff–Van Gieson staining revealed that after 8 weeks of an atherogenic diet the medial elastic lamella of CB2−/−Ldlr−/− and control lesions were largely intact and not perceptibly different; however, after 12 weeks, the medial elastic lamella of CB2−/−Ldlr−/− lesions were noticeably more fragmented compared to their controls (Fig. 4C). Quantification of the number of elastin fiber breaks per lesion showed no significant difference between CB2−/−Ldlr−/− and control lesions (1.8 ± 1.4 versus 2.1 ± 1.8, respectively; P = 0.52) after 8 weeks on the atherogenic diet; however, after 12 weeks, CB2−/−Ldlr−/− lesions contained ∼2-fold more elastin fiber breaks per lesion than control lesions (6.1 ± 4.1 versus 3.1 ± 2.7, respectively; P < 0.05) (Fig. 4D).
Fig. 4.
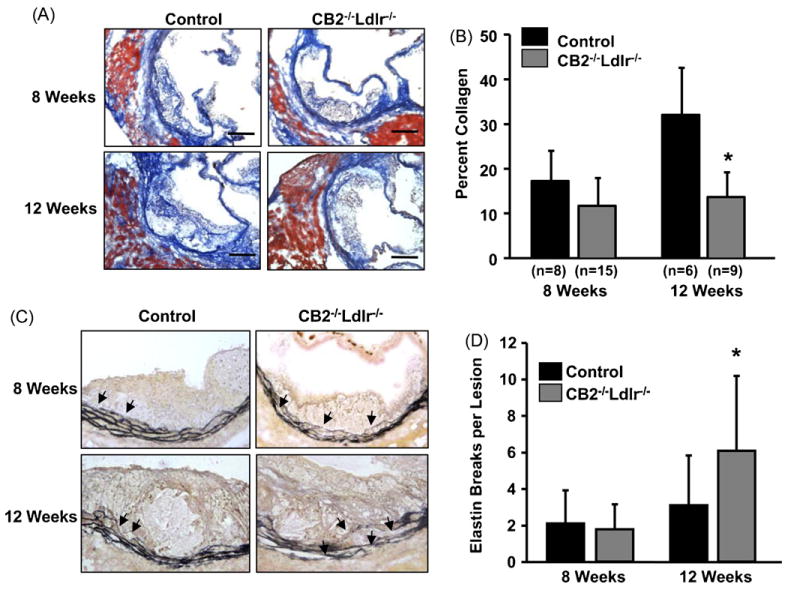
Extracellular matrix composition of atherosclerotic lesions in CB2 receptor-deficient Ldlr−/− mice after 8 and 12 weeks on an atherogenic diet. (A) Representative photomicrographs of atherosclerotic lesions in aortic root cross-sections after Mason's trichrome staining of collagen (blue). Scale bar = 200 μm. (B) Digital quantification of the percentage of aortic root lesion area occupied by collagen. Values are the mean ± SD, n is as indicated, *P < 0.05. (C) Representative images of lesions in aortic root cross-sections after Verhoeff–van Gieson staining of elastin fibers (black). Arrows indicate regions of inner elastic lamina containing elastin fiber breaks. (D) Quantification of the number of elastin fiber breaks present in lesions of each group. Values are mean ± SD, n = 6 for each group, *P < 0.05. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.5. Elevated matrix metalloproteinase 9 levels in lesions of CB2-null mice
MMP-9 immunoreactivity was similar in lesions of both genotypes after 8 weeks on the atherogenic diet (Fig. 5A). In contrast, after 12 weeks, MMP9 immunoreactivity in CB2−/−Ldlr−/− lesions was ∼1.6-fold greater than corresponding wild type lesions (37.1 ± 14.7 versus 23.6 ± 11.0%, respectively; P < 0.05). Peritoneal macrophages produce endocannabinoids which function in autocrine and paracrine signaling mechanisms to modulate a variety of macrophage processes [21,22]. Zymography of media collected from cultured peritoneal macrophages isolated from CB2−/−Ldlr−/− and control mice detected a single band of gelatinase activity corresponding to the approximate molecular weight of MMP9 (92 kDa) (Fig. 5B). Quantification of the zymograms revealed that CB2−/−Ldlr−/− macrophages produced ∼1.8-fold more MMP9 activity compared to wild type macrophages (Fig. 5B). We also examined the effect of CB2 receptor antagonism on MMP9 secretion from wild type peritoneal macrophages. The CB2-selective antagonist, SR144528, dose-dependently increased MMP9 secretion, with the highest doses (1 and 2 μM) producing significantly more MMP9 activity (∼1.4- and 1.5-fold, respectively, P < 0.05) relative to untreated controls (Fig. 5C). A CB1-selective antagonist, AM-251, also produced a dose-dependent trend of increasing MMP9 secretion, however, a statistically significant difference was not achieved (Fig. 5C).
