

Abstract
Mutations of both the IDH1 and IDH2 (isocitratedehydrogenase enzyme 1 and 2) genes have recently been described in cases of human glioma. Since IDH1 mutations have been associated with better clinical outcome, they are suitable predictive markers for adult glioma patients. We have developed a pyrosequencing assay that allows both the sensitive and rapid detection of mutant IDH1 alleles in DNA extracted from formalin-fixed, paraffin-embedded tissues. PCR products that span exon 4 of IDH1 were used as a template for pyrosequencing. For validation, PCR products were additionally cloned and sequenced conventionally by Sanger sequencing. Sensitivity was measured by titration of wild-type and mutant sequences. PCR kinetic experiments were performed to investigate the influences of PCR cycle number on the accuracy of the assay. We found that a minimum of 5% of mutant IDH1 alleles can easily be detected with the pyrosequencing approach. So far, there are few data regarding IDH1 mutation status in high-grade gliomas of childhood. Therefore, we applied this assay to 47 pediatric high-grade glioma samples (age range 6 weeks to 23 years). Mutations were found in 5/14 astrocytoma III and in 6/33 glioblastomas. In conclusion, we have developed a pyrosequencing-based assay for the detection of mutations at the hotspot regions of IDH1 and provide proof for its applicability as a molecular diagnostic assay for clinical samples.
Gliomas are the most common type of brain tumors and range from benign low grade gliomas to aggressive glioblastomas. Glioblastomas are the most common and most malignant tumors of the brain. They may manifest at any age, but preferentially affect adults, with a peak incidence between 45 and 70 years.1,2 Glioblastomas are highly invasive and aggressively growing tumors that respond poorly to radiation therapy and most forms of chemotherapy. The prognosis is correspondingly poor, with most patients dying within one year after diagnosis. Adults glioblastomas may develop from diffuse astrocytomas World Health Organization grade II or anaplastic astrocytomas,3 but more frequently, they manifest after a short clinical history de novo without evidence of a less malignant precursor lesion (primary glioblastoma). Glioblastomas carry complex genetic and epigenetic alterations. Primary glioblastomas of adults frequently show loss of heterozygosity on chromosome 10q (70% of cases), EGFR amplification (36%), p16(INK4a) deletion (31%), and PTEN mutations (25%).3 Secondary glioblastomas as well as their lower grade precursor lesions exhibit frequent mutations of the TP53 gene and epigenetic inactivation of DUSP4.3,4 Pediatric diffuse high-grade gliomas (HGG) differ from GBM of adults. While they are believed to occur de novo, only very few pediatric high grade gliomas display EGFR amplifications5,6,7,8 although the receptor protein is detectable in many cases.8 Inactivation of the p53/MDM2/p14 pathway, in contrast, is also frequently present in pediatric tumors, similar to adult GBM.6 In particular, the TP53 mutational rate is in a similar range.9 The RB pathway seems to be affected less frequently in pediatric HGG.10 Recently somatic mutations at codon 132 of IDH1 gene have been reported in a screening approach of 20,661 protein coding genes in glioblastomas.11,12 Interestingly, all mutations were located at amino acid residue 132 (position 395) an evolutionarily conserved position located within the isocitrate binding site13 with the substitution of Arg -> His (G->A).11 In rare cases mutations of IDH2 have also been described in the homologous region in gliomas lacking IDH1 mutations.1 Isoforms of the enzyme isocitrate dehydrogenase- IDH1 and IDH2 catalyze the oxidative decarboxylation of isocitrate into α-ketoglutarate using either NAD or NADP as cosubstrates.14,15 Isocitrate dehydrogenase enzyme isoform 1 is located in peroxisomes, whereas IDH2 is present in mitochondria.16
IDH1 is mainly involved in metabolic processes and its role in cancer biology is largely unknown. Enzymatic studies with substitution of arginine at residue 132 of IDH1 with a different amino acid (glutamate) have reported that this mutation renders the enzyme catalytically inactive, suggesting a critical role for this residue.17 More recent data, however suggest that the mutant form of the enzyme leads to the production of an alternative metabolite 2-hydroxyglutarate.
