

Summary
We have investigated the Mycobacterium tuberculosis strain types present in the South Asian population of the UK, in which tuberculosis is particularly prevalent. In contrast to the widespread Beijing strains which have the variable number tandem repeats (VNTR) profile 42435, isolates with the VNTR profile 42235, jointly with 02335 or 42234 profiles, appear more frequently in tuberculosis patients of South Asian ethnic origin (SA-strains) in the UK than in any other ethnic group. Using microarray-based comparative genomics to distinguish total or partially deleted genes, we found that three of the common deleted regions in the SA-strains were identical to some deleted genes in the strain CH, which caused an outbreak among South Asian patients in Leicester in 2001 but were different from genomic deletions found in Beijing/W strains. Analysis of some of the deleted regions revealed differences in comparison to the strain CH including the polymorphism in some of the PE/PPE and Esat-6 genes, which may be responsible for the diversity of antigenic variation or differences in the activation of the host immune response. Interrupted genes or the replacement by insertion elements was confirmed in some of the deleted genomic regions. Our results are consistent with the hypothesis that the SA-strains may present common features, implying a common origin for this group of strains.
Keywords: Mycobacterium tuberculosis, PE/PPE, Polymorphism, VNTR 42235, South Asian community
Introduction
According to the World Health Organization report1 one-third of the world's population is infected by Mycobacterium tuberculosis. The bacillus causes 1.6 million deaths each year and more than 8 million new cases annually, with the majority in South East Asia (3 million cases each year). In the UK, despite a low national prevalence of tuberculosis, the incidence is much higher in cities with a large South Asian community.2 A large outbreak of TB was recently reported in 2001 among people of South Asian origin in a school in Leicester, a city with a high overall incidence of tuberculosis.3 Amongst people of South Asian ethnic origin a dominant group has been described.4 This epidemiological profile can be identified by characteristic Exact Tandem Repeat profiles (42235, 02235 and 42234).4 The variable number tandem repeats (VNTR) profile 42235 represented 23% of patient isolates found in Leeds and Bradford, cities in West Yorkshire, UK, and 37% of patient isolates collected in Rawalpindi, Pakistan.4 It is essential to emphasise that this particular group of strains, which we have called South Asian strains (SA-strains), were not related epidemiologically to those in the outbreak in Leicester despite most of the strains having the identical VNTR profile 42235.3
Recent work in San Francisco suggests that patients tend to become infected by strains of M. tuberculosis that have similar genotypes to those associated with their region of birth.5 The susceptibility to intracellular infections in people of South Asian ethnicity cannot be the only factor in the specific host-pathogen interaction with these strains as the combination of host factors with microbial determinants is also likely to play an important role in strain specificity with certain patient populations.
The molecular basis of pathogenicity, virulence and transmissibility in M. tuberculosis is not well known. The study of genetic variability within natural populations of pathogens can provide insight into their evolution and pathogenesis with comparative genomics a powerful tool providing important data that can be used to control transmission, and complements the more extensive studies of variation resulting from insertion sequences such as IS6110.6,7
Many new deleted regions have been found in the genome of different clinical strains of various members of the M. tuberculosis complex. Comparison of the genomes of M. tuberculosis H37Rv and M. bovis Bacilli Calmette-Guérin Pasteur (BCG) identified 14 sequences present in M. tuberculosis H37Rv but absent in M. bovis BCG. These were called regions of difference (RD1-14).8 Similarly, 6 regions were identified, that were absent from the M. tuberculosis H37Rv genome relative to other members of the M. tuberculosis complex: H37Rv relative deletions (RvD1-5) and M. tuberculosis specific deletion 1 (TbD1).8,9 Evolutionary studies have been undertaken to characterise genomic deletions and determine the probable evolution in a large number of different clinical isolates of M. tuberculosis9-11 and some of the genomic deletions of strains of M. tuberculosis studied were suggested as useful markers for defining the Beijing/W family of strains11 and different global lineages of M. tuberculosis.12
The aim of our study was to investigate the genomic characteristics in a set of strains which have been reported predominantly amongst the South Asian community in the UK, in contrast to other strains such as those of the Beijing family which have spread very widely around the world among ethnically mixed populations. We have used micro-array-based comparative genomics to analyse the distribution of the deleted regions around the genome of six clinical strains of M. tuberculosis defined as belonging to the South Asian group by associated VNTR profiles (42235, 02235 and 422344) in comparison to the previously reported CH strain. Examples of clades that are also common but have been isolated from ethnically diverse patients were also included.13,14 Our results indicate that strains of the South Asian group have a common evolutionary origin similar to the strain CH but distinct from members of the Beijing/W clade. The SA-strains appear to be included in the East-African-Indian lineage, defined by the RD750 deletion, which is frequently found in Southeast Asia.12 Recent results of Newton and collegues15 highlights the immunological relevance of this deleted region in the CH strain.
Materials and methods
Strains and growth conditions
Clinical strains 8088, 9375 and 9866 together with isolate 6947 were isolated from South Asian patients in Leeds and Bradford and were from the collection of D. Gascoyne-Binzi (Leeds Teaching Hospitals). The other clinical strains (0135, 2566, 3242) from South Asian patients and 2 strains of “Haarlem family”, 1339 and 7009, were from the collection of P.M. Hawkey (University of Birmingham). The VNTR profiles of these clinical strains are shown in Table 1. The South Indian clinical isolate (TMC120) ATCC 35811 was also included in this study with M. tuberculosis H37Rv used as the reference strain.
