

Abstract
Germline mutations in mismatch repair genes predispose patients to Lynch Syndrome and the majority of these mutations have been detected in two key genes, MLH1 and MSH2. In particular, about a third of the missense variants identified in MLH1 are of unknown clinical significance. Using the PeakPicker software program, we have conducted a proof-of-principle study to investigate whether missense variants in MLH1 lead to allelic imbalances. Lymphocyte RNA extracted from patients harboring known MLH1 variants was used to quantify the ratio of variant to wild-type transcript, while patient lymphocyte DNA was used to establish baseline allelic expression levels. Our analysis indicated that the missense variants c.350C>T, c.793C>T, and c.1852_1853AA>GC, as well as the truncating variant c.1528C>T were all associated with significantly unbalanced allelic expression. However, the variants c.55A>T and c.2246T>C did not demonstrate an allelic imbalance. These results illustrate a novel and efficient method to investigate the pathogenicity of unclassified genetic variants discovered in mismatch repair genes, as well as genes implicated in other inherited diseases. In addition, the PeakPicker methodology has the potential to be applied in the diagnostic setting, which, in conjunction with results from other assays, will help increase both the accuracy and efficiency of genetic testing of colorectal cancer, as well as other inherited diseases.
Lynch syndrome or hereditary nonpolyposis colorectal cancer (HNPCC; MIM 120435), which accounts for about 2% of all colorectal cancer cases, is one of the most common cancer predisposition syndromes. Those affected have about an 80% lifetime risk of developing colorectal cancer.1 In addition, malignancies of the endometrium, skin, bladder, ovaries, kidney, and small intestine are also associated with this syndrome.
Lynch syndrome is an autosomal dominant condition that is characterized by germline mutations in mismatch repair (MMR) genes. The MMR system is a postreplicative DNA repair system that recognizes and repairs base-base mismatches and insertion/deletion loops that occur during DNA replication and escape the proofreading activity of the DNA polymerases. MMR proteins have also been implicated in signaling DNA damage and apoptosis. Germline mutations of the MMR genes MLH1 (MIM 120436), MSH2 (MIM 609309), PMS2 (MIM 600259), PMS1 (MIM 600258), and MSH6 (MIM 600678) have hitherto been implicated in Lynch syndrome. However, about 90% of all known mutations are found in the two MMR genes MLH1 and MSH2,2 which appear to play essential and nonredundant functions of the MMR system.
Tumors with defective MMR function are characterized by expansion/contraction of the microsatellite regions, leading to microsatellite instability (MSI), a hallmark feature seen in about 85% of Lynch syndrome tumors.3 Individuals diagnosed at a younger than average age and/or with familial clustering of colorectal or other Lynch syndrome-associated cancers are generally referred to predictive genetic testing programs. Here, MSI testing is carried out on tumor DNA, which acts as a surrogate marker of defective MMR function. Typically, immunohistochemistry (IHC) is also performed to assess if MMR protein expression is lost. Although MSI testing is a robust assay of MMR activity, it is not a universal indicator of pathogenicity as it is possible for mutations to affect other functions of the MMR pathway.4 Similarly, indication of protein deficiency by IHC does not definitively link the genetic alteration to the loss of protein expression. Additionally, some tumors show equivocal staining patterns with intermediate levels of expression, which cannot be clearly distinguished by IHC. Furthermore, methylation of the MLH1 promoter leads to the MSI phenotype and the loss of MLH1 expression in about 15% of all sporadic colorectal cancers.5,6 Thus, to confirm the presence of Lynch syndrome, the MMR genes of the individuals with MSI tumors are screened for germline mutations. Similar to the other MMR genes, diverse sequence variants, including nonsense, missense, splice site, and frameshift mutations, have been reported throughout the coding region of the MLH1 gene. Many of these mutations are catalogued in the LOVD and MMR Variant Databases (http://chromium.liacs.nl/LOVD2/colon_cancer/home.php and http://www.med.mun.ca/MMRvariants, last accessed April 26, 2010).7 About 24% of mutations detected in MLH1 are missense substitutions.7 The majority of these missense variants are of unknown clinical significance, which are referred to as unclassified genetic variants or variants of uncertain significance. Given that the diagnosis of Lynch syndrome relies primarily on the identification of germline defects in the MMR genes,8 detecting these unclassified genetic variants creates much ambiguity in the clinical setting, as the pathogenicity of these variants cannot be readily ascertained.
On detection of missense variants, several strategies are used to characterize their pathogenic significance. These include linkage analysis, association studies, computational prediction tools and functional assays, each with specific strengths and limitations.8,9,10,11,12,13,14 Linkage analyses is often the most straightforward way to determine the relationship between the disease phenotype and the segregation of the variant, but is often not feasible due to smaller family sizes or unavailability of samples from relevant family members. Computational programs, while helpful in classifying novel variants in the clinical setting, are limited in their predictive value unless a panel of such programs are used.15 However, at present it is important to use a combination of in silico and experimental strategies until improved models of prediction become available. We and other groups have attempted to devise in vitro assays to characterize the pathogenic and functional significance of MLH1 variants. A common approach is to create these alterations in vitro by site-directed mutagenesis, after which these constructs are used to assay expression, MMR function or protein stability.10,12,13,14,16 However, this approach requires a considerable investment of time and effort and cannot be routinely performed in a clinical diagnostic service.
Determining unbalanced allelic expression offers an alternative method of investigating the pathogenic nature of a variant. Several studies in other genes have demonstrated that single nucleotide substitutions are capable of altering the levels of messenger RNA (mRNA) transcripts.17,18,19 Furthermore it has been proposed that such cis-acting functional variants can affect transcription, mRNA processing and mRNA stability.18 We have conducted a proof-of-principle study to investigate whether missense variants in MLH1 lead to an allelic imbalance by measuring levels of transcript present in lymphocytes of individuals carrying the variants c.55A>T (p.I19F), c.350C>T (p.T117M), c.793C>T (p.R265C), c.1528C>T (p.Q510X), c.1852_1853AA>GC (p.K618A), and c.2246T>C (p.L749P), using the PeakPicker method.
