

Abstract
Infection with a high-risk carcinogenic type of human papillomavirus (HPV) is necessary for the development of cervical cancer. The digene HC2 HPV Test (HC2) is an important screening tool but lacks genotyping capability. To address this issue, we developed an assay for the rapid genotyping of HPV in cervical specimens. The three steps of this assay include Hybrid Capture target enrichment, whole-genome amplification, and Luminex XMAP detection. The assay includes the simultaneous detection of two genomic regions from each of 17 high-risk and two low-risk HPV types most associated with disease. The assay performance was tested on HPV plasmids as well as clinical specimens. An analytical limit of detection of 100 copies or less was demonstrated for linear, circular, and integrated HPV DNA. This finding is at least 1 log lower than the HC2 assay limit of detection. There was no cross-reactivity among the HPV types up to 1,000,000 copies. There was also no substantial assay interference from substances in cervical specimens. Although the clinical performance of the assay was not formally tested, the assay had good agreement (Cohen's kappa equal to 0.72) with both a PCR-based HPV genotyping assay (n = 131) and the HC2 assay (n = 502) using representative cervical specimens. This assay may be easy to automate and could be applied for the detection of other targets in future studies.
Infection with a carcinogenic type of human papillomavirus (HPV) is necessary for development of cervical cancer.1,2 There are 13 types of HPV (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59 and 68) confirmed to be of high or moderate risk for cervical cancer and four types (26, 73, 66 and 82) that are probable high-risk. Other HPV types are low-risk and may be associated with genital warts, such as HPV 6 and 11. Molecular-based screening for the most oncogenic types of HPV is being used more frequently to diagnose risk for cervical cancer because it is easier and more reliable than cytological screening.3 For example, the digene HC2 HPV DNA Test (HC2) is widely used for screening4 because it has exceptional clinical performance for a cancer and high-grade disease end point (ie, negative predictive value). Molecular screening, however, is not designed to determine which HPV type is present in an infection. The type of HPV in an infection is important to monitor because the risk of cancer and high-grade lesions increases for those whose HPV infection type is the same over time, especially for HPV 16 and 18.5
Because of the numerous HPV types, genotyping assays require a high level of multiplex. The development of PCR-based assays for HPV genotyping is useful to identify infections with carcinogenic HPV-types, but they have some technical limitations. Many genotyping assays involve PCR with consensus primers that amplify conserved regions of the targeted HPV types.6,7 The efficiency of consensus-PCR is limited because primers have a variable number of nucleotide matches to the various HPV targets. An alternate approach of using multiple primer sets in a single tube is also limited due to interference among multiple primer sets. Multiplex real-time PCRs in a single reaction is also limited by the number of dyes that may be discriminated by the PCR detector instrument. Furthermore, currently described PCRs amplify a relatively small region of HPV, most in the L1 region, so they may miss HPV that has been disrupted due to viral integration.8 Besides integration, many point mutations in the HPV genome have been described, which may decrease the efficiency of any specific primer or probe set.9 The efficiency of HPV genotyping assays may also be reduced if a multitude of human DNA and nontargeted DNA is included in the reaction. This occurs for DNA isolation methods that are not specific to the targeted HPV sequences.
Our HPV genotyping assay was developed to permit rapid and accurate genotyping with a high level of multiplex, while avoiding or mitigating the stated drawbacks of PCR. The assay is called Hybrid Capture (HC)-whole genome amplification (WGA)-Luminex (LMX) and is described as follows. First, there is a sequence-specific sample enrichment process to isolate single-stranded HPV DNAs while removing much of the nonspecific genomic DNA that may interfere with some assays. This sample preparation was devised from HC technology used in the HC2 Test10,11 but requires less time and captures the RNA:DNA hybrids on magnetic beads instead of an assay plate.12 The second, WGA step amplifies the enriched single-stranded DNA using isothermal amplification with φ-29 DNA polymerase. This polymerase is highly processive and has strand-displacement activity, which creates multiple, long linear strands of target in the presence of short random primers or where nicks occur in the DNA.13,14 This step amplifies all sequences present, which permits a high level of multiplex. The long amplicon size allows use of several probes for simultaneous detection of multiple regions of the same HPV target. The final, multiplex detection step utilizes Luminex XMAP technology15 combined with HC signal amplification. LMX technology consists of capturing HPV target amplicons onto oligonucleotides that are fixed to fluorescent beads. For this assay, there are two specific oligonucleotide sequences per bead per HPV target, and each bead has a distinct fluorescent emission. Long complementary HPV RNA probes are bound to the denatured captured DNA creating hybrids on the LMX bead complex. The hybrids are detected using a fluorescent HC antibody (reporter). Multiple reporter antibodies are bound to each hybrid, which provides signal amplification and improves the analytical sensitivity of the assay. The bead and the reporter emissions are analyzed by a LMX flow cytometer instrument. The assay was designed to detect the 17 most common high-risk types detected in cervical cancers and the two most common low-risk types found in genital warts. The assay was tested on samples of HPV DNA plasmids and with clinical specimens. Results were compared with the widely-used clinical diagnostic assay, HC2 test, and with a PCR-based genotyping assay.