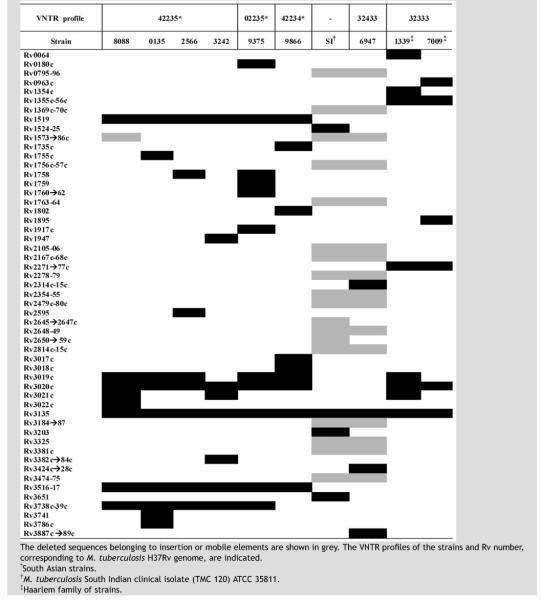
Table 1.
Distribution of deleted genes according to microarray experiments.
|
All the strains were grown at 37 °C in Dubos medium containing 0.05% Tween and supplemented with 0.04% (v/v) Dubos medium Albumin and 0.2% (v/v) glycerol.
DNA isolation and hybridization
Genomic DNA extraction16 and microarray hybridization procedures were performed as previously described,17,18 2–3 μg of DNA was labelled by incorporation of Cy3 and Cy5 dCTP (Amersham) during DNA polymerization. Purification of the final reactions was carried out using a MinElute PCR Purification Kit (Qiagen) before hybridization with the M. tuberculosis genomic microarray.
DNA microarray and analysis
Whole genome DNA microarrays of M. tuberculosis were kindly provided by the BμG@S group at St. George's Hospital Medical School, London. They were constructed by spotting PCR amplicons from partial sequences of the 3924 predicted ORFs of the sequenced strain M. tuberculosis H37Rv onto poly-l-lysine-coated glass microscope slides. Two hybridizations were done for each strain using a different dye each time. The hybridization was carried out at 65 °C overnight. The slides were washed after hybridisation and were scanned (GenePix 4000A, Axon Instruments), analysed by GenePix v.3.0 and normalized using GeneSpring v.6 (Silicon Genetics). The following normalizations were undertaken: after dye swap of the slides, the data were normalized per spot dividing by control channel using a cut off 0.01 and per chip normalizing to the 50th percentile. Genes were only considered to be deleted when the p-value was <0.05.
Confirmation of the deletions by PCR and sequencing
PCR was used to confirm the relevant deletions including: common deletions (Rv1519, Rv3516-17, and Rv3738c-39c) and selected polymorphic regions (Rv1917c, plcC-cut1 and Rv3017c-Rv3022c regions, and Rv3135). Primers were designed using PRIMER 3 software (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi) (Table 2). Ampli-Taq Gold polymerase (Roche-Applied Biosystems) or Expand Long Template PCR System (Roche-Applied Biosystems) was used depending on the genomic region. The oligonucleotides were designed to have annealing temperatures in the range 58–60 °C. The PCR conditions were carried out with 1 μM of primers and 1.5 mM of Mg2+ when AmpliTaq was used, and the following cycles were carried out: 95°/ 1 min; 35 cycles (95°/1 min, 58°/1 min and 72°/1 min) and 72°/10 min. When Expand Long Template PCR System was used, the PCR conditions were carried out with 1 μM of primers and 2 mM of Mg2+ and the cycles were: 94°/2 min; 10 cycles (95°/30 s, 58°/30 s and 68°/5–8 min), 15 cycles (95°/30 s, 58°/30 s and 68°/5–8 min+20 s/cycle) and 68°/7 min. Sequencing reactions were performed with BigDye Terminator v 1.1 (Applied Biosystems) and purified with columns (DyeEx 2.0 Spin Kit, Qiagen). The reactions were purified and analysed by capillary sequencing (Mega-BACE, Amersham). The sequences obtained were compared with TubercuList (http://genolist.pasteur.fr/TubercuList/index.html) and NCBI (http://www.ncbi.nlm.nih.gov).
Table 2.
PCR primers.
| Gene or genomic region affected by deletion | Primer | Sequence (5′–3′) |
|---|---|---|
| Rv1519 | CM1518F | TTCTCACCTGGTTGATCGTG |
| CM1520R | GTCCAGTAATCGTCGCCTTC | |
| CM1518Fb | CGTTTTGAGGATCCCAGTGT | |
| CM1520Rb | GGAATGCCAAATACCGTGAG | |
| RD3 region8 | RD3 intF | TTATCTTGGCGTTGACGATG |
| RD3 intR | CATATAAGGGTGCCCGCTAC | |
| plcD/cut1 and Rvd2 region | CM1755exF | CAGTTCGCTGATGTGACGAT |
| CM1758R | ATTGCCTCCGCTAGAACAGA | |
| ORF3Rvd3F | GATTGCGTTTGTTTTGCTGA | |
| ORF1Rvd2R | TGGTCGCAGTGTTTCCAATA | |
| Rv1917c | CM1916F | ATGACCCTGATCCACCTCTG |
| CM1917exR | TCGATTCCTAAAGCGGCTAA | |
| CM1917R | CCACCAGAGATCAACTC | |
| CM1917F | CGCCACTGTTGAAGAAG | |
| Rv3017c→22c region | CM3017exF | TGGCTGTTCGTCAGTAGGTG |
| CM3022exR | GGAACCTTCACTCGTACACCA | |
| CM3022R | TTGCAGAGTGCGGTGGGGTTT | |
| CM3019exF | CGCTAGCGGAATCAATGTG | |
| IS-R | AGTTTGGTCATCAGCCGTTC | |
| ISup | TACCTCCTCGATGAACCACC | |
| ISdown | CTCTACCAGTACTGCGGCGACG | |
| ISdw | CTGCCTACTACGCTCAAC | |
| Rv3135 | CM3135F | CATATCGCTTGACCCACAGA |
| CM3136R | TCGCTGTTTGCTGTGTCTTT | |
| Rv3516-17 | CM3515F | CCTTGTGTTTGTGGATCGTG |
| CM3518R | TTCGCATGTGTCTCAAGAGG | |
| CM3515Fc | ACCTTGTCGTCCTTTTGCAC | |
| CM3515Rb | CGAAATCCAAACAGCCACTT | |
| Rv3738c-39c | CM3737F | GAGTTCCTCGCCTCACCAT |
| CM3739exR | TCAGTTGACTGACCGGCTTT |
The primers were used to confirm the deletions and polymorphism by PCR and sequencing.