Many of the glioblastomas carrying IDH1 mutations were secondary glioblastomas and contained TP53 mutations.1,3,11 Moreover these mutations were associated with increased overall survival. Therefore, IDH1 mutation status might be a valuable prognostic marker for GBM patients. The assays published so far comprise the use of PCR and direct Sanger sequencing of PCR products. In a clinical environment, it is mandatory to optimize the approach for best sensitivity and accuracy, especially if a clinical decision or prognostic evaluation is desired. Heterogeneity of tumor tissue is a critical aspect for molecular diagnostic approaches, since mutations may only be present in a subset of tumor cells, and the wild type allele is still present in those tumor cells carrying IDH1 mutation. In addition, gliomas often contain significant amounts of other cells from different types. Therefore, a sensitive and quantitative method for the detection of mutant IDH alleles in DNA extracted from fresh frozen as well as formalin-fixed, paraffin-embedded tumor tissues is needed. So far there are few data regarding IDH1 mutation status in high grade gliomas of childhood.1 Therefore, we applied this assay on 47 pediatric HGG samples to detect mutations in IDH1.
Materials and Methods
Tumor Samples
We investigated 47 high-grade glioma tumor patients (22 female and 25 male; mean age at surgery: 10.35 years; range: 4 weeks to 23 years). All tumors were classified according to the World Health Organization (World Health Organization) classification of tumors of the central nervous system.18 The series included 14 pediatric anaplastic astrocytoma III (AAIII) and 33 glioblastomas (World Health Organization grade IV; GBM). Histological assessment of tissue fragments chosen for this study confirmed that all specimens consisted of at least 80% tumor cells.
Collection of Samples and DNA Isolation
DNA was extracted from archival paraffin-embedded tissue using a commercial kit (Qiagen, Hilden, Germany) according to the protocol of the manufacturer. DNA was extracted from a total of 47 paraffin-embedded tissues formerly fixed in 4% formalin derived from the surgical specimens. Ten-micron slices of paraffin-embedded tissues were collected and the sections were dewaxed by xylene wash (3×, 10 minutes at 25°C each) followed by three 100% ethanol washes (10 minutes at 25°C each). The pellets were dried by applying vacuum. Samples were digested with proteinase K (20 mg/ml) at 56°C overnight. DNA was extracted with the QIAmp DNA mini kit (Qiagen).
PCR Amplification
Exon 4 of IDH1 containing the R132 coding region was amplified using three different set of primers, set 1: IDH1-fwd-A-5′-CACCATACGAAATATTCTGG-3′ and IDH1-rev-A-5′-biotin-CAACATGACTTACTTGATCC-3′, set 2: IDH1-fwd-B-5′-GGCTTGTGAGTGGATGGGTAAAA-3′ and IDH1-rev-B-5′-biotin-TTGCCAACATGACTTACTTGATCC-3′, set 3: IDH1-fwd-C-5′-TGGATGGGTAAAACCTATCATCA-3′ and IDH1-rev-C-5′-biotin-TTGCCAACATGACTTACTTGATC-3′ which amplify 135 bp, 76 bp and 66 bp fragments respectively. Amplification of the R132 coding region was carried out using 50 ng genomic DNA as template. The PCR mixture contained 50 mmol/L KCl, 20 mmol/L Tris/HCl (pH 8.4), 200 μmol/L of each deoxyribonucleotide triphosphate, 25 pmol of each primer, 2 mmol/L MgCl2 and 2.5 U of Platinum TaqDNA polymerase (Invitrogen, Carlsbad, California, US Karlsruhe, Germany) in a final volume of 50 μl. PCR was performed using a DNA thermal cycler (Biometra, Göttingen, Germany). The template was denatured initially for 10 minutes at 95°C followed by 40 amplification cycles containing: initial denaturation at 95°C for 10 minutes, followed by 95°C for 30 seconds, annealing for 30 seconds at 54°C and extension at 72°C for 30 seconds. Final extension was carried at 72°C for 10 minutes. PCR products were analyzed by electrophoresis on 2% agarose gels.
Cloning of PCR Products and Cycle Sequencing
PCR products of exon 4 of IDH1 containing R132 coding region were cloned into the TOPO TA cloning/pCR2.1 vector (Invitrogen, Karlsruhe, Germany). Inserts of individual bacterial clones were amplified using vector-specific primers. PCR products were treated with ExoSAP-IT (USB, Staufen, Germany) following manufacturer's instruction and sequenced using the BigDye Prism DNA cycle sequencing kit (Applied Biosystems, 3130 Genetic Analyzer). Ten individual clones were selected for each sample. For the titration and cycle kinetic assays described below bacterial clones carrying wild type and mutant IDH1 alleles were expanded and plasmid DNA containing the respective sequence was isolated by midiprep.