Materials and Methods
Selection of Patients and Families
These probands were referred to the Molecular Genetics Laboratory as part of the Provincial Cancer Genetics Program for the assessment of the possible diagnosis of Lynch syndrome. Predictive genetic testing was offered to clinically affected and at-risk subjects, with pre- and post-test genetic counseling as described previously.20 In addition to complete follow-up information, clinical and histopathological data were collected retrospectively on all affected patients. Informed consent was obtained from all subjects and all studies were performed according to guidelines of the Ethics Committee of the University of Toronto.
Overview of Diagnostic Strategy and Identification of MLH1 Variants
The variants chosen for this study were identified in patients that were screened through the Ontario Predictive Genetic Testing Program at Mt. Sinai Hospital, Toronto, Canada based on a series of predetermined referral criteria. This program screens individuals presenting with either a family history of colorectal cancer, and/or those who develop tumors at a young age of onset (<50). Genetic screening is initiated by performing MSI testing and IHC for the MMR proteins on tumor specimens when available. This is followed by germline mutation testing on lymphocyte DNA. Initially, multiplex ligation-dependent probe amplification is performed to detect large genomic deletions and duplications in the MMR genes. If such changes are not detected, germline DNA is screened by sequencing the MLH1 and MSH2 genes, guided by MSI/IHC results of patient tumors. Through this program, we have identified many established and novel germline mutations, as well as several unclassified variants.
Microsatellite Instability and Immunohistochemistry
MSI testing and IHC analysis for MMR proteins were performed as described previously.21 Matched normal and tumor DNA were assessed using a panel of five microsatellite markers as recommended by the National Cancer Institute.22 These include mononucleotide BAT25 and BAT26 and dinucleotide D2S123, D5S346, and D17S250 microsatellite markers. Each case was designated as either microsatellite unstable (MSI-H; ≥30% markers unstable), MSI-low (MSI-L; <30% markers unstable), or microsatellite stable (MSS; no unstable markers). IHC analysis of the respective MMR proteins was performed on formalin-fixed, paraffin-embedded tissues. Based on the protein expression status, tumors were classified as deficient or intact for the MMR proteins.
DNA Extraction and Mutation Detection
Blood samples were obtained from patients and lymphocytes were isolated using NH4Cl–Tris. DNA was extracted from lymphocytes using the Qiagen (Mississauga, ON, Canada) or saturated salt-out method as described previously.23 DNA from patients, whose tumors showed deficiency for a MMR protein, was subjected to exon by exon sequencing of genomic DNA to screen for alterations in MLH1 and MSH2 on an ABI 377 DNA Sequencer (Applied Biosystems, Foster City, CA). Sequence information of the coding region was derived from RefSeq NM_000249.2 (MLH1), NM_000251.1 (MSH2). PCR conditions and primer sequences are available on request.
RNA Extraction and cDNA Synthesis
RNA extraction was performed using TRIzol according to the manufacturer's protocol (Invitrogen Life Technologies, Burlington, ON, Canada). The RNA was then treated with DNaseI for 30 minutes using the RNase-Free DNase set (Qiagen), which allows for digestion of DNase before purification using the RNeasy MinElute kit (Qiagen). Reverse transcription PCR was performed according to standard techniques as described before,23 and cDNA was generated in triplicate from each RNA sample to minimize variation that may be introduced during the process of reverse transcription.
PeakPicker Analysis
Primer Design and PCR
Primers were designed to amplify the exonic regions flanking the variant of interest so as to allow the same pair of primers to be used for the amplification of both the genomic DNA (gDNA) and cDNA to ensure unbiased amplification. PCR products were then visualized on an ethidium bromide/agarose gel to verify the presence of product. PCR products were treated with 5 units of Shrimp Alkaline Phosphatase (USB Corporation, Cleveland, OH) and 2 units of Exonuclease I (USB) per 15 μl of PCR product for 37°C for 1 hour to remove unincorporated dNTPs and excess amplification primers. The enzymes were then deactivated by incubating at 75°C for 15 minutes.
Sequencing and PeakPicker Analysis
High sensitivity sequencing was carried out as described before using 0.5 μl of Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) with 2 μl of PCR product, 1.75 μl of 5× sequencing buffer, and 10pmol of sequencing primer in a 10 μl reaction using the manufacturer's protocol.24 The products were separated on an Applied Biosystems 3130XL DNA analyzer. Sequencing was carried out with both the forward and reverse primers whenever possible, unless the variant was located too close to the primer binding site.
The PeakPicker software was developed by Ge et al,24,25 for quantitative allele ratio analysis. This software is publicly available at http://genomequebec.mcgill.ca/publications/pastinen/, last accessed April 1, 2010. It relies on multiple alignments of sequence traces generated by high sensitivity sequencing. The reference sequence (gDNA) is used to select the peaks that represent the single nucleotide polymorphism or variant, as well as control peaks. The program then identifies single nucleotide polymorphism and control peaks in all sequences, and analyzes the peaks in parallel. The allelic ratio of the gDNA samples are set to 1 as no allele specific expression is expected. However, by concurrently analyzing the gDNA sample in triplicate, we were able to increase the accuracy of this normalization. A file with single nucleotide polymorphism allele ratios normalized to gDNA peak heights is generated as the output of this program. Allelic ratios were calculated by dividing the peak height of the variant allele by that of wild-type allele. Three independent PCR and sequencing reactions were carried out from each gDNA and RNA (one reaction from each independent cDNA) sample and a two-tailed Student's t-test was used to assess statistically significant differences in allelic expression.