Results
This study used DNA microarray analysis of six clinical isolates of M. tuberculosis representing VNTR profiles commonly associated with people of South Asian origin in the UK to define common and unique deletions in this strain family. In a previous study, Gascoyne-Binzi and colleagues4 found the most predominant VNTR profiles in M. tuberculosis strains isolated from people of South Asian origin in West Yorkshire were 42235 and 02235. The SA-strains (VNTR profiles 42234 and 02235) were chosen since they allowed a genomic comparison of differing clones with similar VNTR profiles to 42235. To validate the comparison of strains causing disease in a mixed population, we also studied additional clinical strains representing VNTR profiles that have been associated with patients coming from different ethnic origins, clade X (local strain 6947 from a South Asian patient, VNTR profile 32433) and Haarlem strains (from a mixed population, VNTR profile 32333) in the UK. The South Indian clinical isolate (TMC120) ATCC 35811 was also included in the study. The deleted regions were compared to those published for the Beijing/W family,11 strain CH (the Leicester outbreak index isolate3) and those reported by Tsolaki and colleagues10
Microarray studies of the clinical strains
The total number of deletions in the SA-strains varied from 10 to 24 deletions. The median number of deleted genes was 12 (range 10–14) when mobile elements were not taken into consideration. Only one South Asian strain, 8088, had an RD3 region deletion (prophage phiRV1) accounting for 14 genes. Excluding strain 8088, strain 9375 had more deletions (14 genes) than any other SA-strain (Table 1). The South Indian clinical isolate (ATCC 35811) and strain 6947 showed a large number of deletions, with 51 and 58 deleted genes, respectively, but most of them (46 genes) were related to mobile elements (Table 1).
Deleted regions were confirmed by PCR and partial or total sequencing in the SA-strains: 8088, 9375 and 9866. The validation of the microarray results was extended to the rest of the strains of the SA group (0135, 2566 and 3242) to confirm commonly deleted genes.
Identification of deleted genomic regions
The deleted regions among SA-strains corresponded to genes encoding PE/PPE proteins, conserved hypothetical proteins, genes involved in intermediary and lipid metabolism or in cell wall/cell processes. Some of them were esx genes, which could contribute to interactions with the host.19-21 Partial deletions which might affect the expression of adjacent genes were detected such as Rv3017c-Rv3022A and Rv3738-Rv3739c regions.
According to studies by Gagneux and colleagues12 the deletion of Rv1519, designated by the large sequence polymorphism (LSP) RD750 included our SA-strains in the East-African-Indian lineage, the South Indian clinical isolate ATCC 35811 in the Indo-Oceanic lineage (deleted Rv3651 gene, LSP defined by RD239) which includes “ancestral” strains with the TbD1 genomic region which is deleted in “modern” strains of M. tuberculosis,9 and Haarlem strains and the local strain 6947 in the Euro-American lineage (deleted Rv2270-80 and Rv2313c-15c genomic regions, LSPs defined by RD182 and RD183, respectively).12
Common deletions
The analysis of the deletion events in the SA-strains investigated showed that these strains presented some common deletions present in all strains (Rv1519, Rv3516-Rv3517 and Rv3738c-Rv3739c). These were found to be indistinguishable after examination by PCR and sequencing to some of the deletions reported in the strain CH causing the outbreak in Leicester.3 The deletion Rv3738c-Rv3739c (PPE66-PPE67) was associated with all the SA-strains except 9866 (Tables 1 and 3). None of these common deletions have been described in Beijing/W strains11 or in Tsolaki studies.10
Table 3.
Comparison of the affected genes by deletions in the CH strain3 with those of the SA-strains.
| South Asian strains (East African Indian lineage) |
|||||||
|---|---|---|---|---|---|---|---|
| VNTR | CH3 42235 |
8088* 42235 |
0135 42235 |
2566 42235 |
3242 42235 |
9375* 02235 |
9866* 42234 |
| Deleted | |||||||
| genes | |||||||
| Rv0180c | − | + | + | + | + | − | + |
| Rv1519† | − | − | − | − | − | − | − |
| Rv1995-96 | − | + | + | + | + | + | + |
| Rv3017c | + | + | + | + | + | + | − |
| Rv3018c | + | + | + | + | + | + | − |
| Rv3019c | − | − | − | − | + | − | − |
| Rv3020c | − | − | − | − | − | − | − |
| Rv3021c | + | − | + | + | − | + | + |
| Rv3022c | + | − | + | + | − | + | + |
| Rv3135 | + | − | − | − | − | − | − |
| Rv3516-17 | − | − | − | − | − | − | − |
| Rv3738c-39c | − | − | − | − | − | − | + |
Polymorphic deletions
The SA-strains also showed additional deletions which distinguished them from the CH strain and from other strain families3,10 which gave rise to genetic variability in this specific group of strains (Tables 1 and 3). The sites of genetic variation among M. tuberculosis isolates could contribute to the putative differences in the phenotypic and biological properties of the strains.
Previous studies have shown polymorphism in the deletions of some PE/PPE genes22,23 and were found among the M. tuberculosis isolates studied here. Four polymorphic deletions in genes or regions were studied in these SA-strains: Rv3135 (PPE50), Rv1917c (PPE34), Rv1755c-Rv1758 (plcC-cut1) region, and Rv3017c-Rv3022c region (esx-QPPE46-PE27A-esxR-esxS-PPE47-PPE48).