Materials and Methods
Specimens
Cervical specimens were collected in Specimen Transport Medium (STM, QIAGEN, Gaithersburg, MD) using an endocervical brush. The specimens were collected from a routine HPV screening population by a health care clinic and sent to QIAGEN for analysis. The HPV status of these specimens was determined using the HC2 assay. Most of the HPV-positives and a subset of HPV-negatives were selected for assay testing. The cytological and histological diagnoses are not known nor are they within the scope of this report.
RNA Probes
A set of 19 type-specific HPV RNA probes were prepared by in vitro transcription of near full-length HPV DNA clones. Two 500–800 base deletions were engineered into two separate regions of each RNA template plasmid using cloning techniques (ExSite PCR-based site-directed mutagenesis kit, Stratagene, La Jolla, CA). The deleted sequences were located in regions of low homology between HPV types, as determined by multiple sequence alignment. The 19 type-specific probes included HPV 6, 11, 16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, 73, and 82.
HC Sample Preparation
A 50-μl aliquot of plasmid DNA or specimen (1 ml STM) was denatured (50°C, 15 m) with an alkaline Denaturation Reagent (QIAGEN) in a microtitre plate. A neutralizing Probe Diluent (QIAGEN) was added, which included the 19 RNA probes (5 ng each). A solution of paramagnetic beads (approximately 2 × 106, Protein-G, Dynal, Life Technologies, Carlsbad, CA) coupled with a monoclonal antibody specific to RNA:DNA hybrids were added. RNA:DNA hybrids formed and bound to the antibody-beads during a hybridization step (65°C, 30 m, 1100 rpm rotation). For clinical specimens, after hybridization, the magnetic beads with attached hybrids were washed 8× with a warm buffer (60°C; 40 mmol/L Tris, pH 8.2, 100 mmol/L NaCl, 0.05% Sodium Azide, 0.05% Tween-20) and a final wash with a second buffer (62.5 mmol/L Tris-HCl, pH 8, 0.75 mmol/L MgCl2, 12.5 mmol/L (NH4)2 SO4). A 96-well plate was used and a magnetic plate (Ambion, Austin, TX) was used to retain the beads during washing.
Whole Genome Amplification
After aspiration of the last wash buffer, an amplification buffer (20 μl) was added to the target mixture and was incubated for 2 hours in a heat block (30°C). The amplification buffer contained 62.5 mmol/L Tris-HCl, pH 7.5, 0.75 mmol/L MgCl2, 12.5 mmol/L (NH4)2 SO4, 5 mmol/L DTT, 250 μg/ml BSA, 5 mmol/L dNTP, 312.5 μmol/L random pentamers and 1 U of φ-29 DNA polymerase (New England Biolabs, Ipswitch, MA). The pentamers (IDT, Coralville, IA) were synthesized with 3′ ultimate and penultimate phosphorothioate bonds to prevent degradation by φ-29 polymerase nuclease activity.
Multiplex Detection Using LMX Technology
The amplified DNA sample was alkali-denatured (60°C, 15 minutes) with Denaturation Reagent (QIAGEN). A neutralizing Probe Diluent (QIAGEN) that contained the 19 type-specific RNA probes (20 ng each) was added. A solution of 19 HPV type-specific LMX beads (3000 beads per type per assay) was added. Each type-specific LMX bead was coupled to two oligonucleotide capture probes that were specific for an HPV type. The oligonucleotides were designed to be specific using the BLAST algorithm, and they were located in a HPV genome approximately in the middle of a RNA probe deletion. The detection oligonucleotides were manufactured (IDT, Coralville, IA) with amino-links, and they were coupled to the carboxyl-groups of LMX beads according to manufacturer's protocol. A RNA:DNA hybrid-specific monoclonal antibody conjugated with the phycoerythrin fluorophore (4 ng per assay with added goat serum [2% final], Gibco) was added to the mixture. The components were hybridized (65°C, 30 m, 1100 rpm rotation) and then diluted 1:3 in pure water. This mixture was then analyzed in a Luminex 100 flow cytometer (1.7 Luminex software) which measured median fluorescence intensity (MFI).
HPV Screening Assay
The FDA-approved, digene HC2 Test (QIAGEN) for cervical cancer screening was used to detect 13 high-risk HPV types in samples, according to the manufacturer's instructions.
Gp5+−6+ PCR Genotyping Assay
A published HPV genotyping protocol based on PCR using consensus primers was used.12 Briefly, DNA was isolated using the DNeasy purification kit (QIAGEN) and amplified using the GP5+ and GP6+ (biotin-labeled) primers.16 The biotin-labeled amplicons were captured by 19 type-specific oligonucleotides bound to LMX beads, streptavidin-phycoerythrin was used as the reporter bound to the target, and the HPV genotypes were analyzed using the LMX instrument. The type-specific oligonucleotides bound to LMX beads were not identical to those used to capture HPV DNA for the HC-WGA.