Allele-Specific (Asymmetric) PCR
Allele-specific PCR was carried out determine the variant allele that was in linkage with the each of the alleles of the c.655A>G (p.I219V, rs1799977) polymorphism. Primers were designed to amplify the exonic regions surrounding the variant and the c.655A>G polymorphism in the MLH1 gene. Differential amplification of alleles was achieved by designing two unique reverse primers that contained either of variant bases at its 3′ end. Given the stringent requirement for complementarity at this location, this allowed us to preferentially amplify each of the two alleles separately. PCR was then carried out as described above using a common forward primer, except that up to twice as much reverse primer was used as forward primer to ensure successful amplification of the product. PCR products were then purified with Shrimp Alkaline Phosphatase and Exonuclease and sequenced using the Applied Biosystems 3130XL DNA analyzer.
In Silico Analysis of Effect on Splicing
We used the computational programs NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html, last accessed April 13, 2010.) and GeneScan 1.0 (http://genes.mit.edu/GENESCAN.html, last accessed April 13, 2010.) to determine the predicted effect of the splice site mutation. Default settings were used for all programs.
Results
In this study we determined the allelic expression of MLH1 in lymphocytes obtained from individuals carrying missense variants c.55A>T, c.350C>T, c.793C>T, c.1852_1853AA>GC, c.2246T>C, and the truncating variant c.1528C>T in MLH1. Genomic DNA (gDNA) was used as a reference to establish baseline levels of expression, and cDNA generated from three independent reverse transcription reactions was used to measure mRNA transcript levels. Identical primer pairs were used to PCR amplify the region surrounding the variant in both gDNA and cDNA samples, followed by high sensitivity sequencing of the amplicons. The sequence information was then analyzed using the PeakPicker program. This program quantifies peak heights of the polymorphic bases and uses this information to compute their relative abundance in the sample (Figure 1). As a component of this step, this program compares the heights of adjacent peaks to control for the efficiency of the sequencing reaction. The ratio of the variant bases is set to 1 in the gDNA samples and the heights of the peaks are normalized to the values derived from gDNA. The allelic ratio is computed by dividing the average expression of variant/wild-type allele in cDNA by the average expression of variant/wild-type allele for the gDNA samples. In accordance with previously published studies, we set ≤0.5 as a threshold value below which allelic ratios were considered abnormal.26,27 This ratio corresponds to a situation where twice as much wild-type transcript is present compared to the transcript carrying the variant base in the patient's cDNA.
Figure 1.
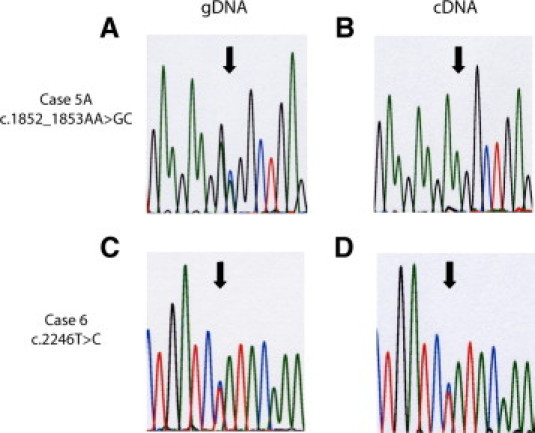
Representative chromatograms for cases 5A and 6. The left panels (A and C) indicate genomic DNA (gDNA) and the right panels (B and D) indicate complementary DNA (cDNA). Case 5A demonstrates decreased levels of the variant (GC) allele relative to the wild-type (AA) allele, while case 6 shows no change in the levels of the variant (C) allele relative to the wild-type (T) allele. Arrows highlight the heterozygous peaks. Both chromatograms shown were generated using the respective forward primer. (Blue: C, Green: A, Red: T, Black: G).
By exonic sequencing, we identified the missense variant c.55A>T in exon 1 of an individual who developed endometrial cancer at the age of 47 (case 1). PeakPicker analysis demonstrated that the allelic ratio of the variant/wild-type allele was 0.85 (P = 0.564), which was not found to be statistically significant when assessed using a two-tailed Student's t-test. The clinicopathological variables relating to each case are summarized in Table 1.
Table 1.
Summary of Allelic Ratios and Clinical Characteristics
| Case | Alteration | Predicted effect on protein | Normalized allelic ratio | Clinical diagnosis | MLH1 IHC status | Tumor MSI status |
|---|---|---|---|---|---|---|
| 1 | c.55A>T | Ile19Phe | 0.85 | Endometrial cancer at 47 | ND | ND |
| 2 | c.350C>T | Thr117Met | 0.19*,0.07*† | At-risk testing‡ | N/A | N/A |
| 3 | c.793C>T | Arg265Cys | 0.50*† | CRC at 55 | Deficient | MSI-H |
| 4 | c.1528C>T | Gln510Ter | 0.38* | CRC at 42 | Deficient | MSI-H |
| 5A | c.1852_1853AA>GC | Lys618Ala | 0.05*,0.13*† | Small bowel cancer at 34 | Intact | MSI-H |
| 5B | c.1852_1853AA>GC | Lys618Ala | 0.50* | At-risk testing | N/A | N/A |
| 6 | c.2246T>C | Leu749Pro | 1.02 | CRC at 24 | Intact | MSI-H |
IHC, immunohistochemistry; MSI-H, microsatellite instability-high; ND, not determined (no access to tumor sample); CRC, colorectal cancer; N/A, not applicable.
Indicates statistically significant values as determined by two-tailed Student's t-test.
Indicates analysis carried out using primers that flank the common MLH1 polymorphism c.655A>G.
Case 2's father (the proband) carried the same variant and presented with a MLH1-deficient MSI-H tumor, but his DNA/RNA was not available for analysis. Cases 5A and 5B are members of the same kindred.