Deletions in Rv3135 were observed in all of the strains examined in this study (Table 1). A characteristic partial deletion was identified in all of the SA strains studied and the deletion differed from those present in other strains (Figure 1A); this deletion was not found in the strain CH.3 The deletion in this gene for this group of strains was situated in part of the probe in the array so depending on the experiment the p-value obtained in this deletion was variable among SA-strains (p-value = 0.0271−0.168) but the validation by PCR and sequencing confirmed the identical partial deletion in all the strains. The deletion of 135 nucleotides in the sequence of the gene caused the loss of 45 amino acids in the sequence of the protein (Figure 1B).
Figure 1.

(A) Variation in the Rv3135 gene (PPE). The gene in H37Rv is shown with a grey box and the orientation of the gene is indicated by an arrow. The line with dots represents the deleted sequence in the strains compared to H37Rv. The new sequence in some of the strains is shown as a black box. (B) Sequence comparison of different Rv3135 variants: 1. H37Rv; 2. South Asian strains (clinical strains: 8088, 0135, 2566, 3242, 9375 and 9866); and 3. SI-ATCC; and 4. Haarlem-6947-Erdman (Acc. no. AE007137)-CDC1551 (Acc. no. Y17598). The deleted nucleotides are indicated by dashes. The new sequence with respect to H37Rv is in lower case. The deleted amino acids in SA-strains are indicated by a box in the sequence of H37Rv.
In order to confirm the variability of this gene, the deletion of Rv3135 was also confirmed in the two Haarlem strains, the clade X strain (6947) and the South Indian strain ATCC 35811, both of them with a p-value <0.05. The gene Rv3135 in the clade X strain 6947 and the two Haarlem strains had an insertion of 20 nucleotides at the 5′ end of the deleted genomic sequence. The sequences in these strains were 100% homologous with those in the Erdman and CDC1551 strains of M. tuberculosis (http://www.ncbi.nlm.nih.gov/). The clinical isolate South Indian ATCC 35811 showed a large deletion in Rv3135 with almost the entire region deleted including the promoter sequences.
Isolate 9375 (VNTR profile 02235) was the only isolate that possessed a partial deletion in Rv1917c (PPE34) that was detected by the microarray. Rv1917c encodes a surface-exposed protein, highly polymorphic in clinical isolates.24 Most of the polymorphism detected in this gene is caused by variation in the number of tandem DNA repeats in locus ETR-A (positions 2165204-2165611; www.sanger.ac.uk) of the VNTR profile.24 However, no evidence of the ETR-A region was detected in Rv1917c of the 02235 strain and the deleted region was replaced by IS6110. Subsequent analyses by PCR and sequencing also showed polymorphism in the SA-strains 8088 (VNTR profile 42235) and 9866 (VNTR profile 42234) with the interruption of the gene by IS6110 (Figure 2) that was not detected by microarray. The orientation of IS6110 in all 3 strains was opposite that of the direction of gene transcription. The insertion of IS6110 in the genome was also found in the strain CH but the deletion affecting this gene (Rv1917c) was not detected by the authors3,25 similar to SA-strains 8088 and 9866.
Figure 2.

Polymorphism in the Rv1917c gene: (A) Rv1917c region amplified by PCR. H, H37Rv; (a) clinical strain 8088; (b) clinical strain 9375 and (c) clinical strain 9866. (B) Graphic representation of the location of the insertion of IS6110 in Rv1917c. The gene is represented by a dark grey box and the IS6110 are indicated by a light grey box. The insertion points are indicated with lines. The scale in base pairs is indicated.
A notable microarray result was the detection of one polymorphic region from Rv3017c to Rv3022c among the three SA-strains. All three strains have at least esxR (Rv3019c) and esxS (Rv3020c) deleted, with the deletion of adjacent genes in this region dependant on the strain. This region contains genes with highly repetitive sequences; PE/PPEs like Rv3018c (PPE46), Rv3018A (PE27A), Rv3021c (PPE47), Rv3022c (PPE48) and Rv3022A (PE29), and three genes encoding Esat-6 like esxQ (Rv3017c), esxR (Rv3019c) and esxS (Rv3020c). One IS6110 sequence has been found inserted in the intergenic region of Rv3018c and Rv3019c of the CH strain25 but the deletion of Rv3019c-Rv3020c located by microarrays was not confirmed.3 The variability of the deletions in this genomic region has previously been shown in some M. tuberculosis and M. microti strains22 but this is the first report of replacement by an insertion element or transposase in this region (Figure 3) in closely related strains.
Figure 3.

Polymorphism in the region Rv3017c→Rv3022c. The orientation of the genes and the genes are shown. The insertion of IS6110 is indicated by a black arrow, the insertion of the transposase (second ORF of the IS6110) is represented by a striped arrow. The scale in base pairs is indicated.
To confirm deletions, the region from Rv1755c (plcD, phospholipase) to Rv1758 (cut1, cutinase) was analysed by PCR. These genes are interrupted by a single IS6110 element in H37Rv and correspond to the RvD2 region that is deleted in H37Rv but present in M. bovis, H37Ra and other clinical strains.8,26 Insertion and deletion events in this region result in notably high diversity among strains.27 The region Rvd2 is located downstream of the plcD gene (Rv1755c) and seems to be present in the strains 9375 and 9866 but not in 8088. In strains 8088 and 9866, the plcD (Rv1755c) and cut1 (Rv1758) genes were not interrupted by a copy of IS6110, whereas they are in the M. tuberculosis H37Rv genome (Figure 4). The plcD (Rv1755c) gene also seems to be interrupted by a copy of IS6110 in the genome of the CH strain.25 Rv1759c (wag22, PE-PGRS member) is only absent in isolate 9375 where the deletion extends to the adjacent genes, as detected by microarray analysis. The size of the PCR fragment seems to indicate the presence of the region Rvd2 in this strain and sequencing using the internal primers of Rvd2 confirmed its presence.