Results
Assay Design
The genotypes of 17 high- or intermediate-risk (16, 18, 26, 31, 33, 35 39, 45, 51, 52, 56, 58, 59, 66, 68, 73, and 82) and two low-risk (6 and 11) were detected by the three-step, HC-WGA-LMX multiplex assay (Figure 1). In the HC enrichment step, endogenous RNA:DNA hybrids in the sample were degraded and the target DNA strands were separated. Then, RNA:DNA hybrids were formed in the neutralizing hybridization mixture between the synthetic RNA probes and the complementary DNA target strands. These hybrids were simultaneously isolated with the magnetic beads bearing the hybrid-specific antibody. Single DNA target strands were enriched and nonspecific DNA, and potential interfering substances were removed during washing. In the whole genome amplification step, the entire sample of magnetic beads was reacted. Although not directly tested, no evidence of nonuniform amplification was observed. In the LMX detection step, amplified DNA was alkali-denatured then hybridized with the 19 HPV-type specific RNA probes in the neutralizing hybridization buffer. The deletions in the RNA probes provided gaps in the hybrid that enabled efficient annealing of detection oligonucleotides that were bound to the LMX beads. The discrimination of HPV types resulted from the specificity of the two detection oligonucleotides for its HPV type (Table 1). Each bead could bind to an HPV target at two loci due to having two specific oligonucleotides from separate regions. The hybrid-specific antibody with phycoerythrin reporter provided signal amplification. A 96-well plate was analyzed in 40 minutes by the LMX instrument and software, and the time to run the entire 96-well assay was less than 5 hours.
Figure 1.
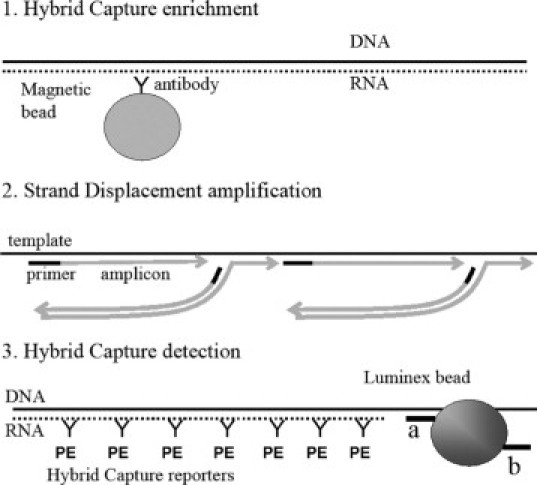
Schematic diagram of the HC-WGA-LMX assay. 1: Hybrid Capture enrichment shows a double-deleted RNA (dotted line) single-stranded target DNA (solid line) hybrid captured by a hybrid-specific antibody (Y-shape) bound to a paramagnetic bead (shaded circle). 2: Isothermal, whole-genome, strand displacement amplification by φ-29 shows the DNA template (long black line) with single strands being polymerized (gray lines with arrowheads) at random priming sites (primers are short black lines) and displacing complementary amplicon strands. The arrowheads indicate the polymerization direction. 3: Hybrid Capture detection shows a double-deleted RNA probe (dotted line)-DNA amplicon (solid line) hybrid captured onto a detection oligonucleotide (short black line) of a LMX bead. Two detection oligonucleotide probes in separate genomic regions are labeled a and b. There is one LMX bead set for each HPV type, each with a distinct color and a distinct pair of oligonucleotide capture probes. The hybrid is decorated with hybrid capture antibodies (Y shapes) conjugated with phycoerythrin (PE), which acts as the reporter in the LMX assay (see text).
Table 1.