Case 2 presented for at-risk testing and is a carrier of the c. 350C>T variant, which occurs in exon 4. Her father (the proband) was diagnosed with MSI-H, MLH1 deficient adenocarcinoma of the colon and was found to carry c. 350C>T. However, his lymphocyte DNA and RNA was not available for analysis. This variant led to an allelic ratio of 0.19 (P = 0.015). Case 2 was also heterozygous for the c.655A>G (p.I219V) polymorphism. We therefore reanalyzed this case using a primer set that flanked this polymorphism to rule out the possibility that the observed allele ratio was primer specific. When PeakPicker analysis was carried out using primers that flanked this polymorphism in exon 8 of MLH1, the allelic ratio was found to be 0.07(P = 0.0004), which is in agreement with the results obtained using primers that flank the variant in exon 4.
The variant c.793C>T, which occurs in exon 10 was detected in an individual who developed colorectal cancer at age 55(case 3). Given the location of this substitution in the third nucleotide at the beginning of the exon, it was not possible to use identical primers to amplify the region surrounding this variant from both gDNA and cDNA. However, case 3 was heterozygous for the c.655A>G polymorphism, which enabled us to use primers flanking this region to assay its allelic ratio. Using allele-specific PCR we were able to show that c.655A occurs on the wild-type allele (c.793C, results not shown). PeakPicker analysis demonstrated that this variant led to an allelic ratio 0.5 (P = 0.025).
Case 4 was diagnosed with colorectal cancer at the age of 42. Exonic sequencing indicated that this individual carries the nonsense mutation c.1528C>T in exon 13 of MLH1. This mutation led to an allelic ratio of 0.38 (P = 0.0023).
The MLH1 variant c.1852_1853AA>GC was the only genetic alteration discovered in a proband that developed small bowel cancer at the age of 34 years (Case 5A). This variant represents a two nucleotide substitution in exon 16 of MLH1. PeakPicker analysis revealed that the proband's allelic ratio was reduced to 0.05 (P = 0.0009) (Figure, 1A and 1B). This proband was also heterozygous for a common MLH1 polymorphism c.655A>G (where c.655A occurs on the wild-type allele). Using primers that flank this polymorphic region the MLH1 allelic ratio was determined to be 0.13 (P = 0.0004), confirming our previous finding. This proband's father (case 5B), who presented for at-risk testing, is also a carrier of c.1852_1853AA>GC variant. By PeakPicker analysis case 5B's MLH1 allelic ratio was demonstrated to be 0.5 (P = 0.0135). In keeping with the results from case 5A, this variant appears to be associated with an allelic imbalance. However, as this individual was not heterozygous for the c.655A>G polymorphism we were unable to repeat the analysis as with case 5A.
The c.2246T>C variant was discovered in a proband that developed colorectal cancer at the age of 24 (case 6). This variant occurs in the final exon (19) of MLH1 and led to an allelic ratio of 1.02 (P = 0.8253, Figure 1, C and D).
Discussion
The discovery of a predisposing mutation in Lynch syndrome families and the concomitant increase in surveillance has been shown to significantly reduce mortality.28,29 Therefore it is of paramount importance to classify the pathogenicity associated with unclassified variants to determine their role in cancer predisposition. PeakPicker provides a relatively rapid and robust method of differentiating potentially pathogenic variants that lead to unbalanced allelic expression. Furthermore, no specialized tools or complex technical skills are required to perform this analysis, thus this technique has the potential to be implemented in a diagnostic setting for the routine assessment of unclassified genetic variants.
The c.55A>T variant has been reported in two previous independent studies; however, no functional studies have been carried out to-date.30,31 Although PeakPicker analysis demonstrated that this variant generated an allelic ratio that was less than 1, this value was not found to be statistically significant. In this study, we considered an allelic ratio ≤0.5, combined with statistical significance to be indicative of altered allelic expression/stability. A previous study documented that the normal range of allelic variation in MLH1 is between 0.81 and 1.23,32 which is in line with our interpretation that this variant is not associated with altered allelic expression/stability. It is possible that some genetic variants may only mildly affect mRNA levels and thus lead to intermediate allelic ratios (1 to 0.5). The accurate interpretation of these variants in the clinical setting will require that the arbitrary cut off established in this study be considered in the light of other available evidence (ie, MSI/IHC information of the proband's tumor). In this case, we did not have supplementary data from MSI and IHC. However, we observed expression levels comparable to that of wild-type MLH1 protein by in vitro expression assays (Perera, S and Bapat, B, unpublished data). Thus, taken together these observations suggest that altered mRNA expression/stability is unlikely to be the mechanism by which this variant mediates its pathogenicity, but highlights the utility of PeakPicker analysis in cases where incomplete clinical information is present.
Based on phylogenetic, structural and functional evidence, as well as segregation data, the c.350 C>T variant is considered to be a deleterious mutation.33 This variant has been shown to affect the efficiency of MMR function by several studies.34,35 Results from our study that indicates that this variant is associated with unbalanced allelic expression, further supports these findings. However, by conversion analysis technology this variant did not show decreased allelic expression.8 While this may be due to the differences between these two methodologies, another possibility is that other unique genetic influences may have led to the different allelic profiles observed in the two individuals.
The c.793C>T variant has been reported to lead to reduced MMR efficiency in several assays.34,35,36,37 Furthermore, in a previous study using pulse chase assays and cycloheximide treatments, we were able to show that this variant decreased the stability of the MLH1 protein.14 This variant has also been associated with abnormal splicing, with two independent studies demonstrating that it induced partial skipping of exon 10.33,38 Given its proximity to the intron–exon border, this highlights the role of cis-acting elements in flanking sequences in regulating splicing and defining exons. Our study indicates that this variant is associated with an allelic imbalance. Interestingly, conversion analysis also indicated that this variant decreased the expression of the variant allele.8 Despite the distinctions between the two assays, these similar findings provide evidence of the sensitivity and accuracy of the PeakPicker methodology.