Development of IDH1 (R132) Pyrosequencing Assays
Assay Design
IDH1 PCR amplification primers flanking the R132 mutation hotspot within exon 4 of IDH1 were amplified as mentioned above. The reverse primer contained biotin at the 5′ position. For the product amplified using primer set 1, pyrosequencing primer IDH1-Py1-5′-GTGAGTGGATGGATGGGTAAAACC-3′ was used with the nucleotide dispensation order NGT->-AGT/TGT/GGT (first base) CNT-> CAT/CTT/CCT (second base) (Figure 1). IDH1-Py2-5′-TGATCCCCATAAGCA-3′ reads complementary strand and was used for the products amplified from set 2 and 3, with the nucleotide dispensation order NCA-> TCA/ACA/CCA (first base) GNA-> GTA/GAA/GGA (second base) (see supplemental Figures S1 and S2 at http://jmd.amjpathol.org). All three primer sets performed equally well on DNA extracted from formalin-fixed, paraffin-embedded tissues.
Figure 1.

Schematic representation and primer localization. Map of the pyrosequencing assay for detection of codon transversion at positions 394 and 395 within exon 4, leading to the IDH1 R132 mutation. Primer-binding sites are depicted in lower case and the sequence to be analyzed in upper case. The mutation site assayed is highlighted by a box, below are the codons with the expected changes at the first and second positions, respectively. Arrows indicate the annealing positions of primer sequences from 5′ to 3′.
Pyrosequencing Reaction
Single-stranded DNA templates were immobilized on streptavidin-coated Sepharose high-performance beads (GE Health care, Uppsala, Sweden) using the PSQ Vacuum Prep Tool and Vacuum Prep Worktable (Biotage, Uppsala, Sweden), according to the manufacturer's instructions, then incubated at 80°C for 2 minutes and allowed to anneal to 0.4 mmol/L sequencing primer at room temperature. Pyrosequencing was performed using PyroGold Reagents (Biotage) on the Pyromark Q24 instrument (Biotage), according to the manufacturer's instructions. Controls in which the sequencing primer or template were omitted were used to detect background signal. Pyrogram outputs were analyzed by the PyroMark Q24 software (Biotage) using the allele quantification (AQ) software to determine the percentage of mutant versus wild-type alleles according to percentage relative peak height.
For the pyrosequencing primer-1, the assay was designed to begin sequence analysis from 12 bp upstream (381) of the hotspot containing R132 mutation (394 and 395) and for pyrosequencing primer 2, 2 bp downstream (398) of the hotspot. A sequential nucleotide dispensation protocol was used that reflects the expected order of nucleotide incorporation and the potential base change within the first or second positions of codon 132 (IDH1). Peak heights are proportional to the number of nucleotides that are incorporated with each dispensation.
Titration of Mutant and Wild-Type IDH1 R132 Alleles
Purified plasmid DNA cloned with R132H mutant coding sequence (CAT), the most frequent mutation in gliomas and wild type (CGT) were diluted to generate mixtures containing 100%, 75%, 50%, 25%, 10%, 5%, and 0% of either wild type or mutant DNA in the final concentration. Fifty ng of each of the above mixtures was PCR amplified using one biotinylated primer and analyzed on 2% agarose gels. The resulting products were subjected to pyrosequencing and actual percentage of ‘A/T’ (Pyroprimer-1/Pyroprimer-2) at the second position of the hot spot was quantified by applying the allele quantification method (AQ).
PCR Cycle Kinetics
PCR amplification of a DNA mixture comprising wild type and mutant alleles in the ratio 1:1 (20 ng each) was carried out in a 50 μl reaction volume with varying number of amplification cycles 20, 30, 40, 50, and 60 cycles. The PCR product was analyzed on 2% agarose gels. The pyrosequencing reaction was performed by using identical conditions for all of the products.
Results
Comparison of Conventional Sequencing and Pyrosequencing Results
To detect R132 mutations we performed both pyrosequencing and conventional cycle sequencing of cloned PCR products for IDH1 on 47 genomic DNA samples extracted from paraffin-fixed tumor specimens of pediatric high-grade gliomas.
Figure 2 shows representative pyrograms from direct pyrosequencing reactions of PCR products and chromatograms from cloned PCR products analyzed by conventional cycle sequencing. Out of the 47 DNA samples analyzed, we identified 11 cases carrying the point mutation (G –> A) at the hotspot (R132) at the second position of the codon (CGT) thereby causing the amino acid sequence alteration R132H. Mutations were found in 5/14 AAIII (age range 5 to 16 years) and in 6/33 GBM (age range 6 weeks to 17 years). No correlation with a particular age group could be detected.
Figure 2.