Fig. 5.
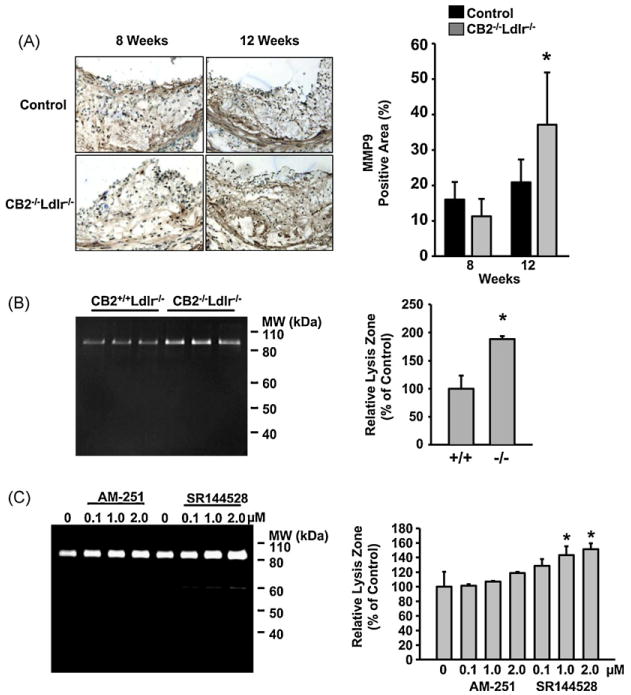
CB2 receptor deficiency upregulates MMP9 in atherosclerotic lesions of Ldlr-null mice. (A) Representative photomicrographs of aortic root lesions from CB2−/−Ldlr−/− and control mice after 8 and 12 weeks on an atherogenic diet immunohistochemically stained with anti-MMP9. Right panel shows the results of quantification of the MMP9-positive areas for each group. Values are mean ± SD, n = 7 for each group, *P < 0.05. (B) Zymogram showing MMP9 activity in the conditioned media of peritoneal macrophages isolated from CB2−/−Ldlr−/− and control mice (left panel). Digital quantification of the MMP9 zones of lysis in the zymogram (right panel). Values are the mean ± SD, n = 3 for each genotype, *P < 0.05. (C) Zymogram showing MMP9 activity secreted by wild type macrophages incubated for 24 h in the presence of increasing concentrations of a CB2 antagonist (SR144528) or a CB1 antagonist (AM-251) as indicated (left panel). Digital quantification of MMP9 activity in the zymogram (right panel). Data are reported as percentage of the relative integrated densities of the lysis zones for each treatment normalized to the mean of untreated controls. Data are representative of 2 independent experiments with triplicate samples. *P < 0.05.
4. Discussion
Pharmacological studies suggest that the CB2 receptor plays a role in atherosclerosis [14,17]. In the current study, we directly examined the role of the CB2 receptor in atherosclerosis by investigating the effects of systemic CB2 receptor deficiency on atherosclerosis in Ldlr-null mice. We observed that CB2 receptor deficiency did not affect the size of lesions in Ldlr−/− mice; however, it did increase lesional macrophage accumulation and SMC infiltration, in addition to reducing lesional apoptosis and altering the ECM of lesions.
The lack of an effect of CB2 receptor gene deficiency on the size of atherosclerotic lesions in Ldlr-null mice is somewhat unexpected as administration of exogenous cannabinoids decreases the size of atherosclerotic lesions in ApoE−/− mice by mechanisms sensitive to CB2 receptor-selective antagonism [14,17]. There are several possible explanations for these somewhat contradictory results. First, activation of CB2 receptors by endogenous ligands during atherogenesis may not be sufficient to significantly affect lesion size, but when exogenous cannabinoids are supplemented, CB2 receptor signaling is augmented to levels sufficient to produce an affect on lesion size. Second, a novel cannabinoid receptor, which compensates for the loss of CB2 in CB2−/−Ldlr−/− mice, may be responsible for the effects of exogenous cannabinoid supplementation on lesion size. Pharmacological evidence for novel cannabinoid receptors, some of which display sensitivity to CB1 - and CB2-selective antagonists, lends some support to this suggestion [23,24]. Lastly, these results may be due to differences between the Ldlr−/− and ApoE−/− mouse strains, which are well-known to vary in lipoprotein characteristics and other parameters that determine the pathophysiology of atherosclerosis [25].