Capture Oligonucleotides Designed to be Specific for HPV Types
| Name | Sequence |
|---|---|
| HPV6-1 | 5′-TTTCCATGAAATTCTAGGCAGCACGCGCAGGCT-3′ |
| HPV6-2 | 5′-GGGTGTTGGGTAGGGTTAATGCCAGGTTCAAAAGA-3′ |
| HPV11-1 | 5′-AATATTTAGTGTGCCCAGCAAAAGGTCTTGTAGTTG-3′ |
| HPV11-2 | 5′-GCAGACGTCCGTCCTCGATATCCACTTTGCAAT-3′ |
| HPV16-1 | 5′-CAGCTCTGTGCATAACTGTGGTAACTTTCTGGG-3′ |
| HPV16-2 | 5′-GAAGTAGGTGAGGCTGCATGTGAAGTGGTAG-3′ |
| HPV18-1 | 5′-GAACGCGATGGTACAGGCACTGCAGGGTCC-3′ |
| HPV18-2 | 5′-ACACGCACACGCTTGGCAGGTTTAGAAGAC-3′ |
| HPV26-1 | 5′-CCTACACAGTACAAGTGGAGGCCATCACCCG-3′ |
| HPV26-2 | 5′-GTCTGACACATACTGTTGTAACCCATAGTTAAACACAGG-3′ |
| HPV31-1 | 5′-GAAGTAGTAGTTGCAGACGCCCCTAAAGGTTGC-3′ |
| HPV31-2 | 5′-GAGGTCTTCTCCAACATGCTATGCAACGTCCTG-3′ |
| HPV33-1 | 5′-GTTCTACACGGGTTTGCAGCACGATCAACAACG-3′ |
| HPV33-2 | 5′-CGCTGCTTGTGGTGGTCGGTTATCGTTGTCTG-3′ |
| HPV35-1 | 5′-CTCGCTTGGTGGGGTTGTAGGGGAGCTCGG-3′ |
| HPV35-2 | 5′-GACGTAGTGTCGCCTCACATTTACAACAGGAC-3′ |
| HPV39-1 | 5′-GTGAGCCTGTGTTATATGTAGTGCCCGAATCCC-3′ |
| HPV39-2 | 5′-GCTGTAGTTGTCGCAGAGTATCCCGTGAGG-3′ |
| HPV45-1 | 5′-CCTCCTGCGTCCACTACACCTAGCACTA-3′ |
| HPV45-2 | 5′-TGCGTGCGTGTATGTATGAATGTGCCTTG-3′ |
| HPV51-1 | 5′-GGCCAATGTAGTACCTTCAACCTTATTAACAACATCAGG-3′ |
| HPV51-2 | 5′-TTACGTTGTCGTGTACGTTGCCAGCAATTAGCG-3′ |
| HPV52-1 | 5′-CGAATTGTG TGAGGTGCTGGAAGAATCGGTGC-3′ |
| HPV52-2 | 5′-GATCGTTCACAACTTTTACCTGCACCGGATCC-3′ |
| HPV56-1 | 5′-CTGTCGGTATTGTCTGTGTCGCTGATGTGTG-3′ |
| HPV56-2 | 5′-CTAGGTTCTCTAGATGTTTGTCTCCAGCACCCC-3′ |
| HPV58-1 | 5′-GATACACACACATTTGCAGCCCGGTCCACACA-3′ |
| HPV58-2 | 5′-GGTGGCAAAGGACGTATGTGAGTGCAGAGGAC-3′ |
| HPV59-1 | 5′-GCGTTGCGGAGGGGTATGATAGTTGCTCAGAAG-3′ |
| HPV59-2 | 5′-GTCTAGGCGTGTAGGAGGAAACAAGATGGGG-3′ |
| HPV66-1 | 5′-GTAAGGAACACCACCTAACCTGACACACACTGCCCA-3′ |
| HPV66-2 | 5′-GCTGTCTCCCTGTCTTCCTGTGTATTGTTTATAAGTG-3′ |
| HPV68-1 | 5′-CTGAACACAGCAGTTCTCTATACCAATGGCGCTATTTC-3′ |
| HPV68-2 | 5′-TTGGTTGCCCCTGAGCAGTCGGACCCTATGGATA-3′ |
| HPV73-1 | 5′-GCCATGTACTTCACAAACTGTTAATACTGGTGATTGTCCC-3′ |
| HPV73-2 | 5′-CACGAAGTGTCAGTGCACAGTATGCCTTGC-3′ |
| HPV82-1 | 5′-GCGCCGCATTGCTGCACCTCGTTTATATAGCAGGGCATTTTC-3′ |
| HPV82-2 | 5′-CCTGGCGCATGTCATACACACCACATTACTC-3′ |
Two probes are designed for each HPV.
Analytical Sensitivity and Specificity
The assay reproducibly detected 100 copies of HPV plasmid DNA mixed with 5 μg of Herring Sperm DNA (Figure 2). The nonspecific DNA was added to mimic the upper end of human DNA typically present in a cervical specimen. The added DNA did not interfere with the limit-of-detection of the assay when using the sequence-specific hybrid capture sample preparation. The assay signal was robust for approximately 100 copies of HPV from a positive clinical specimen diluted into a HPV-negative pool (Figure 2). Copy number was determined using the HC2 assay by comparing the signal for the specimen with the signal for the positive calibrator (1 pg/ml). The assay detected 100 copies of HPV 16 targets that existed either as circular plasmids, linearized plasmids (SalI digestion of an HPV 16 plasmid), or integrated genomic DNA of CasKi cells (Figure 3). The assay signal was lower, however, for linear and integrated DNA than for circular DNA. The assay signal was not equal for each of the 19 HPV plasmid targets (Figure 4), for example, the lowest signal for HPV 6 was approximately sixfold lower than the highest signal for HPV 66. The background signal (no target) for the various HPV bead-types was between 15–100 MFI. The assay detected all 19 HPV types with no substantial cross-reactivity between HPV types (Figure 4), even for 1,000,000 copies of plasmid target. A signal of 200–250 MFI was chosen as a cut-off threshold for positive samples. The assay signal tended to decrease with extremely high DNA target input, as may be seen by comparing Figure 4, A and B. This high-end hook-effect may be due to an excess of complementary DNA strands competing with the detection probes. This is seen in many nucleic acid assays including PCR; however, for this assay, it was never severe enough to cause a false-negative result.