Previous functional studies carried out with the c.1852_1853AA>GC variant have produced conflicting pieces of evidence regarding its pathogenicity,10 making it difficult to ascertain if c.1852_1853AA>GC is a pathogenic or benign variant. In a previous study we demonstrated that the c.1852_1853AA>GC variant drastically decreases the stability of the MLH1 protein.14 However, these assays are time consuming and therefore not practical to be applied to the clinical setting. Furthermore, results from in vitro findings do not always mirror the effect of the variant in vivo. The results of the PeakPicker analysis demonstrate that this substitution is associated with decreased expression/stability of the variant allele in the individual's lymphocyte RNA. The differential effect we see in these two family members could be attributed to unique genetic background or environmental exposures that might have influenced the levels of MLH1 transcribed or the stability of this transcript. However, it is not possible to rule out if the unbalanced allelic expression profile we observed is the result of a hitherto undiscovered mutation that lies in regulatory regions or deep within intronic regions that is in linkage with the variant allele.
The c.2246T>C variant lies within the c-terminal homology domain that is thought to be important in stabilizing the interaction between MLH1 and PMS2,39 and while this variant has been previously reported, no functional studies have been done to date.40,41,42 Previous studies carried out in our laboratory have demonstrated that another missense variant affecting the same nucleotide, MLH1 c.2246 T>A, did not affect protein expression or stability but showed decreased efficiency of heterodimerizing with PMS2.14 Given our finding that this variant is not associated with an allelic imbalance, it is likely that this variant affects heterodimerization of MLH1 rather than its expression or stability.
Truncating mutations are the most common type of mutation identified in both the MLH1 and the MSH2 genes.43 Truncating mutations lead to transcripts with premature termination codons that are generally degraded by nonsense mediated RNA decay, which functions to eliminate truncated proteins that may have deleterious effects.44 However, depending the location of the termination codon, truncating mutations may or may not lead to decreased mRNA levels. The finding that the truncating mutation, c.1528 C>T is associated with diminished allelic expression/stability, confirms PeakPicker's sensitivity and its versatility in recognizing the effects of diverse types of genetic alterations.
A limitation of the PeakPicker strategy is that it is difficult to assay variants that occur close to exon boundaries or those that occur in relatively small (<80 bp) exons. Furthermore, heterozygosity of the variant allele is required to carry out this analysis. However, these issues can be circumvented if the individual carrying the unclassified variant is heterozygous for a known, common polymorphism such as c.655A>G. This polymorphism is suitable for PeakPicker analysis as it has the highest level of heterozygosity among exonic MLH1 polymorphisms,32 and occurs within exon 9 of MLH1 away from intron/exon boundaries. In this situation, primers flanking the polymorphic region can be used to confirm the effect of the uncharacterized variant. This is more cost efficient than designing unique primers surrounding each exon, and can be adopted when a patient population shows a high frequency of a common single nucleotide polymorphism. Alternatively, in the case of short exonic regions, it may also be possible to use primers with linker sequences so that useful sequence information is not lost in the process of sequencing. A short linker sequence was added on to the primers flanking c.350C>T, which enabled us to assay this variant, which occurs in a relatively short exon. Importantly, PeakPicker cannot be used as a stand-alone test, as there may be several variants that do not lead to allelic imbalances but may possibly affect other functions. Thus, this analysis should be used in conjunction with a panel of assays that will help determine the putative effect of genetic variants on expression and/or function. Additionally, this should be combined with other relevant clinical and family history information indicating tumor MMR deficiency status to obtain a more accurate understanding of a variant's pathogenicity.
Another strength of the PeakPicker strategy is that it will aid in discovering mutations in individuals who were classified to be mutation-negative using conventional diagnostic techniques. A major problem that arises in the detection of mutations is the possible masking of alterations in one allele by the normal sequence present on the other allele. Techniques such as conversion technology and methods based on single nucleotide primer extension are able to analyze the maternal and paternal alleles separately and have been applied to the detection of MMR alterations.8,26,32 However, conversion technology is cumbersome and cannot be readily implemented in the diagnostic setting. Using an allelic expression assay based on single nucleotide primer extension, Renkonen and colleagues26 were able to identify the MLH1 R100X nonsense mutation in a mutation-negative family. PeakPicker, which is very similar in principle to single nucleotide primer extension assays, offers a very efficient method of detecting hitherto unidentified genetic changes by measuring imbalances in allelic expression. Importantly, PeakPicker analysis and the benefits it offers can be extended to the study of other genes implicated in inherited cancers, as well as other diseases. BRCA1 and BRCA2 are examples of two such genes that can benefit from this type of analysis as the Breast cancer Information Core (BIC) database lists about 1500 variants, mostly missense substitutions of unknown clinical significance.45
Unclassified variants in MLH1 have been shown to alter protein expression, stability, and turnover.10,14 However, not much attention has been paid to the role of unclassified variants in leading to allelic imbalances, despite its potential to be a noteworthy cause of MMR deficiency. Allelic imbalances could arise due to genetic alterations that affect the efficiency of transcription, alter transcript expression, or affect mRNA stability, and this study provides initial evidence that a subset of unclassified missense variants in MLH1 affect these processes. Similarly, a previous study showed that a missense variant in RB1 was able to significantly impair the allelic balance in a pedigree with retinoblastoma.19 Another mechanism by which unclassified variants may potentially contribute to altered mRNA levels is through abnormal splicing. Several missense variants in MLH1, including the c.793C>T variant that result in splicing abnormalities have been shown to be associated with RNA defects.46 However, the splicing prediction tool, NNSPLICE and the gene prediction program, GeneScan did not predict that these variants would lead to splicing abnormalities. Furthermore, these variants may also affect splicing by altering exonic splice enhancer/silencer sites that aid in exon definition. Overall, while the extent of the effect may depend on the missense alteration in question and its mechanism of action, measuring allelic imbalances offers another important clue regarding the pathogenicity of unclassified variants.