The observation that lesional macrophage content is unaffected by CB2 receptor deficiency in Ldlr−/− mice in lesions after 8 weeks of atherogenic diet intervention but is increased in lesions after 12 weeks indicates that CB2 receptor-mediated signaling does not play a significant role in monocyte/macrophage recruitment during initial lesion formation but instead, becomes sufficient to reduce macrophage recruitment as lesions progress. This is consistent with the prior study by Steffens et al. [14] demonstrating that Δ9-tetrahydrocannabinol-induced lesion regression in ApoE−/− mice was, at least partly, a consequence of reduced leukocyte recruitment. In this scenario, exogenous cannabinoid supplementation augments endogenous CB2 receptor activation resulting in a greater impairment of monocyte/macrophage recruitment.
Although no variation in the level of CB1 receptor expression in CB2-null macrophages was observed (Supplemental Fig. 1), we can not rule out the possibility that the lack of more pronounced effects of CB2 receptor deficiency on lesion formation is not due to compensatory activation of CB1 receptors or compensatory expression of a novel cannabinoid receptor(s). Evidence of novel cannabinoid receptors in the vasculature lend some support to this suggestion [23,24]. Future studies examining the effect of cannabinoid supplementation on lesional macrophage content in CB2−/−Ldlr−/− mice should further clarify the role of CB2 receptors in cannabinoid-induced lesion regression.
Proliferation and migration of vascular SMCs are important events in atherogenesis which could be affected by CB2 receptor signaling. In vitro, SMC proliferation and migration induced by TNF-α is attenuated by CB2 receptor agonists [26], while SMC proliferation and migration in response to PDGF is attenuated by SR141716, a CB1 receptor antagonist [27]. This suggests that CB1 and CB2 receptors may differentially modulate the proliferative and migratory responses of SMCs to cytokines and chemokines. The increased SMC immunoreactivity observed in CB2−/−Ldlr−/− lesions is consistent with an inhibitory role of CB2 receptors in SMC proliferation and migration during atherosclerosis. Indirect effects brought about by the interaction between CB2-null macrophages and SMCs may also contribute to the increased SMC content of CB2−/−Ldlr−/− lesions. The inability to ascribe the observed effects of CB2 receptor deficiency to any one particular cell type is an inherent limitation of the current experimental approach. Experiments employing adoptive transfer of CB2-deficient bone marrow derived cells in to Ldlr-null mice should provide insight into the relevant cell types involved in CB2 receptor-modulated events during atherosclerosis.
Diminished lesional apoptosis in CB2 receptor-null mice is in agreement with our prior in vitro study demonstrating reduced susceptibility of CB2-null macrophages to oxLDL-induced apoptosis [15], and our previous study associating reduced lesional apoptosis (due to Bax gene deficiency) with increased lesional macrophage content [3]. It is important to note that the CB2 receptor and Bax likely play different roles in apoptosis. While Bax functions in the execution phase of apoptosis which is common to a broad range of apoptosis inducers, CB2 receptors modulate the response of macrophages to specific apoptosis inducers, the presence of which likely varies within atherosclerotic lesions as they progress. The observation that reduced apoptosis was not observed in lesions of CB2-null mice after 8 weeks on an atherogenic diet, but was evident after 12 weeks, suggest that a threshold level of a specific apoptosis inducer, and/or CB2 ligand, must accumulate before CB2-mediated modulation of apoptosis occurs [15]. Although TUNEL-positive nuclei were largely localized to macrophage-enriched regions, it is possible that CB2 receptor deficiency also alters apoptosis of cell types in addition to macrophages in atherosclerotic lesions.