Figure 2.
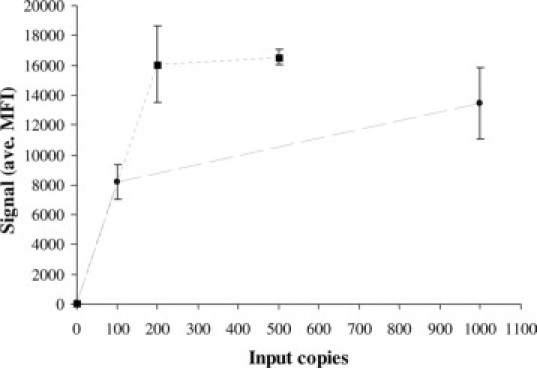
Plot of the average (n = 4) HC-WGA-LMX assay signal (median fluorescence intensity, MFI) for various HPV 16 inputs (clinical HPV DNA diluted in clinical-negative specimen pool, dotted line; HPV plasmids diluted in STM with 5 μg herring DNA, dashed line). Error bars show SD of signal. The no target (0 copies) control is less than 200 MFI.
Figure 3.
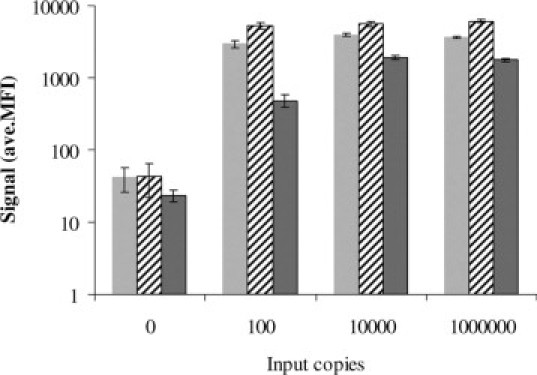
Plots of the HC-WGA-LMX assay signal (average MFI, n = 2) for various structures of HPV 16 DNA targets; HPV 16 plasmid DNA linearized with SalI (gray bars); circular HPV 16 plasmid DNA (cross-hatched bars); and integrated (genomic) HPV 16 DNA from CasKi cells (dark gray bars). Targets were diluted in STM. Error bars indicate SD.
Figure 4.
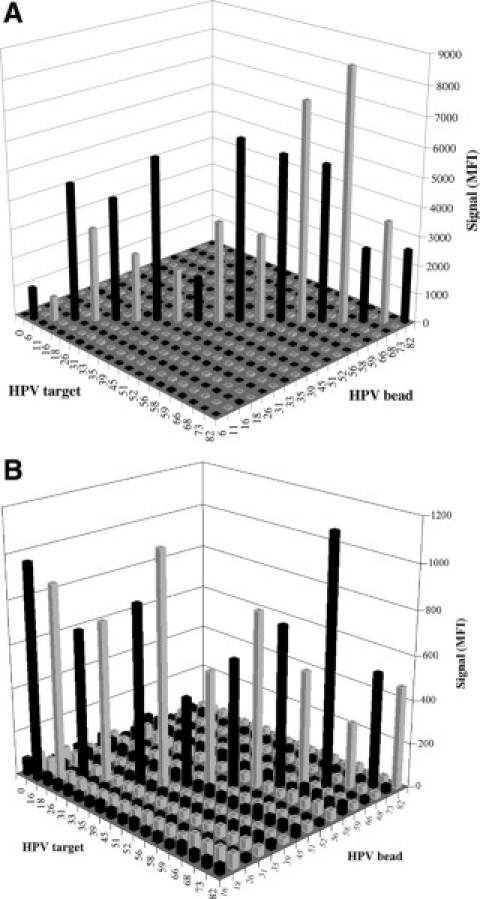
Assay signal-to-noise (S/N, average, n = 3) was unequal for each of the 19 HPV plasmid types (500 copies, A; 1,000,000 copies, B). Each HPV plasmid was assayed separately by the multiplex HC-WGA-LMX (19 types) assay. Background signal of controls with no targets was low.
The sequence-specific sample preparation, or HC enrichment, was absolutely required for adequate analytical sensitivity with clinical specimens in this assay. The percent capture of the specific DNA targets was approximately 50–70% (not shown), and the percent removal of nontarget DNA was not determined. However, when total double-stranded DNA was isolated by other methods, as opposed to HC enrichment, the sensitivity was 2-logs lower (not shown).
The HPV targets were amplified up to 107-fold by the 2-hour isothermal amplification step (not shown). The amplification was efficient enough so that only a fraction (1/3) of the amplicon volume was required for the detection step of the assay. The assay signal was reduced if more than 33% of the amplicon reaction was used in the assay detection step (not shown). This reduction may be due to competition between the LMX-probes and the multitude of amplified complementary DNA target strands.