Figure 4.

Graphical representation of the Rv1755c-Rv1765c region: (A) the orientation of the genes is represented by arrows. The IS6110 in H37Rv are shown by black arrows. The deleted sequences in each strain were confirmed by PCR. The assumed substitution by Rvd2 in the strains 9375 and 9866 is shown in the figure. The asterisks indicate the truncated genes in H37Rv by IS6110 insertion. (B) Region amplified by PCR. H, H37Rv; (a) clinical strain 8088; (b) clinical strain 9866 and L, ladder.
The deletion or presence of prophages is variable in clinical strains of M. tuberculosis. Strain 8088 possessed a deletion in the RD3 region (phiRV1 prophage). This prophage is present in the genome of M. tuberculosis H37Rv but deleted in M. bovis BCG and some strains of M. tuberculosis.8 This deletion also appeared in the clade X strain 6947 and in the South Indian clinical isolate ATCC 35811 (our results) and has been described in the Beijing/W strains.11
Discussion
Our results demonstrate that the SA-strains tested have a common deletion profile which is different from the deletion profile in other clinical isolates3,10 by microarrays. The 42235 strains analysed in this study are members of the Gagneux East-African Indian LSP Group as defined by the deletion in RD750 (Rv1519)12 whereas in previous Spoligotyping studies on strains in India28 it can be extrapolated that 42235 strains are part of the CAS/Delhi Spoligotyping group with other strains assigned to the EAI or CAS/Delhi family. Definition of which VNTR profiles are members of the Gagneux Indo-Oceanic LSP Group are outside the scope of this study as only the SI isolate in this study possesses the defining RD239 (Rv3651) deletion. In the publication on Indian strains other VNTR profiles are predominantly members of the Spoligotyping EAI family but membership of the Gagneux Indo-Oceanic LSP Group has not been defined.
It is interesting to note that some of the deleted regions in the SA-strains studied here were also found in the index strain CH from the outbreak in Leicester in 2001, where the strains were isolated from patients that were predominantly of South Asia origin.3 The exact cause of the latter outbreak is still unknown but the particular environment in the school could be a reason for the pattern of the outbreak.
The deletion of LSP designated RD75012 involving the Rv1519 and Rv1520 genes, that encode conserved hypothetical proteins with unknown function, has been found to be involved in the persistence in human populations of the CH strain.15 An understanding of the pathogenicity of this group of strains might be aided by an in-depth study of genetic similarities and differences of an expanded collection of strains with similar epidemiological and genomic characteristics.
Our results show genetic alterations in some of the PE/PPE genes of SA-strains, in which major variability is seen in genes belonging to the PE/PPE family that represent a major portion of the M. tuberculosis H37Rv genome.19 The biological function of most of these proteins is unknown, however it has been suggested that they participate in antigenic variation or interfere with host immune responses.19-21 Moreover, genes of the esat-6 family,19 which are closely linked in the genome to genes encoding PE/PPEs,29 have been demonstrated to encode several immunodominant molecules that are strongly recognized by the immune system in different animal models of TB as well as by T-cells from humans exposed to M. tuberculosis.30 This family of proteins has been shown to be immunogenic in a human peripheral blood mononuclear cell model.31 Some Mycobacterium determinants, such as PE members, can influence the type of host response and can alter the composition of the infiltrates and the cellular composition of lesions.32 These potential antigens for host immunity may play an important role during host–pathogen interactions. The antigenic variability in the analysed strains could contribute, with a combination of other factors, to a special interaction with specific hosts, in this case people originating from South Asia.
Perhaps another major source of polymorphism among M. tuberculosis isolates seems to be defined by deletions in Rv3135, as the gene is larger in Beijing strains than in H37Rv.23,33 The commonly deleted regions of Beijing/W clade do not extend to this family of proteins.11 The Rv3135 gene coding for a PPE50 protein, which was found to be uniquely variable by Musser and colleagues23 could be a useful marker to differentiate epidemiologically related groups of strains, since there was a common deletion of this gene in the SA-strains. However, the full extent of variability within this region needs to be determined in a larger collection of isolates as this gene was not affected by deletion in the CH strain3 (Table 3). Despite having an unknown function, Rv3135 was considered an essential gene by Sassetti and colleagues34 and is closely linked to a two-component system (dosR/dosS) in the genome of H37Rv. However, the effect of this deletion in these strains remains unknown, as a defect in this PPE gene may be complemented by another gene absent in the reference strain H37Rv or by another PPE due to the functional similarities among these members. It would be interesting to investigate the putative role of this gene.
The presence of an insertion in Rv1917c (PPE34) could contribute to genetic variability, which is not only important in genomic evolution but also in the expression of these genes and in antigenic variation. The similar point of insertion of the IS6110 among our SA-strains and the CH strain suggest a hot spot in this gene. Polymorphisms in this gene are used as an epidemiological tool because it contains one of the more discriminatory loci (ETR-A) in VNTR typing.35
Deletion of esat-6-like genes might confer a selective advantage during certain stages of infection or transmission. The role provided by the selective pressure of the host's immunological system in the deletion of some genes is not well understood; however Musser and collaborators23 suggested there was limited selective pressure because of little genetic diversity in a large number of genes encoding essential antigens which are recognized by the host immune system. Recently the binding capability of the closely related Esat-6 proteins has been reported,36 but the loss of two of these genes (Rv3019c and Rv3020c) could decrease the opportunity for plasticity in the SA-strains. The strain 9866 (with VNTR 42234) is the only strain that shows deletion of three of the esx genes (Rv3017c, Rv3019c and Rv3020c); further analysis is necessary to understand the implication of this.