CB2 receptor deficiency could reduce collagen content by slowing collagen synthesis and/or accelerating collagen degradation. SMCs are a primary source of collagen deposition in atherosclerotic lesions [28,29], however, the observed increase in SMC content in lesions of CB2-null mice suggest that collagen reduction is not due to lower numbers of intimal SMCs; although, a reduction of collagen synthesis by CB2-null SMCs can not be ruled out. The observation of increased MMP9 immunoreactivity in CB2−/−Ldlr−/− lesions, as well as increased secretion of MMP9 activity from CB2-null macrophages, suggests that upregulation of MMP9 in CB2−/−Ldlr−/− lesions is, at least partially, responsible for the reduction of collagen. In support of this hypothesis, MMP9 is suppressed in immune cells by cannabinoid compounds [30,31] and was induced by CB2 receptor antagonism (Fig. 5C). Increased disruption and fragmentation of medial elastic fibers in CB2−/−Ldlr−/− lesions may also be the result of increased MMP9 activity. Increased degradation of elastin may also facilitate the migration of SMCs from the media into the intima and contribute to the increase in SMC content of CB2−/−Ldlr−/− lesions. Collectively, these results suggest that CB2 receptor signaling contributes to the establishment and maintenance of more stable atherosclerotic lesions, in part, by suppressing MMP9. It should also be noted that while zymography reveals the total enzymatic activity, it may not reflect the net in vivo activity of MMP9 which is subject to the levels of tissue inhibitors of matrix metalloproteinases. Future studies examining MMP activities in atherosclerotic lesions of CB2−/−Ldlr−/− mice should clarify the role of CB2 receptors in modulating MMP activities and extracellular matrix remodeling of lesions.
Macrophage death is common in the shoulders of rupture-prone human plaques and is considered a destabilizing factor in plaque rupture [5]. Therefore, even though degradation of ECM components may promote lesion destabilization, the absence of CB2 receptor signaling might also provide a beneficial effect due to reduced lesional apoptosis in more advanced lesions. Determining the extent to which any modulation of macrophage apoptosis associated with CB2 receptor deficiency, or treatments with CB2-selective agonists and antagonists, affects plaque stability, will require studies using ApoE−/− mice in which plaque rupture can be dependably observed in the innominate artery [32].
In conclusion, the present study provides the first direct evidence that CB2 receptor signaling is not significantly involved in the initial formation of atherosclerotic lesions, but may play an important role in modulating cellular composition and stability of lesions as they progress. These results support the idea that pharmacologic compounds with activity at CB2 receptors may be useful in modifying atherosclerosis and underscore the need for further studies aimed at more fully elucidating the role of CB2 receptor signaling in plaque composition and vulnerability.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Institutes of Health (grant No. HL085137 to Dr. Thewke). The authors are grateful to Dr. Zhihua Han for his technical help and Dr. Michael Sinensky for his valuable assistance in preparing the manuscript.
Footnotes
Conflict of interest: The authors have no conflict of interest to disclose.
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:l0.1016/j.atherosclerosis.2010.07.060.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Brown AJ, Jessup W. Oxysterols and atherosclerosis. Atherosclerosis. 1999;142:1–28. doi: 10.1016/s0021-9150(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Thewke DP, Su YR, et al. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25:174–9. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arai S, Shelton JM, Chen M, et al. A role for the apoptosis inhibitory factor aim/spalpha/api6 in atherosclerosis development. Cell Metab. 2005;1:201–13. doi: 10.1016/j.cmet.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Tabas I. Apoptosis and plaque destabilization in atherosclerosis: the role of macrophage apoptosis induced by cholesterol. Cell Death Differ. 2004;11(Suppl. 1):S12–6. doi: 10.1038/sj.cdd.4401444. [DOI] [PubMed] [Google Scholar]
- 6.Matsuda LA, Lolait SJ, Brownstein MJ, et al. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–4. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 7.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–5. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 8.Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci. 2001;22:565–72. doi: 10.1016/s0165-6147(00)01805-8. [DOI] [PubMed] [Google Scholar]