A second type of competitive inhibition resulted when a small amount of one HPV plasmid type was co-amplified with a large amount of a second type of plasmid. The assay signal for 100 or 1000 copies of HPV plasmid dropped to near background levels when amplified with a 3–4 log excess of a second plasmid type. A similar reduction also occurred with the consensus PCR method (Figure 5).
Figure 5.
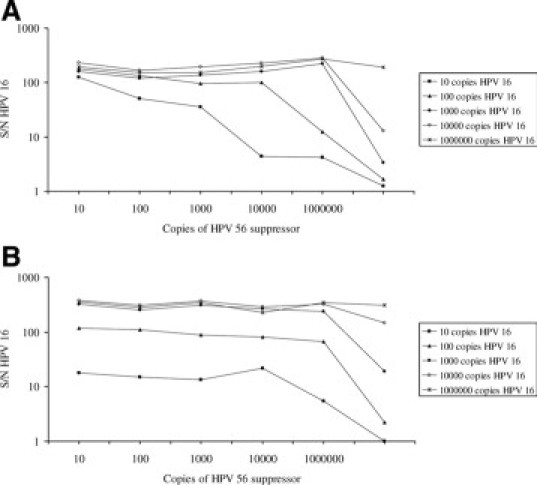
Suppression of the HC-WGA-LMX assay (A) and the gp-PCR assay (B) with variable amounts of HPV 16 and HPV 56. The traces represent the assay S/N (y axis; n = 3 replicates) for increasing amounts of HPV 16 plasmid copies (square, 10; triangle, 100; solid star, 1000; open star, 10000; x-shape, 1000,000) amplified with various HPV 56 copies (x axis).
Specimen Interference
The assay signal was not substantially reduced by interfering substances found in cervical specimens, such as blood and mucus. Aliquots of a specimen (approximately 500 copies) that contained a HPV 16 infection were diluted into HPV-negative specimens (n = 65) with various coloration and opacity due to blood and other specimen components. The assay signal for each sample was not equal, but all samples were detectable well above background. The raw signals (MFI) for the group of 65 samples were 1939 for minimum, 4995 for median, 4738 for mean, 5847 for maximum, and 815 for SD; the 500-copy positive control signal was 5595, and the negative background signal was 40. The multiple warm saline buffer washes reduced background signal and increased signal for some clinical specimens.
Comparison of the HC-WGA-LMX Assay with HC2 and Consensus PCR
The limit-of-detection for the HC-WGA-LMX assay is approximately 50-fold lower than the HC2 assay. This was determined by preparing serial dilutions of a HPV 16–positive cervical specimen in a HPV-negative specimen pool and then, testing the dilutions in both the new assay and in the HC2 assay (Figure 6).
Figure 6.
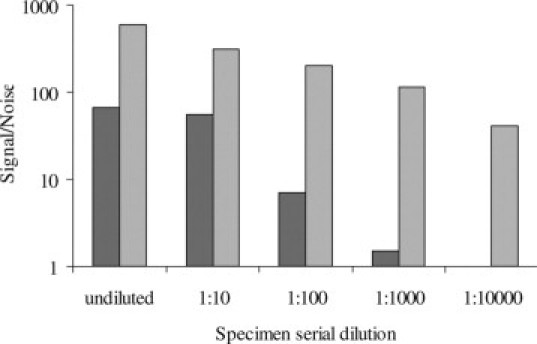
Analytical limit-of-detection of the HC-WGA-LMX assay is approximately 2 logs lower than for the HC2 screening assay. The signal for a serial dilution (n = 2 replicates) of HPV 16–positive signal is shown for two assays; dark bars, HC2 assay; light bars, HC-WGA-LMX assay.
Two specimen sets were selected to compare assay performance of the HC-WGA-LMX assay with the HC2 assay (n = 502) and with a consensus PCR assay (n = 131). The specimens were archived, and cervical specimens were collected in STM from normal populations. The 2 × 2 interrater reliability test was used for comparison (Table 2). No cytological or histological data are reported for these specimens. The HC-WGA-LMX assay was more likely to be positive than the HC2 or gp-PCR assay. There were no discrepancies between the HPV types detected by PCR and HC-WGA-LMX when both assays were positive. The 229 specimens that were HPV-positive consisted of 24.3% HPV 16, 3.8% HPV 18, 1.1% HPV 31, 8.1% HPV 33, 4.7% HPV 35, 2.1% HPV 39, 5.3% HPV 45, 3.2% HPV 51, 7.0% HPV 52, 8.7% HPV 56, 4.8% HPV 58, 13.2% HPV 59, 6.6% HPV 66 and 5.4% HPV 68. No representative HPV 73, HPV 82, HPV 6, or HPV 11 specimens were encountered. Some specimens (18%) were positive for multiple types (usually two, but for five types in one case).
Table 2.