The insertion elements replacing deletions in SA-strains could modify the expression of adjacent PE/PPE genes in the region from Rv3017c to Rv3022c. Taking into account that IS6110 can upregulate downstream genes,37,38 the replacement event in this region demonstrates the important role of insertion elements in the evolution of the genome and in the contribution to phenotypic diversity. The presence of insertion elements could contribute to this variability, which is not only important in genomic evolution but also in expression of these genes and in antigenic variation. In M. microti the deletion MiD4 removes the esx genes Rv3019c and Rv3020c, but the strains which possess this deletion and RD5 (deletion in the plcC region) were still able to produce disease.39 However, the protective efficacy of Rv3019c and its suitability as promising candidate TB vaccine, showed by Hogarth and colleagues40 suggests that the deficiency of members of the esat-6 gene family may affect the immunological response in the patient.
In strain 8088, one IS6110 element was inserted in the plcC gene (data not shown) but this gene was not interrupted by insertion elements in 9375 or 9866 strains. Mutation of gene plcC confers attenuation,44 with further studies required to confirm this effect in SA-strains. In addition the Rvd2 region has been found in two of the SA-strains in this study. Located close to this region is Rv1759c (wag22), which encodes a member of the PE-PGRS glycine-rich protein family.41 This gene is expressed in tuberculosis infections and the protein is recognized by sera from patients and seems to have a biological role in the interaction of the bacillus with the host.41 Vera-Cabrera and collaborators42 have already shown that the phospholipase genomic region is a preferential locus for IS6110 transposition. However, in the SA-strains studied Rv1755c (plcD) is not interrupted by IS6110.
The polymorphic deletions detected in SA-strains in this study (Rv3135, Rv1917c, Rv1755c-Rv1758 and Rv3017c-Rv3022c) were polymorphic among previously analysed clinical strains of M. tuberculosis22-24,26 and are not specific to the SA-strains. Locus Rv3135 has been recently assigned to a group of large-sequence polymorphisms (LSP Group C) by Alland and colleagues43 in which it has been suggested that sequence alterations in this region occur under selective pressure.
This study indicates that both SA strains 8088 and 9375 (VNTRs 42235 and 02235, respectively) showed the same deletion in the Rv3738-Rv3739c region which was not present in the 9866 strain (VNTR profile 42234) (Table 1). This genomic deletion involved a partial removal of the Rv3737 gene, a probable conserved transmembrane protein close to a transcriptional regulatory protein (Rv3736) belonging to the AraC/XylS family.19 The implication that deletions of Rv3737, Rv3738 and Rv3739 affect expression of Rv3736 will need further studies.
We have demonstrated variability in the genome of strains of M. tuberculosis which are associated with patients of South Asian origin. The reasons for the host selection of this group of strains cannot be explained only by the deletion pattern, but must be a combination of events which are involved in the interaction with the cell, even by factors arising from the host. It would be interesting to investigate the relationship between the host interaction profile and some of the mycobacterial genes affected by the deletion or deletion-replacement events, especially with the recent results published by Newton and colleagues15 with the Rv1519 gene. Studies have shown the presence of deleted genes in a family of strains widespread around the world, with Tsolaki and collaborators11 showing that the Beijing/W strains lack the deletion in genes involved in antigenic variation such as PE/PPE members or Esat-6 like proteins.
Some of these genes, such as Rv3135 or Rv3738-Rv3739, are very closely linked to genes known to be important during infection. The high levels of variability in particular regions like Rv3017c-Rv3022c in the M. tuberculosis SA strains need further investigation in order to understand the differences in the pathogenesis, virulence and development of this prevalent global strain.
Acknowledgments
We acknowledge BμG@S (The Bacterial Microarray Group at St. George's Hospital Medical School) and particularly Philip Butcher, for the supply of the Mycobacterium tuberculosis microarray and advice.
Funding: This research was supported at NIMR by the award of a Marie Curie Fellowship of the European Community programme Human Potential under contract number HPMF-CT-2002-016015 to M. del Carmen Menéndez, and by the Medical Research Council. We acknowledge the Wellcome Trust for funding the multi-collaborative microbial pathogen microarray facility at St. George's under its Functional Genomics Resources Initiative.
Footnotes
Publisher's Disclaimer: This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author's institution, sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
Accession numbers: The nucleotide sequences of deleted regions of SA-strains are in the EMBL Data Bank with the accession numbers: AJ878456, AJ878457, AJ878458, AJ878459, AJ878460, AJ878461, AJ879166, AJ879167, AJ879168, AJ879169, AJ879170, AJ879171, AJ879172, AJ879173, AJ879174, AJ879175, AJ879176, AJ879177, AJ879178, AJ879179, AJ879180, and AJ879181.
Competing interests: None declared
Ethical approval: Not required
References
- 1.WHO Tuberculosis, fact sheet no. 104. World Health Organization. 2007. 〈 http://www.who.int/mediacentre/factsheets/fs104/en/ 〉 .