- 9.Galiegue S, Mary S, Marchand J, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 10.Buckley NE, McCoy KL, Mezey E, et al. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;396:141–9. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- 11.Mach F, Montecucco F, Steffens S. Cannabinoid receptors in acute and chronic complications of atherosclerosis. Br J Pharmacol. 2008;153:290–8. doi: 10.1038/sj.bjp.0707517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pacher P, Ungvari Z. Pleiotropic effects of the CB2 cannabinoid receptor activation on human monocyte migration: implications for atherosclerosis and inflammatory diseases. Am J Physiol Heart Circ Physiol. 2008;294:H1133–4. doi: 10.1152/ajpheart.00020.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montecucco F, Burger F, Mach F, et al. CB2 cannabinoid receptor agonist jwh-015 modulates human monocyte migration through defined intracellular signaling pathways. Am J Physiol Heart Circ Physiol. 2008;294:H1145–55. doi: 10.1152/ajpheart.01328.2007. [DOI] [PubMed] [Google Scholar]
- 14.Steffens S, Veillard NR, Arnaud C, et al. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature. 2005;434:782–6. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- 15.Freeman-Anderson NE, Pickle TG, Netherland CD, et al. Cannabinoid (CB2) receptor deficiency reduces the susceptibility of macrophages to oxidized ldl/oxysterol-induced apoptosis. J Lipid Res. 2008;49:2338–46. doi: 10.1194/jlr.M800105-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugamura K, Sugiyama S, Nozaki T, et al. Activated endocannabinoid system in coronary artery disease and antiinflammatory effects of cannabinoid 1 receptor blockade on macrophages. Circulation. 2009;119:28–36. doi: 10.1161/CIRCULATIONAHA.108.811992. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Y, Yuan Z, Liu Y, et al. Activation of cannabinoid CB2 receptor ameliorates atherosclerosis associated with suppression of adhesion molecules. J Cardiovasc Pharmacol. 2010 doi: 10.1097/FJC.0b013e3181d2644d. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 18.Paigen B, Morrow A, Holmes PA, et al. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–40. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 19.Sukhova GK, Shi GP, Simon DI, et al. Expression of the elastolytic cathepsins s and k in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–83. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeman NE, Rusinol AE, Linton M, et al. Acyl-coenzyme a: cholesterol acyltransferase promotes oxidized ldl/oxysterol-induced apoptosis in macrophages. J Lipid Res. 2005;46:1933–43. doi: 10.1194/jlr.M500101-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid PC, Kuwae T, Krebsbach RJ, et al. Anandamide and other nacylethanolamines in mouse peritoneal macrophages. Chem Phys Lipids. 1997;87:103–10. doi: 10.1016/s0009-3084(97)00032-7. [DOI] [PubMed] [Google Scholar]
- 22.Kuwae T, Shiota Y, Schmid PC, et al. Biosynthesis and turnover of anandamide and other n-acylethanolamines in peritoneal macrophages. FEBS Lett. 1999;459:123–7. doi: 10.1016/s0014-5793(99)01226-0. [DOI] [PubMed] [Google Scholar]
- 23.Mackie K, Stella N. Cannabinoid receptors and endocannabinoids: evidence for new players. Aaps J. 2006;8:E298–306. doi: 10.1007/BF02854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Begg M, Pacher P, Batkai S, et al. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;106:133–45. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Daugherty A, Rateri DL. Development of experimental designs for atherosclerosis studies in mice. Methods. 2005;36:129–38. doi: 10.1016/j.ymeth.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 26.Rajesh M, Mukhopadhyay P, Hasko G, et al. CB2 cannabinoid receptor agonists attenuate tnf-alpha-induced human vascular smooth muscle cell proliferation and migration. Br J Pharmacol. 2008;153:347–57. doi: 10.1038/sj.bjp.0707569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajesh M, Mukhopadhyay P, Hasko G, et al. Cannabinoid cb1 receptor inhibition decreases vascular smooth muscle migration and proliferation. Biochem Biophys Res Commun. 2008;377:1248–52. doi: 10.1016/j.bbrc.2008.10.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newby AC, Zaltsman AB. Fibrous cap formation or destruction – the critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc Res. 1999;41:345–60. [PubMed] [Google Scholar]
- 29.Newby AC. Metalloproteinases. vulnerable atherosclerotic plaques. Trends Cardiovasc Med. 2007;17:253–8. doi: 10.1016/j.tcm.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh S, Preet A, Groopman JE, et al. Cannabinoid receptor CB2 modulates the cxcl12/cxcr4-mediated chemotaxis of t lymphocytes. Mol Immunol. 2006;43:2169–79. doi: 10.1016/j.molimm.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Tauber S, Scheider-Stock R, Ullrich O. Investigating immunomodulatory mechanisms of cannabinoids: the role of mmp-9. Cell Commun Signal. 2009;7(Suppl. 1):A89. [Google Scholar]
- 32.Rosenfeld ME, Polinsky P, Virmani R, et al. Advanced atherosclerotic lesions in the innominate artery of the apoe knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587–92. doi: 10.1161/01.atv.20.12.2587. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.