Interrater Reliability Data in 2 × 2 Tables Comparing the Hybrid-Specific Typing Assay with Either the HC2 or the gp-PCR Assay
| HC2; n = 502 | Positive HC2 | Negative HC2 |
|---|---|---|
| Positive new assay | 64 | 31 |
| Negative new assay | 9 | 398 |
| Positive agreement | 0.88 | |
| Negative agreement | 0.93 | |
| Cohen's kappa | 0.72 | |
| gp-PCR; n = 131 | Positive PCR | Negative PCR |
| Positive new assay | 98 | 9 |
| Negative new assay | 3 | 21 |
| Positive agreement | 0.97 | |
| Negative agreement | 0.7 | |
| Cohen's kappa | 0.72 | |
Each comparison was done with a different set of specimens.
Discussion
The HC-WGA-LMX assay determined the genotypes of 19 types of HPV in samples and in clinical specimens and was rapid and easy to perform. The analytical sensitivity is higher than for HC2 and is approximately similar to PCR. The sensitivity may be further optimized to achieve a given clinical performance objective by adding more, or less, enzyme or extending the amplification time. For this work, the assay sensitivity enabled detection of all of the HC2-positive specimens plus additional ones that were verified by PCR. It is important to note that the clinical performance of this assay was not determined by this study. The assay amplified all of the various forms of HPV, including circular, linear, and integrated. Thus, the assay does not depend exclusively on rolling circle amplification.17 The HPV DNA in clinical STM specimens was intact and long enough to provide a sufficient template for φ-29 amplification. In some experiments (not shown), we found that DNA templates shorter than 500 bp were not detected well by these assay conditions. The difference in assay signal intensity for each HPV target at low copy number may be due to various hybridization efficiencies for each of the 19 targets with their respective capture probes. The HC-WGA-LMX assay signal reaches a plateau after a 3- to 4-log increase in target concentration from baseline. Thus, the assay, as described here, may not quantify HPV targets over a larger dynamic range.
Design of LMX capture probes is critical for assay specificity, and it also may impact the signal intensity over background for each type of target. Having two (or more) specific capture oligonucleotide sequences for each target bound to the same bead allows detection of HPV targets that are mutated or integrated. When combined with isothermal amplification of the entire target region, this approach may detect mutated target sequences better than methods that use sequence-specific primers and relatively short amplicon regions. Further design and optimization of the capture probes may allow addition of more HPV targets to the assay and may also resolve inequalities in signal:noise for each target type.
Multiple infections may occur in 20–35% of specimens. The abundance and distribution of HPVs in multiple infections are not well known, and widely different target loads in a multiple infection may be rare. However, a limitation of the assay is the reduction of signal that occurs for a plasmid target in low abundance when it is amplified with another target that is 2 or 3 logs higher in abundance. Thus, a false-negative result might occur if the lower abundance target is less than 1000 copies and the other target(s) are greater than 1,000,000 copies. The reduction of signal for the lesser of two targets is approximately the same for the assay using consensus-PCR. This suppression may be due to a limitation in some assay component, such as polymerase. Thus, further optimization with components and polymerase may resolve the problem.
Model experiments indicate that there are no substances in clinical STM specimens that cause substantial assay interference. This was dependent on the stringent heated-wash procedure of the HC sample preparation process. Some HC2-positive specimens were not detected if a less stringent wash was used (not shown). In these cases, the “false-negative” specimens were either dark brown due to blood or somewhat cloudy due possibly to foreign substances (eg, gels).
The HC-WGA-LMX assay detected HPV in a greater number of clinical specimens than the HC2 Test. This was expected due to the lower limit-of-detection of the HC-WGA-LMX assay. The discrepant HC2-positive but HC-WGA-LMX–negative specimens may result from a lower analytical specificity of HC2.18 The HC-WGA-LMX was comparable to the PCR genotyping assay. The clinical relevance and specificity of the HC-WGA-LMX assay is beyond the scope of this article. The assay, however, may be flexible enough to achieve a given clinical end point.
In summary, the enrichment of single-stranded targets by Hybrid Capture technology in solution was absolutely necessary for the described analytical performance of the assay. This sample preparation combined with whole-genome amplification is adaptable to high-throughput instrumentation. This assay is efficient and flexible by design and may be expanded to include hundreds of other targets including HPVs or other pathogens. These proposed assays may be useful in clinical diagnostics.
Acknowledgements
We thank Nina Brown, Kim Keating, Sameera Rangwala, Hiam Salim, Julie Giles, and others in R&D at QIAGEN for their dedicated efforts.
Footnotes
Supported by QIAGEN, Inc.
The authors submit that they all are employees of QIAGEN, Inc. except A.L. who is an advisor to QIAGEN.