In summary, we demonstrate that the publicly available software tool, PeakPicker was able to identify MLH1 variants that led to unbalanced allelic expression in a time and cost efficient manner, making it amenable to implementation in the diagnostic setting. Information on allelic ratio in conjunction with other assays will help increase confidence in ascertaining the pathogenicity of a variant, especially if limited clinical data are present. This technique essentially generates a snapshot of the gene expression profile present in the individual's cells. Therefore, unlike in vitro assessment strategies this assay takes into consideration the unique environmental or genetic influences that may be specific to the individual. The PeakPicker strategy will also open avenues to further analyze apparently mutation-negative cases. Moreover, this strategy can be used to help classify genetic alterations identified in several hereditary diseases, allowing for the improved accuracy and efficiency of predictive genetic testing programs.
Acknowledgements
We thank the patients and their families for participating in this study. We are grateful to Dr. Tomi Pastinen (McGill University, Montreal, QC, Canada) and Dr. Bing Ge for providing us with the PeakPicker software and for helping us set up the program. We also gratefully acknowledge the assistance provided by the staff of the Molecular Pathology Laboratory, the Biospecimen Repository and the Familial GI Cancer Registry of the Mt. Sinai Hospital for their help with this study.
Footnotes
Supported (S.P.) by the Canadian Institutes of Health Research/Canadian Digestive Health Foundation Doctoral Award and graduate studentships from the University of Toronto (Frank Fletcher Memorial Fund, Kristii Pia Callum Award).
CME Disclosure: None of the authors disclosed any relevant financial relationships.
References
- 1.Umar A, Risinger JI, Hawk ET, Barrett JC. Testing guidelines for hereditary non-polyposis colorectal cancer. Nat Rev Cancer. 2004;4:153–158. doi: 10.1038/nrc1278. [DOI] [PubMed] [Google Scholar]
- 2.Chung DC, Rustgi AK. The hereditary nonpolyposis colorectal cancer syndrome: genetics and clinical implications. Ann Intern Med. 2003;138:560–570. doi: 10.7326/0003-4819-138-7-200304010-00012. [DOI] [PubMed] [Google Scholar]
- 3.Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, Petersen GM, Kinzler KW, Vogelstein B, de la Chapelle A. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 4.Barnetson RA, Cartwright N, van Vliet A, Haq N, Drew K, Farrington S, Williams N, Warner J, Campbell H, Porteous ME, Dunlop MG. Classification of ambiguous mutations in DNA mismatch repair genes identified in a population-based study of colorectal cancer. Hum Mutat. 2008;29:367–374. doi: 10.1002/humu.20635. [DOI] [PubMed] [Google Scholar]
- 5.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 6.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woods MO, Williams P, Careen A, Edwards L, Bartlett S, McLaughlin JR, Younghusband HB. A new variant database for mismatch repair genes associated with Lynch syndrome. Hum Mutat. 2007;28:669–673. doi: 10.1002/humu.20502. [DOI] [PubMed] [Google Scholar]
- 8.Casey G, Lindor NM, Papadopoulos N, Thibodeau SN, Moskow J, Steelman S, Buzin CH, Sommer SS, Collins CE, Butz M, Aronson M, Gallinger S, Barker MA, Young JP, Jass JR, Hopper JL, Diep A, Bapat B, Salem M, Seminara D, Haile R. Conversion analysis for mutation detection in MLH1 and MSH2 in patients with colorectal cancer. JAMA. 2005;293:799–809. doi: 10.1001/jama.293.7.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipkin SM, Rozek LS, Rennert G, Yang W, Chen PC, Hacia J, Hunt N, Shin B, Fodor S, Kokoris M, Greenson JK, Fearon E, Lynch H, Collins F, Gruber SB. The MLH1 D132H variant is associated with susceptibility to sporadic colorectal cancer. Nat Genet. 2004;36:694–699. doi: 10.1038/ng1374. [DOI] [PubMed] [Google Scholar]
- 10.Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lonnqvist KE, Holinski-Feder E, Sutter C, McKinnon W, Duraisamy S, Gerdes AM, Peltomaki P, Kohonen-Ccorish M, Mangold E, Macrae F, Greenblatt M, de la Chapelle A, Nystrom M. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–549. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Shimodaira H, Filosi N, Shibata H, Suzuki T, Radice P, Kanamaru R, Friend SH, Kolodner RD, Ishioka C. Functional analysis of human MLH1 mutations in Saccharomyces cerevisiae. Nat Genet. 1998;19:384–389. doi: 10.1038/1277. [DOI] [PubMed] [Google Scholar]
- 12.Trojan J, Zeuzem S, Randolph A, Hemmerle C, Brieger A, Raedle J, Plotz G, Jiricny J, Marra G. Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology. 2002;122:211–219. doi: 10.1053/gast.2002.30296. [DOI] [PubMed] [Google Scholar]