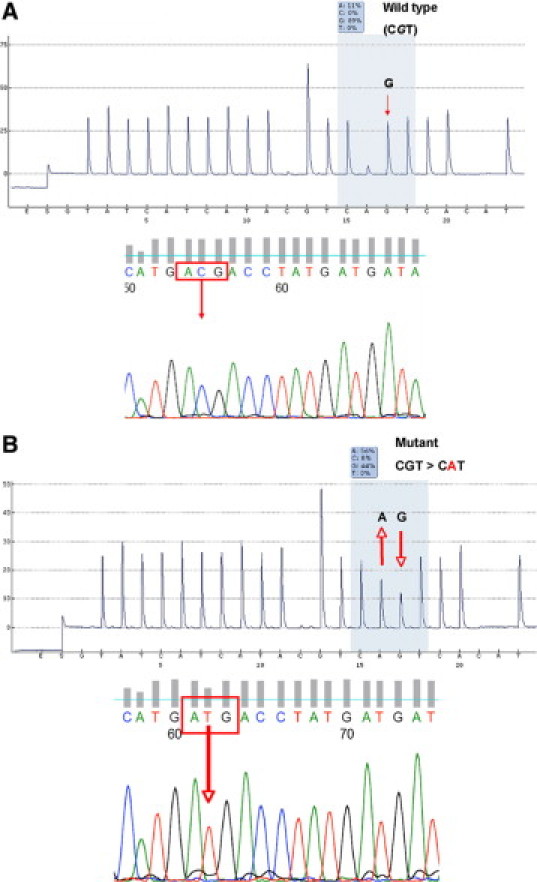
Pyrogram and conventional sequencing traces. Detection of IDH1 R132H mutations in DNA samples isolated from HGG specimens. (A, top) Pyrogram of a sample that contained no mutation at the second position of the hotspot codon 395 (CGT) and the chromatogram of the same fragment obtained by cycle sequencing of single clone of a cloned PCR product (A, bottom), confirming the result of the pyrosequencing assay. Of 10 clones sequenced, no mutants were found. Below (B, top), pyrosequencing result of a DNA sample that exhibits a mutation at position (395), resulting in the R132H mutation and a chromatogram obtained by cycle sequencing of a single clone of a cloned PCR product (B, bottom). Three clones out of 10 were found to be mutated. Supplemental Figure S2 (available at http://jmd.amjpathol.org) shows pyrograms of the same tumor samples generated using an alternative primer set, using a reverse pyrosequencing primer and leading to virtually identical results. Codon 132 is highlighted as boxed text. Arrows indicate the wildtype (G) and the mutant (A) base.
Both technologies yielded identical results. The pyrosequencing assay reliably detects all other possible mutations at position 132 (determined by investigation of adult secondary GBM as positive controls, data not shown), even though no other variation of position 132 have been found in HGG (See supplemental Figure S3 at http://jmd.amjpathol.org).
Samples were analyzed by conventional sequencing of cloned PCR products (Figure 2, A and B, bottom chromatograms) and pyrosequencing (Figure 2, A and B, top pyrograms) in triplicate independent experiments. The relative abundance of adenine at the second position was significantly increased (25 to 58%) in mutated samples (Figure 2B) when compared to the samples (2 to 20%) which did not have R132 mutation (Figure 2A). The results are listed in Table 1 and in supplemental Table S1 at http://jmd.amjpathol.org. Statistical analysis showed that the increased ratio of adenine in the mutated samples was statistically significant (P ≤ 0.001).
Table 1.
Allele Quantification Values at the 132 Position of IDH1
| Mutant IDH1 R132H |
Wild-type IDH1 R132 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| (CGT > CAT) |
(CGT) |
||||||||
| Sample ID | Diagnosis | Sex | Age (years) | % A | Sample ID | Diagnosis | Sex | Age (years) | % A |
| 550 | GBM | M | 9 | 23.33 ± 6.69 (n = 3) | 546 | GBM | F | 10 | 7.66 ± 1.02 (n = 3) |
| 608 | AA III | F | 16 | 47.00 ± 4.00 (n = 2) | 548 | GBM | F | 14 | 12.33 ± 3.84 (n = 3) |
| 640 | AA III | M | 5 | 55.00 + 2.57 (n = 3) | 549 | GBM | F | 7 | 8.33 + 0.66 (n = 3) |
| 730 | AA III | M | 13 | 50.0 ± 34.39 (n = 3) | 564 | GBM | F | 17 | 11.33 ± 1.33 (n = 3) |
| 677 | GBM | M | 17 | 34.00 + 10.40 (n = 3) | 596 | AA III | M | 13 | 9.66 ± 3.48 (n = 3) |
| 726 | AA III | M | 7 | 29.33 ± 1.20 (n = 3) | 646 | GBM | F | 14 | 10.00 ± 3.60 (n = 3) |
| 719 | GBM | M | 4 | 8.66 ± 0.88 (n = 3) | |||||
| 738 | AA III | M | 23 | 9.66 + 0.88 (n = 3) | |||||
| 751 | AA III | M | 7 | 9.66 ± 2.18 (n = 3) | |||||
| 698 | GBM | F | 10 | 15.33 ± 0.66 (n = 3) | |||||
IDH1 gene analysis of six pediatric high-grade glioma samples at position 395, resulting in the R132H mutation, along with 10 tumor samples exclusively carrying wild-type alleles. The numerical values represent the percentage of the mutated nucleotide determined by the allele quantification method. Values between the groups were statistically significant at p < 0.001 when compared by one-way ANOVA. M, male; F, female. The wild type and the mutant codon 132 of IDH1 appears in parentheses with the respective wild type and mutant base in bold (left). On the right, the wild type codon 132 is in bold.