References
- 1.Lorincz AT, Reid R, Jenson AB, Greenberg MD, Lancaster W, Kurman RJ. Human papillomavirus infection of the cervix: relative risk associations of 15 common anogenital types. Obstet Gynecol. 1992;79:328–337. doi: 10.1097/00006250-199203000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJF, Meijer CJLM, for the International Agency for Research on Cancer Multicenter Cervical Cancer Study Group Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 3.Castle PE, Rodriguez AC, Burk RD, Herrero R, Wacholder S, Alfaro M, Morales J, Guillen D, Sherman ME, Solomon D, Schiffman M. Short term persistence of human papillomavirus and risk of cervical precancer and cancer: population based cohort study. Br Med J. 2009;339:b2569. doi: 10.1136/bmj.b2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sankaranarayanan R, Nene BM, Shastri SS, Jayant K, Muwonge R, Budukh AM, Hingmire S, Malvi SG, Thorat R, Kothari A, Chinoy R, Kelkar R, Kane S, Desai S, Keskar VR, Rajeshwarkar R, Panse N, Dinshaw KA. HPV screening for cervical cancer in rural India. N Engl J Med. 2009;360:1385–1394. doi: 10.1056/NEJMoa0808516. [DOI] [PubMed] [Google Scholar]
- 5.Xi LF, Hughes JP, Edelstein ZR, Kiviat NB, Koutsky LA, Mao C, Ho J, Schiffman M. Human papillomavirus (HPV) type 16 and type 18 dna loads at baseline and persistence of type-specific infection during a 2-year follow-up. J Infect Dis. 2009;200:1789–1797. doi: 10.1086/647993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castle PE, Porras C, Quint WG, Rodriguez AC, Schiffman M, Gravitt P, Gonzalez P, Katki HA, Silva S, Freer E, van Doorn LJ, Jimenez S, Herrero R, Hildesheim A. A comparison of two PCR-based human papillomavirus genotyping methods. J Clin Microbiol. 2008;46:3437–3445. doi: 10.1128/JCM.00620-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klug SJ, Molijn A, Schopp B, Holz B, Iftner A, Quint W, Snijders PJF, Petry K-U, Kjaer SK, Munk C, Iftner T. Comparison of the performance of different HPV genotyping methods for detecting genital HPV types. J Med Virol. 2008;80:1264–1274. doi: 10.1002/jmv.21191. [DOI] [PubMed] [Google Scholar]
- 8.Ziegert C, Wentzensen N, Vinokurova S, Kisseljov F, Einenkel J, Hoeckel M, von Knebel Doeberitz M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene. 2003;22:3977–3984. doi: 10.1038/sj.onc.1206629. [DOI] [PubMed] [Google Scholar]
- 9.Yamada T, Wheeler CM, Halpern AL, Stewart AC, Hildesheim A, Jenison SA. Human papillomavirus type 16 variant lineages in United States populations characterized by nucleotide sequence analysis of the E6. L2, and L1 coding segments. J Virol. 1995;69:7743–7753. doi: 10.1128/jvi.69.12.7743-7753.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorincz AT. Hybrid Capture™ method for detection of human papillomavirus DNA in clinical specimens: a tool for clinical management of equivocal Pap smears and for population screening. J Obstet Gynaecol Res. 1996;22:629–636. doi: 10.1111/j.1447-0756.1996.tb01081.x. [DOI] [PubMed] [Google Scholar]
- 11.Lazar JG, Cullen AP, Mielzynska I, Meijide MG, Lorincz AT. Hybrid Capture®: a sensitive signal amplification-based chemiluminescent test for the detection and quantitation of human viral and bacterial pathogens. J Clin Ligand Assay. 1999;22:139–151. [Google Scholar]
- 12.Nazarenko I, Kobayashi L, Giles J, Fishman C, Chen G, Lorincz A. A novel method of HPV genotyping using Hybrid Capture sample preparation method combined with GP5+/6+ PCR and multiplex detection on Luminex XMAP. J Virol Methods. 2008;154:76–81. doi: 10.1016/j.jviromet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Meijer WJJ, Horcajadas JA, Salas M. φ29 family of phages. Microbiol Mol Biol Rev. 2001;65:261–287. doi: 10.1128/MMBR.65.2.261-287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lage JM, Leamon JH, Pojovic T, Hamann S, Lacey M, Dillon D, Segraves R, Vossbrinck B, Gonzalez A, Pinkel D, Albertson DG, Costa J, Lizardi PM. Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res. 2003;13:294–307. doi: 10.1101/gr.377203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunbar SA. Applications of Luminex® xMAP™ technology for rapid, high-throughput multiplexed nucleic acid detection. Clin Chim Acta. 2006;363:71–82. doi: 10.1016/j.cccn.2005.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van den Brule AJ, Pol R, Fransen-Daalmeijer N, Schouls LM, Meijer CJ, Snijders PJ. GP5+/6+ PCR followed by reverse line blot analysis enables rapid and high-throughput identification of human papillomavirus genotypes. J Clin Microbiol. 2002;40:779–787. doi: 10.1128/JCM.40.3.779-787.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat Genet. 1998;19:225–232. doi: 10.1038/898. [DOI] [PubMed] [Google Scholar]
- 18.Castle PE, Solomon D, Wheeler CM, Gravitt PE, Wacholder S, Schiffman M. Human papillomavirus genotype specificity of Hybrid Capture 2. J Clin Microbiol. 2008;46:2595–2604. doi: 10.1128/JCM.00824-08. [DOI] [PMC free article] [PubMed] [Google Scholar]