- 13.Brieger A, Trojan J, Raedle J, Plotz G, Zeuzem S. Transient mismatch repair gene transfection for functional analysis of genetic hMLH1 and hMSH2 variants. Gut. 2002;51:677–684. doi: 10.1136/gut.51.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perera S, Bapat B. The MLH1 variants p.Arg265Cys and p.Lys618Ala affect protein stability while p.Leu749Gln affects heterodimer formation. Hum Mutat. 2008;29:332. doi: 10.1002/humu.9523. [DOI] [PubMed] [Google Scholar]
- 15.Chan PA, Duraisamy S, Miller PJ, Newell JA, McBride C, Bond JP, Raevaara T, Ollila S, Nystrom M, Grimm AJ, Christodoulou J, Oetting WS, Greenblatt MS. Interpreting missense variants: comparing computational methods in human disease genes CDKN2A. MLH1, MSH2, MECP2, and tyrosinase (TYR) Hum Mutat. 2007;28:683–693. doi: 10.1002/humu.20492. [DOI] [PubMed] [Google Scholar]
- 16.Raevaara TE, Gerdes AM, Lonnqvist KE, Tybjaerg-Hansen A, Abdel-Rahman WM, Kariola R, Peltomaki P, Nystrom-Lahti M. HNPCC mutation MLH1 P648S makes the functional protein unstable, and homozygosity predisposes to mild neurofibromatosis type 1. Genes Chromosomes Cancer. 2004;40:261–265. doi: 10.1002/gcc.20040. [DOI] [PubMed] [Google Scholar]
- 17.Pinsonneault J, Nielsen CU, Sadee W. Genetic variants of the human H+/dipeptide transporter PEPT2: analysis of haplotype functions. J Pharmacol Exp Ther. 2004;311:1088–1096. doi: 10.1124/jpet.104.073098. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Wang D, Johnson AD, Papp AC, Sadee W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem. 2005;280:32618–32624. doi: 10.1074/jbc.M504942200. [DOI] [PubMed] [Google Scholar]
- 19.Murakami Y, Isogai K, Tomita H, Sakurai-Yageta M, Maruyama T, Hidaka A, Nose K, Sugano K, Kaneko A. Detection of allelic imbalance in the gene expression of hMSH2 or RB1 in lymphocytes from pedigrees of hereditary, nonpolyposis, colorectal cancer, and retinoblastoma by an RNA difference plot. J Hum Genet. 2004;49:635–641. doi: 10.1007/s10038-004-0201-0. [DOI] [PubMed] [Google Scholar]
- 20.Soravia C, Sugg SL, Berk T, Mitri A, Cheng H, Gallinger S, Cohen Z, Asa SL, Bapat BV. Familial adenomatous polyposis-associated thyroid cancer: a clinical, pathological, and molecular genetics study. Am J Pathol. 1999;154:127–135. doi: 10.1016/S0002-9440(10)65259-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woods MO, Hyde AJ, Curtis FK, Stuckless S, Green JS, Pollett AF, Robb JD, Green RC, Croitoru ME, Careen A, Chaulk JA, Jegathesan J, McLaughlin JR, Gallinger SS, Younghusband HB, Bapat BV, Parfrey PS. High frequency of hereditary colorectal cancer in Newfoundland likely involves novel susceptibility genes. Clin Cancer Res. 2005;11:6853–6861. doi: 10.1158/1078-0432.CCR-05-0726. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, Srivastava S. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89:1758–1762. doi: 10.1093/jnci/89.23.1758. [DOI] [PubMed] [Google Scholar]
- 23.Charames GS, Ramyar L, Mitri A, Berk T, Cheng H, Jung J, Bocangel P, Chodirker B, Greenberg C, Spriggs E, Bapat B. A large novel deletion in the APC promoter region causes gene silencing and leads to classical familial adenomatous polyposis in a Manitoba Mennonite kindred. Hum Genet. 2008;124:535–541. doi: 10.1007/s00439-008-0579-4. [DOI] [PubMed] [Google Scholar]
- 24.Ge B, Gurd S, Gaudin T, Dore C, Lepage P, Harmsen E, Hudson TJ, Pastinen T. Survey of allelic expression using EST mining. Genome Res. 2005;15:1584–1591. doi: 10.1101/gr.4023805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee PD, Ge B, Greenwood CM, Sinnett D, Fortin Y, Brunet S, Fortin A, Takane M, Skamene E, Pastinen T, Hallett M, Hudson TJ, Sladek R. Mapping cis-acting regulatory variation in recombinant congenic strains. Physiol Genomics. 2006;25:294–302. doi: 10.1152/physiolgenomics.00168.2005. [DOI] [PubMed] [Google Scholar]
- 26.Renkonen E, Zhang Y, Lohi H, Salovaara R, Abdel-Rahman WM, Nilbert M, Aittomaki K, Jarvinen HJ, Mecklin JP, Lindblom A, Peltomaki P. Altered expression of MLH1. MSH2, and MSH6 in predisposition to hereditary nonpolyposis colorectal cancer. J Clin Oncol. 2003;21:3629–3637. doi: 10.1200/JCO.2003.03.181. [DOI] [PubMed] [Google Scholar]
- 27.Renkonen ET, Nieminen P, Abdel-Rahman WM, Moisio AL, Jarvela I, Arte S, Jarvinen HJ, Peltomaki P. Adenomatous polyposis families that screen APC mutation-negative by conventional methods are genetically heterogeneous. J Clin Oncol. 2005;23:5651–5659. doi: 10.1200/JCO.2005.14.712. [DOI] [PubMed] [Google Scholar]
- 28.Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomaki P, De La Chapelle A, Mecklin JP. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 29.Mecklin JP, Aarnio M, Laara E, Kairaluoma MV, Pylvanainen K, Peltomaki P, Aaltonen LA, Jarvinen HJ. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology. 2007;133:1093–1098. doi: 10.1053/j.gastro.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Andrew SE, Whiteside D, Buzin C, Greenberg C, Spriggs E. An intronic polymorphism of the hMLH1 gene contributes toward incomplete genetic testing for HNPCC. Genet Test. 2002;6:319–322. doi: 10.1089/10906570260471868. [DOI] [PubMed] [Google Scholar]