Titration of Mutant IDH1 DNA Against the Wild-Type Sequence
To determine the analytic sensitivity of the assay, mixing studies of wild-type and mutant IDH1 alleles were performed. PCR products were generated using set-3 primers from mixtures of plasmid DNA containing IDH1 mutant allele and wild type in different ratios (100:0, 75:25, 50:50, 25:75, 10:90, 5:95, 0:100) and used as templates for sequencing. Pyrosequencing was performed using pyroprimer-2.
We observed that the pyrosequencing detection method was sensitive to the decreasing concentrations of the mutated allele in the mixture (Figure 3, A–G). The presence of mutant alleles was represented by an adenine peak at the second nucleotide position of the pyrogram. The abundance of mutant alleles (T) relative to wild type alleles (C) was given as a relative peak height. A strong linearity of AQ values with the expected values according to the dilution series ranging from 99% “T- signal” in a dilution that contained 100% mutant allele to 96% “C- signal” in a dilution having 100% wild type alleles. The titration results from the triplicate experiments clearly proved the sensitivity of the pyrosequencing assay to identify R132 mutations in the IDH1 gene even if only 5% of the mutant alleles are present in the DNA sample (Figure 3H).
Figure 3.
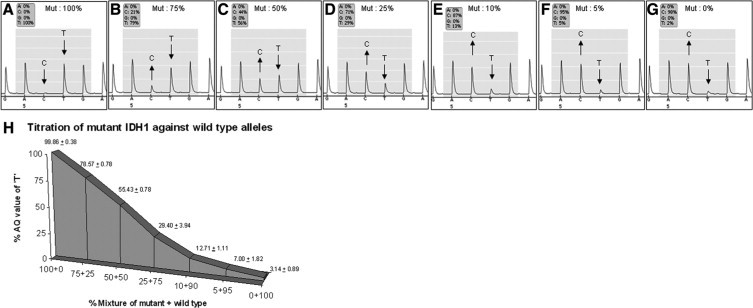
Titration of mutant and wild type IDH1 alleles. Results of the titration assay investigating the sensitivity of pyrosequencing analysis to detect IDH1 R132H mutations. Plasmids containing mutant and wild-type alleles were mixed to generate templates of 100%, 75%, 50%, 25%, and 0% mutant alleles and 0%, 25%, 50%, 75%, and 100% wild-type alleles. A–G: Pyrograms showing the percentage of adenine and guanine at the second position of codon 132 at different concentrations of mutant and wild-type alleles. H: Graphical representation of the AQ values of triplicate titration experiments. Ratio of the mutant allele to wild type is indicated on the x axis, and the percentage of adenine values is given on the y axis. Arrows indicate the wildtype (C) and the mutant (T) base.
PCR Cycle Kinetics
To monitor the influence of number of amplification cycles on the identification of mutant or wild type alleles, which might give rise to artifacts due to nonlinear amplification, a mixture of 1:1, plasmids containing mutant and wild type allele was used as template for PCR amplification with primer set 1. Products were amplified with different numbers of cycles (20, 30, 40, 50, and 60) from the above template as well as from wild type template. The products were subjected to pyrosequencing analysis using pyrosequencing primer-1 and AQ percentages were determined. The AQ values for mutant alleles of three sets of experiments showed no significant change during different cycle numbers (20 cycles, 43.66 ± 1.15; 30 cycles, 43.66 ± 0.57; 40 cycles, 45.50 ± 7.81; 50 cycles, 48.30 ± 4.9; 60 cycles, 44.66 ± 2.08). The results depicted in Figure 4, A–E clearly indicate that the number of amplification cycles did not alter the quantitative evaluation of mutated and wild type IDH1 alleles.