- 2.Ormerod LP, Charlett A, Gilham C, Darbyshire JH, Watson JM. Geographical distribution of tuberculosis notifications in national surveys of England and Wales in 1988 and 1993: report of the Public Health Laboratory Service/British Thoracic Society/Department of Health Collaborative Group. Thorax. 1998;53:176–81. doi: 10.1136/thx.53.3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajakumar K, Shafi J, Smith RJ, Stabler RA, Andrew PW, Modha D, et al. Use of genome level-informed PCR as a new investigational approach for analysis of outbreak-associated Mycobacterium tuberculosis isolates. J Clin Microbiol. 2004;42:1890–6. doi: 10.1128/JCM.42.5.1890-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gascoyne-Binzi DM, Barlow RE, Essex A, Gelletlie R, Khan MA, Hafiz S, et al. Predominant VNTR family of strains of Mycobacterium tuberculosis isolated from South Asian patients. Int J Tuberc Lung Dis. 2002;6:492–6. doi: 10.5588/09640569512995. [DOI] [PubMed] [Google Scholar]
- 5.Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci USA. 2004;101:4871–6. doi: 10.1073/pnas.0305627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Soolingen D, de Haas PE, Hermans PW, van Embden JD. DNA fingerprinting of Mycobacterium tuberculosis. Methods Enzymol. 1994;235:196–205. doi: 10.1016/0076-6879(94)35141-4. [DOI] [PubMed] [Google Scholar]
- 7.Shamputa IC, Rigouts L, Eyongeta LA, El Aila NA, van Deun A, Salim AH, et al. Genotypic and phenotypic heterogeneity among Mycobacterium tuberculosis isolates from pulmonary tuberculosis patients. J Clin Microbiol. 2004;42:5528–36. doi: 10.1128/JCM.42.12.5528-5536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon SV, Brosch R, Billault A, Garnier T, Eiglmeier K, Cole ST. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol Microbiol. 1999;32:643–55. doi: 10.1046/j.1365-2958.1999.01383.x. [DOI] [PubMed] [Google Scholar]
- 9.Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. 2002;99:3684–9. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsolaki AG, Hirsh AE, DeRiemer K, Enciso JA, Wong MZ, Hannan M, et al. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Pro Natl Acad Sci USA. 2004;101:4865–70. doi: 10.1073/pnas.0305634101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsolaki AG, Gagneux S, Pym AS, Goguet de la Salmoniere Y-OL, Kreiswirth BN, van Soolingen D, et al. Genomic deletions classify the Beijing/W strains as a distinct genetic lineage of Mycobacterium tuberculosis. J Clin Microbiol. 2005;43:3185–91. doi: 10.1128/JCM.43.7.3185-3191.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:2869–73. doi: 10.1073/pnas.0511240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soini H, Pan X, Amin A, Graviss EA, Siddiqu A, Musser JM. Characterization of Mycobacterium tuberculosis isolates from patients in Houston, Texas, by spoligotyping. J Clin Microbiol. 2000;38:669–76. doi: 10.1128/jcm.38.2.669-676.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kremer K, van Soolingen D, Frothingham R, Haas WH, Hermans PWM, Martín C, et al. Comparison of methods based on different molecular epidemiological markers for typing of Mycobacterium tuberculosis complex strains: interlaboratory study of discriminatory power and reproducibility. J Clin Microbiol. 1999;37:2607–18. doi: 10.1128/jcm.37.8.2607-2618.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newton SM, Smith RJ, Wilkinson KA, Nicol MP, Garton NJ, Staples KJ, et al. A deletion defining a common Asian lineage of Mycobacterium tuberculosis associates with immune subversion. Proc Natl Acad Sci USA. 2006;103:15594–8. doi: 10.1073/pnas.0604283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis EO, Sedgwick SG, Colston MJ. Novel structure of the recA locus of Mycobacterium tuberculosis implies processing of the gene product. J Bacteriol. 1991;173:5653–62. doi: 10.1128/jb.173.18.5653-5662.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, et al. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology. 2002;148:3129–38. doi: 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- 18.Frota CC, Hunt DM, Buxton RS, Rickman L, Hinds J, Kremer K, et al. Genome structure in the vole bacillus, Mycobacterium microti, a member of the Mycobacterium tuberculosis complex with a low virulence for humans. Microbiology. 2004;150:1519–27. doi: 10.1099/mic.0.26660-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 20.Banu S, Honore N, Saint-Joanis B, Philpott D, Prevost MC, Cole ST. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol Microbiol. 2002;44:9–19. doi: 10.1046/j.1365-2958.2002.02813.x. [DOI] [PubMed] [Google Scholar]
- 21.Brennan MJ, Delogu G, Chen YP, Bardarov S, Kriakov J, Alavi M, et al. Evidence that mycobacterial PE_PGRS proteins are cell surface constituents that influence interactions with other cells. Infect Immun. 2001;69:7326–33. doi: 10.1128/IAI.69.12.7326-7333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marmiesse M, Brodin P, Buchrieser C, Gutierrez C, Simoes N, Vincent V, et al. Macro-array and bioinformatic analyses reveal mycobacterial ‘core’ genes, variation in the ESAT-6 gene family and new phylogenetic markers for the Mycobacterium tuberculosis complex. Microbiology. 2004;150:483–96. doi: 10.1099/mic.0.26662-0. [DOI] [PubMed] [Google Scholar]
- 23.Musser JM, Amin A, Ramaswamy S. Negligible genetic diversity of Mycobacterium tuberculosis host immune system protein targets: evidence of limited selective pressure. Genetics. 2000;155:7–16. doi: 10.1093/genetics/155.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sampson SL, Lukey P, Warren RM, van Helden PD, Richardson M, Everett MJ. Expression, characterization and subcellular localization of the Mycobacterium tuberculosis PPE gene Rv1917c. Tuberculosis. 2001;81:305–17. doi: 10.1054/tube.2001.0304. [DOI] [PubMed] [Google Scholar]