- 31.Kurzawski G, Suchy J, Kladny J, Safranow K, Jakubowska A, Elsakov P, Kucinskas V, Gardovski J, Irmejs A, Sibul H, Huzarski T, Byrski T, Debniak T, Cybulski C, Gronwald J, Oszurek O, Clark J, Gozdz S, Niepsuj S, Slomski R, Plawski A, Lacka-Wojciechowska A, Rozmiarek A, Fiszer-Maliszewska L, Bebenek M, Sorokin D, Stawicka M, Godlewski D, Richter P, Brozek I, Wysocka B, Jawien A, Banaszkiewicz Z, Kowalczyk J, Czudowska D, Goretzki PE, Moeslein G, Lubinski J. Germline MSH2 and MLH1 mutational spectrum in HNPCC families from Poland and the Baltic States. J Med Genet. 2002;39:E65. doi: 10.1136/jmg.39.10.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tournier I, Raux G, Di Fiore F, Marechal I, Leclerc C, Martin C, Wang Q, Buisine MP, Stoppa-Lyonnet D, Olschwang S, Frebourg T, Tosi M. Analysis of the allele-specific expression of the mismatch repair gene MLH1 using a simple DHPLC-based method. Hum Mutat. 2004;23:379–384. doi: 10.1002/humu.20008. [DOI] [PubMed] [Google Scholar]
- 33.Tournier I, Vezain M, Martins A, Charbonnier F, Baert-Desurmont S, Olschwang S, Wang Q, Buisine MP, Soret J, Tazi J, Frebourg T, Tosi M. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum Mutat. 2008;29:1412–1424. doi: 10.1002/humu.20796. [DOI] [PubMed] [Google Scholar]
- 34.Plotz G, Welsch C, Giron-Monzon L, Friedhoff P, Albrecht M, Piiper A, Biondi RM, Lengauer T, Zeuzem S, Raedle J. Mutations in the MutSalpha interaction interface of MLH1 can abolish DNA mismatch repair. Nucleic Acids Res. 2006;34:6574–6586. doi: 10.1093/nar/gkl944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi M, Shimodaira H, Andreutti-Zaugg C, Iggo R, Kolodner RD, Ishioka C. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 2007;67:4595–4604. doi: 10.1158/0008-5472.CAN-06-3509. [DOI] [PubMed] [Google Scholar]
- 36.Ellison AR, Lofing J, Bitter GA. Functional analysis of human MLH1 and MSH2 missense variants and hybrid human-yeast MLH1 proteins in Saccharomyces cerevisiae. Hum Mol Genet. 2001;10:1889–1900. doi: 10.1093/hmg/10.18.1889. [DOI] [PubMed] [Google Scholar]
- 37.Drost M, Zonneveld JB, van Dijk L, Morreau H, Tops CM, Vasen HF, Wijnen JT, de Wind N: A cell-free assay for the functional analysis of variants of the mismatch repair protein MLH1. Hum Mutat, 31: 247–253 [DOI] [PubMed]
- 38.Lastella P, Surdo NC, Resta N, Guanti G, Stella A. In silico and in vivo splicing analysis of MLH1 and MSH2 missense mutations shows exon- and tissue-specific effects. BMC Genomics. 2006;7:243. doi: 10.1186/1471-2164-7-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohd AB, Palama B, Nelson SE, Tomer G, Nguyen M, Huo X, Buermeyer AB. Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair (Amst) 2006;5:347–361. doi: 10.1016/j.dnarep.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Colombino M, Cossu A, Arba A, Manca A, Curci A, Avallone A, Comella G, Botti G, Scintu F, Amoruso M, D'Abbicco D, d'Agnessa MR, Spanu A, Tanda F, Palmieri G. Microsatellite instability and mutation analysis among southern Italian patients with colorectal carcinoma: detection of different alterations accounting for MLH1 and MSH2 inactivation in familial cases. Ann Oncol. 2003;14:1530–1536. doi: 10.1093/annonc/mdg402. [DOI] [PubMed] [Google Scholar]
- 41.de Leon MP, Benatti P, Di Gregorio C, Losi L, Pedroni M, Ponti G, Genuardi M, Viel A, Lucci-Cordisco E, Rossi G, Roncucci L. Genotype-phenotype correlations in individuals with a founder mutation in the MLH1 gene and hereditary non-polyposis colorectal cancer. Scand J Gastroenterol. 2007;42:746–753. doi: 10.1080/00365520601026681. [DOI] [PubMed] [Google Scholar]
- 42.Losi L, Di Gregorio C, Pedroni M, Ponti G, Roncucci L, Scarselli A, Genuardi M, Baglioni S, Marino M, Rossi G, Benatti P, Maffei S, Menigatti M, Roncari B, Ponz de Leon M. Molecular genetic alterations and clinical features in early-onset colorectal carcinomas and their role for the recognition of hereditary cancer syndromes. Am J Gastroenterol. 2005;100:2280–2287. doi: 10.1111/j.1572-0241.2005.00223.x. [DOI] [PubMed] [Google Scholar]
- 43.Peltomaki P, Vasen HF. Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer Gastroenterology. 1997;113:1146–1158. doi: 10.1053/gast.1997.v113.pm9322509. [DOI] [PubMed] [Google Scholar]
- 44.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 45.Distelman-Menachem T, Shapira T, Laitman Y, Kaufman B, Barak F, Tavtigian S, Friedman E. Analysis of BRCA1/BRCA2 genes' contribution to breast cancer susceptibility in high risk Jewish Ashkenazi women. Fam Cancer. 2009;8:127–133. doi: 10.1007/s10689-008-9216-6. [DOI] [PubMed] [Google Scholar]
- 46.Sharp A, Pichert G, Lucassen A, Eccles D. RNA analysis reveals splicing mutations and loss of expression defects in MLH1 and BRCA1. Hum Mutat. 2004;24:272. doi: 10.1002/humu.9267. [DOI] [PubMed] [Google Scholar]