Figure 4.

PCR cycle kinetics of the IDH1 pyrosequencing assay. Plasmids containing mutant (CAT) and wild-type (CGT) IDH1 alleles were used in a 1:1 ratio. The R132 hotspot region was amplified by PCR with different cycle numbers (20, 30, 40, 50, and 60). Pyrosequencing of the PCR products and subsequent allele quantification showed that the relative abundance of mutant allele (A, C) and wild type (B, D) were unaffected by increasing PCR cycle numbers. E: Graphical representation of the AQ values for IDH1 mutant and wild-type alleles at different cycle numbers. Arrows indicate the wildtype (G) and the mutant (A) base.
Discussion
IDH mutations represent a frequent event in diffuse gliomas World Health Organization grade II and World Health Organization grade III in adults and can serve as a diagnostic tool.12 A fraction of adult GBM (World Health Organization grade IV) also shows this alteration and these patients seem to show a better clinical course. All of the mutations of IDH1 and IDH2 occur at the same hotspot site so that a robust and reproducible assay to monitor the mutational status of this residue would represent a valuable tool. Here we have established a pyrosequencing assay and applied it on a panel of pediatric high grade gliomas since only few data exist on this entity.
Pediatric high-grade gliomas represent approximately 10% of all pediatric brain tumors. Similar to adult high-grade gliomas, they behave very aggressively. However, the overall prognosis seems slightly better than in adults with a higher percentage of long-term survivors, but nevertheless, survival remains poor in general despite of a variety of therapies that include chemotherapy and radiotherapy. Pediatric gliomas do not fit in the genetic models of adult high-grade gliomas. While alterations of the p53/MDM2/p14 pathway were detectable in a similar frequency compared to adults, EGFR amplifications are uncommon in contrast to adult primary GBM although it is believed that pediatric HGG also arise de novo.19
In studies on high-grade gliomas of adults IDH1 has gained major attention as a novel marker for diffuse gliomas World Health Organization grade II and III as well as secondary GBM (sGBM) World Health Organization grade IV. Here IDH1 mutations have been associated with a better clinical outcome.1,20,21
To date, six types of the IDH1 codon 132 mutations (R132H, R132C, R132S, R132G, R132L, and R132V) have been detected in human cancers.11,22 In glial tumors, R132H is the most common IDH1 mutation followed by R132C.14,15 In pilocytic astrocytomas (World Health Organization grade I) no genetic alterations were identified in IDH1 and it has been suggested that IDH1 together with structural alterations of the BRAF gene may be used to discriminate pilocytic astrocytomas and glioblastomas of adults.11,23
To date the classical way to detect mutations of the hot spot region is the direct sequencing of a PCR product of the respective sequence.24 Due to heterogeneity of glioma samples, as well as wild-type alleles present in the tumor cells, however this procedure bears the risk to overlook mutated alleles in the presence of wild-type sequences and, therefore, might be less suitable as a molecular diagnostic tool.25,26,27,28 Therefore we optimized a pyrosequencing based detection system29,30 for the R132 hotspot mutation of IDH1 and applied this approach to DNA extracted from archival tumor specimens of pediatric patients with high grade gliomas. Pyrosequencing has the advantage to accurately measure the amount of wild-type and mutant bases on a given position in a rapid and sensitive reaction without the need for cloning and sequencing of individual clones.
To calibrate the pyrosequencing assays for IDH1 mutation, we serially diluted a plasmid carrying a mutant IDH1 (R132H) and a plasmid containing the wild type IDH1 exon 4. Mutation analysis by pyrosequencing to detect ‘A/G’ or ‘T/C’ signals revealed a consistent figure with respect to dilutions. This titration assay indicated that a minimum of 5% of mutated DNA in a background of normal DNA could be easily detected and therefore allows confident screening for mutations. The reproducibility was analyzed in three different sets of experiments with identical experimental settings and confirmed the accuracy of the method with the reading of 99.86 ± 0.38% ‘A’ in a mixture containing 100% mutant alleles and 7.00 ± 1.82% ‘A’ in the mixture containing 5% of the mutant allele. Results of the PCR cycle kinetics experiment carried out to check the effect of increasing number of amplification cycles (20 to 60 cycles with the regular amplification being carried out with 40 cycles) clearly rules out the chance of artifacts due to preferential amplification of mutant or wild-type alleles.