- 25.Yesilkaya H, Forbes KJ, Shafi J, Smith R, Dale JW, Rajakumar K, et al. The genetic portrait of an outbreak strain. Tuberculosis. 2006;86:357–62. doi: 10.1016/j.tube.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 26.Brosch R, Philipp WJ, Stavropoulos E, Colston MJ, Cole ST, Gordon SV. Genomic analysis reveals variation between Mycobacterium tuberculosis H37Rv and the attenuated M. tuberculosis H37Ra strain. Infect Immun. 1999;67:5768–74. doi: 10.1128/iai.67.11.5768-5774.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho TB, Robertson BD, Taylor GM, Shaw RJ, Young DB. Comparison of Mycobacterium tuberculosis genomes reveals frequent deletions in a 20 kb variable region in clinical isolates. Yeast. 2000;17:272–82. doi: 10.1002/1097-0061(200012)17:4<272::AID-YEA48>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gutierrez MC, Ahmed N, Willery E, Narayanan S, Hasnain SE, Chauhan DS, et al. Predominance of ancestral lineages of Mycobacterium tuberculosis in India. Emerg Infect Dis. 2006;12:1367–74. doi: 10.3201/eid1209.050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tekaia F, Gordon SV, Garnier T, Brosch R, Barrell BG, Cole ST. Analysis of the proteome of Mycobacterium tuberculosis in silico. Tuberc Lung Dis. 1999;79:329–42. doi: 10.1054/tuld.1999.0220. [DOI] [PubMed] [Google Scholar]
- 30.Skjøt RLV, Brock I, Arend SM, Munk ME, Theisen M, Ottenhoff THM, et al. Epitope mapping of the immunodominant antigen TB10.4 and the two homologous proteins TB10.3 and TB12.9, which constitute a subfamily of the esat-6 gene family. Infect Immun. 2002;70:5446–53. doi: 10.1128/IAI.70.10.5446-5453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skeiky YA, Ovendale PJ, Jen S, Alderson MR, Dillon DC, Smith S, et al. T cell expression cloning of a Mycobacterium tuberculosis gene encoding a protective antigen associated with the early control of infection. J Immunol. 2000;165:7140–9. doi: 10.4049/jimmunol.165.12.7140. [DOI] [PubMed] [Google Scholar]
- 32.Cosma CL, Sherman DR, Ramakrishnan L. The secret lives of the pathogenic mycobacteria. Annu Rev Microbiol. 2003;57:641–76. doi: 10.1146/annurev.micro.57.030502.091033. [DOI] [PubMed] [Google Scholar]
- 33.Mokrousov I, Narvskaya O, Otten T, Vyazovaya A, Limeschenko E, Steklova L, et al. Phylogenetic reconstruction within Mycobacterium tuberculosis Beijing genotype in northwestern Russia. Res Microbiol. 2002;153:629–37. doi: 10.1016/s0923-2508(02)01374-8. [DOI] [PubMed] [Google Scholar]
- 34.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 35.Sola C, Filliol I, Legrand E, Lesjean S, Locht C, Supply P, et al. Genotyping of the Mycobacterium tuberculosis complex using MIRUs: association with VNTR and spoligotyping for molecular epidemiology and evolutionary genetics. Infect Genet Evol. 2003;3:125–33. doi: 10.1016/s1567-1348(03)00011-x. [DOI] [PubMed] [Google Scholar]
- 36.Lightbody KL, Renshaw PS, Collins ML, Wright RL, Hunt DM, Gordon SV, et al. Characterisation of complex formation between members of the Mycobacterium tuberculosis complex CFP-10/ESAT-6 protein family: towards an understanding of the rules governing complex formation and thereby functional flexibility. FEMS Microbiol Lett. 2004;238:255–62. doi: 10.1016/j.femsle.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 37.Safi H, Barnes PF, Lakey DL, Shams H, Samten B, Vankayalapati R, et al. IS6110 functions as a mobile, monocyte-activated promoter in Mycobacterium tuberculosis. Mol Microbiol. 2004;52:999–1012. doi: 10.1111/j.1365-2958.2004.04037.x. [DOI] [PubMed] [Google Scholar]
- 38.Soto CY, Menéndez MC, Pérez E, Samper S, Gómez AB, García MJ, et al. IS6110 mediates increased transcription of the phoP virulence gene in a multidrug-resistant clinical isolate responsible for tuberculosis outbreaks. J Clin Microbiol. 2004;42:212–9. doi: 10.1128/JCM.42.1.212-219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.García-Pelayo MC, Caimi KC, Inwald JK, Hinds J, Bigi F, Romano MI, et al. Microarray analysis of Mycobacterium microti reveals deletion of genes encoding PE-PPE proteins and ESAT-6 family antigens. Tuberculosis. 2004;84:159–66. doi: 10.1016/j.tube.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Hogarth PJ, Logan KE, Vordermeier HM, Singh M, Hewinson RG, Chambers MA. Protective immunity against Mycobacterium bovis induced by vaccination with Rv3109c—a member of the esat-6 gene family. Vaccine. 2005;23:2557–64. doi: 10.1016/j.vaccine.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 41.Espitia C, Laclette JP, Mondragón-Palomino M, Amador A, Campuzano J, Martens A, et al. The PE-PGRS glycine-rich proteins of Mycobacterium tuberculosis: a new family of fibronectin-binding proteins? Microbiology. 1999;145:3487–95. doi: 10.1099/00221287-145-12-3487. [DOI] [PubMed] [Google Scholar]
- 42.Vera-Cabrera L, Hernández-Vera MA, Welsh O, Johnson WM, Castro-Garza J. Phospholipase region of Mycobacterium tuberculosis is a preferential locus for IS6110 transposition. J Clin Microbiol. 2001;39:3499–504. doi: 10.1128/JCM.39.10.3499-3504.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alland D, Lacher DW, Hazbon MH, Motiwala AS, Qi W, Fleischmann RD, et al. Role of large sequence polymorphisms (LSPs) in generating genomic diversity among clinical isolates of Mycobacterium tuberculosis and the utility of LSPs in phylogenetic analysis. J Clin Microbiol. 2007;45:39–46. doi: 10.1128/JCM.02483-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raynaud C, Guilhot C, Rauzier J, Bordat Y, Pelicic V, Manganelli R, et al. Phospholipases C are involved in the virulence of Mycobacterium tuberculosis. Mol Microbiol. 2002;45:203–17. doi: 10.1046/j.1365-2958.2002.03009.x. [DOI] [PubMed] [Google Scholar]