In this study 47 pediatric high-grade glioma samples comprising of 14 anaplastic astrocytoma grade III (AA III) and 33 glioblastoma (GBM) samples were analyzed to detect the mutations in hot spot codons of IDH1. In contrast to a previous study on 15 pediatric GBM samples,1 we observed mutations in IDH1 in 6 out of 33 (18%) pediatric GBM. In addition our collection of pediatric high grade gliomas also contained 14 anaplastic astrocytomas of which 5 were found to be mutant (35%). The correlation with clinical data, available for 8 samples with mutation of IDH1 and 27 cases with wt IDH1, however, did not show a significant correlation of IDH1 mutation with better clinical outcome. Interestingly all mutations detected in our study were of the CGT-CAT type causing R132H, which was reported to be the most frequent variant in adult gliomas as well.1,12,21,22,23,31,32 Technically, our approach would also pick up any other sequence variation at position one and two of IDH1 codon 132 as described in the supplemental Figures S1, S2, and S3 (http://jmd.amjpathol.org). The age of the patients carrying mutations in their tumors ranged from 6 weeks to 17 years so there is no obvious association to specific age groups.
To prove the principal applicability of the pyrosequencing assay to detect all possible mutation variation at R132, we have also applied the assay to reference adult glioma samples with confirmed IDH1 mutational status. We have established the pyrosequencing assay to target the particular site of mutation with different sets of primers for amplification and pyrosequencing (see Figure 2 and supplemental Figures S1 and S2 at http://jmd.amjpathol.org) yielding identical results. Our assay can be used for the sensitive and quantitative detection of all mutant and wild type IDH1 R132 alleles. This approach has several advantages over conventional sequencing, especially if DNA is derived from archival material and significantly degraded because small fragments can be analyzed in short span of time, whereas conventional sequencing does not allow analysis of the first few bases of the amplified fragment due to noise and therefore relies on larger PCR products. In addition sensitive and quantitative data can be obtained without cloning and sequencing of individual clones.
In conclusion, we have designed a novel sensitive pyrosequencing assay for the rapid and accurate identification of R132 mutations of the IDH1 genes in clinical samples. This approach was applied to a cohort of 47 pediatric high grade gliomas and uncovered IDH1 R132H mutations in 11/47 cases.
Footnotes
Supported by National Genome Research Network NGFNplus, Brain Tumor Net (grant 01GS08187, SP8 and SP9b), the Kinderkrebsstiftung (grant DKS 2006.03), German Ministry for Education and Research BMBF, Competence Network Pediatric Oncology and Hematology, Project Embryonal Tumors (grant 01GI0418), and BONFOR program of the Medical Faculty of the University of Bonn.
None of the authors disclosed any relevant financial relationships.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Web Extra Material
{kind=link}
{kind=link}
{kind=link}
References
- 1.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–225. doi: 10.1093/jnen/61.3.215. discussion 226–229. [DOI] [PubMed] [Google Scholar]
- 3.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waha A, Felsberg J, Hartmann W, von dem Knesebeck A, Mikeska T, Joos S, Wolter M, Koch A, Yan PS, Endl E, Wiestler OD, Reifenberger G, Pietsch T, Waha A. Epigenetic downregulation of mitogen-activated protein kinase phosphatase MKP-2 relieves its growth suppressive activity in glioma cells. Cancer Res. 2010;70(4):1689–1699. doi: 10.1158/0008-5472.CAN-09-3218. [DOI] [PubMed] [Google Scholar]
- 5.Raffel C, Frederick L, O'Fallon JR, Atherton-Skaff P, Perry A, Jenkins RB, James CD. Analysis of oncogene and tumor suppressor gene alterations in pediatric malignant astrocytomas reveals reduced survival for patients with PTEN mutations. Clin Cancer Res. 1999;5:4085–4090. [PubMed] [Google Scholar]
- 6.Sung T, Miller DC, Hayes RL, Alonso M, Yee H, Newcomb EW. Preferential inactivation of the p53 tumor suppressor pathway and lack of EGFR amplification distinguish de novo high grade pediatric astrocytomas from de novo adult astrocytomas. Brain Pathol. 2000;10:249–259. doi: 10.1111/j.1750-3639.2000.tb00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Y, Ng HK, Zhang SF, Ding M, Pang JC, Zheng J, Poon WS. Genetic alterations in pediatric high-grade astrocytomas. Hum Pathol. 1999;30:1284–1290. doi: 10.1016/s0046-8177(99)90057-6. [DOI] [PubMed] [Google Scholar]
- 8.Bredel M, Pollack IF, Hamilton RL, James CD. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res. 1999;5:1786–1792. [PubMed] [Google Scholar]
- 9.Pollack IF, Hamilton RL, Finkelstein SD, Campbell JW, Martinez AJ, Sherwin RN, Bozik ME, Gollin SM. The relationship between TP53 mutations and overexpression of p53 and prognosis in malignant gliomas of childhood. Cancer Res. 1997;57:304–309. [PubMed] [Google Scholar]
- 10.Rickert CH, Strater R, Kaatsch P, Wassmann H, Jurgens H, Dockhorn-Dworniczak B, Paulus W. Pediatric high-grade astrocytomas show chromosomal imbalances distinct from adult cases. Am J Pathol. 2001;158:1525–1532. doi: 10.1016/S0002-9440(10)64103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174:1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 2004;279:33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- 14.Illingworth JA, Tipton KF. Purification and properties of the nicotinamide-adenine dinucleotide phosphate-dependent isocitrate dehydrogenase from pig liver cytoplasm. Biochem J. 1970;118:253–258. doi: 10.1042/bj1180253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geisbrecht BV, Gould SJ. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem. 1999;274:30527–30533. doi: 10.1074/jbc.274.43.30527. [DOI] [PubMed] [Google Scholar]
- 16.Thompson CB. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med. 2009;360:813–815. doi: 10.1056/NEJMe0810213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jennings GT, Minard KI, McAlister-Henn L. Expression and mutagenesis of mammalian cytosolic NADP+-specific isocitrate dehydrogenase. Biochemistry. 1997;36:13743–13747. doi: 10.1021/bi970916r. [DOI] [PubMed] [Google Scholar]
- 18.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pietsch T, Taylor MD, Rutka JT. Molecular pathogenesis of childhood brain tumors. J Neurooncol. 2004;70:203–215. doi: 10.1007/s11060-004-2750-7. [DOI] [PubMed] [Google Scholar]
- 20.Ducray F, El Hallani S, Idbaih A. Diagnostic and prognostic markers in gliomas. Curr Opin Oncol. 2009;21:537–542. doi: 10.1097/CCO.0b013e32833065a7. [DOI] [PubMed] [Google Scholar]
- 21.Sonoda Y, Kumabe T, Nakamura T, Saito R, Kanamori M, Yamashita Y, Suzuki H, Tominaga T. Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci. 2009;100:1996–1998. doi: 10.1111/j.1349-7006.2009.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116:597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 23.Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009;11:341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68:1319–1325. doi: 10.1097/NEN.0b013e3181c391be. [DOI] [PubMed] [Google Scholar]
- 25.Mane SM, Meltzer SJ, Gutheil JC, Kapil V, Lee EJ, Needleman SW. RAS gene activation in acute myelogenous leukemia: analysis by in vitro amplification and DNA base sequence determination. Genes Chromosomes Cancer. 1990;2:71–77. doi: 10.1002/gcc.2870020113. [DOI] [PubMed] [Google Scholar]
- 26.Cogswell PC, Morgan R, Dunn M, Neubauer A, Nelson P, Poland-Johnston NK, Sandberg AA, Liu E. Mutations of the ras protooncogenes in chronic myelogenous leukemia: a high frequency of ras mutations in bcr/abl rearrangement-negative chronic myelogenous leukemia. Blood. 1989;74:2629–2633. [PubMed] [Google Scholar]
- 27.Ball ED, Mills LE, Neubauer A, Liu E. Detection of minimal acute myeloid leukemia cells in bone marrow by probing for mutated ras oncogenes using the polymerase chain reaction. Prog Clin Biol Res. 1990;333:499–506. [PubMed] [Google Scholar]
- 28.Kato Y, Jin G, Kuan CT, McLendon RE, Yan H, Bigner DD. A monoclonal antibody IMab-1 specifically recognizes IDH1R132H, the most common glioma-derived mutation. Biochem Biophys Res Commun. 2009;390:547–551. doi: 10.1016/j.bbrc.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ronaghi M, Uhlen M, Nyren P. A sequencing method based on real-time pyrophosphate. Science. 1998;281:363–365. doi: 10.1126/science.281.5375.363. [DOI] [PubMed] [Google Scholar]
- 30.Fakhrai-Rad H, Pourmand N, Ronaghi M. Pyrosequencing: an accurate detection platform for single nucleotide polymorphisms. Hum Mutat. 2002;19:479–485. doi: 10.1002/humu.10078. [DOI] [PubMed] [Google Scholar]
- 31.Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M, Molinari F, Knowles M, Cerrato A, Rodolfo M, Scarpa A, Felicioni L, Buttitta F, Malatesta S, Marchetti A, Bardelli A. IDH1 mutations at residue p.R132 IDH1(R132) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 2009;30:7–11. doi: 10.1002/humu.20937. [DOI] [PubMed] [Google Scholar]
- 32.Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.