

Abstract
Mitochondria are key organelles in cell life, whose dysfunction is associated with a variety of diseases. Their crucial role in intermediary metabolism and energy conversion makes them a preferred target in tissues, like the heart, where the energetic demands are very high. In the cardiomyocyte, the spatial organization of mitochondria favors their interaction with the sarcoplasmic reticulum, thereby offering a mechanism for Ca2+mediated crosstalk between these two organelles. Recently, the molecular basis for this interaction has started to be unraveled, and we are learning how ER-mitochondrial interactions are often exploited by death signals, like proapoptotic Bcl-2 family members, to amplify the cell death cascade. Here we will review our current understanding of the structural basis and the functional consequences of the close interaction between sarcoplasmic reticulum and mitochondria on cardiomyocyte function and death.
Keywords: Mitochondrial fusion, endoplasmic reticulum, calcium, bcl2 proteins, apoptosis, mitochondrial permeability transition
Introduction
Individually, sarcoplasmic reticulum and mitochondria direct critical functions essential to maintaining myocardial homeostasis and producing cardiac contraction. The sarcoplasmic reticulum (SR) is the major intra-cardiomyocyte storage depot for calcium. During excitation-contraction coupling, calcium influx through plasma membrane L-type voltage -gated calcium channels (LCC) stimulates calcium-induced, ryanodine receptor-mediated release of SR calcium into the cytosol, which induces myofibrillar contraction. In diastole the sarcoplasmic-reticulum calcium ATPase (SERCA) pumps calcium back into the SR, reversing the rise in cytosolic calcium. Functional relationships between cardiomyocyte membrane calcium channels, SR, and myofibrillar elements are facilitated by a highly organized sub-cellular architecture in which deep transverse tubular plasma membrane invaginations (t-tubules) enforce proximity of membrane LCC to intracellular SR located deep within sarcomeres. SR calcium reuptake in diastole in an energy intensive process, requiring large amounts of mitochondrially-generated ATP. The heart is therefore one of the most mitochondrial-rich organs, with mitochondria occupying approximately 30% of the volume of a ventricular cardiomyocyte. As with the SR, mitochondrial localization within cardiomyocytes is highly ordered.
Consistent with their importance to normal cardiac function, SR and mitochondrial dysfunction are associated with heart failure. Mitochondrial ATP production is impaired in heart failure, which predisposes to cardiomyocyte autophagy. Furthermore, mitochondria are the gatekeepers of apoptotic and necrotic cardiomyocyte death, which is increased in heart failure. SR dysfunction in heart failure is revealed by characteristic prolongation of the calcium transient, mechanistically attributable to impaired SR calcium reuptake. Accordingly, there are ongoing efforts to treat heart failure using gene therapy to increase SERCA or impair the function of its endogenous inhibitor protein, phospholamban.
Accumulating evidence indicates that in addition to their separate functions, SR-mitochondrial cross talk is critically important to cardiac health. Cardiomyocyte ATP production and cardiomyocyte calcium signaling require communications between mitochondrial and SR (reviewed in: 1–3). Mitochondrial-SR tethering may also contribute to maintaining the highly constrained myofibrillar mitochondrial sub-cellular organization that is characteristic of cardiac myocytes. However, too much of a good thing can have adverse consequences, and this review discusses emerging evidence favoring mitochondrial-SR interactions in programmed cardiac myocyte death. We address the role of mitochondria and ER in the control of cardiac myocyte death and examine the relevance of their interplay for amplification and modulation of noxious signals. Here, we use the term “programmed cell death” to include cell elimination through apoptosis, necrosis, or any other mechanism subsequent to an internal decision or death program. As is described below, apoptosis is distinguished by the essential role of Bcl2 family proteins that target mitochondrial outer membranes to initiate the process, and by its requirement for ATP to fuel the mechanisms of cell death 4–7. In contrast, necrosis is initiated by calcium signaling to the mitochondria, and results when ATP production is reversed and cellular ATP is insufficient to drive basal metabolism. Because of recent new findings in the field, we discuss in detail the functional implications of specialized, recently discovered protein organelle tethers to mitochondrial-mitochondrial and mitochondrial-ER signaling in the cardiomyocyte.
Mitochondria and cell death: an overview
Mitochondria not only ensure most of the ATP required by the cell, but are central for several signalling cascades. They modulate cytosolic Ca2+transients and participate in anabolic and catabolic reactions essential to normal turnover of essential cellular metabolites. As a byproduct of their respiratory activity, mitochondria produce reactive oxygen species that can act as second messengers. Finally, mitochondria are core components that amplify signals for programmed cell death 8. In mammalian cells, there are two major downstream apoptotic signalling pathways that culminate in the activation of caspases and are linked in some cells: the death receptor pathway and the mitochondrial pathway 9. Cleavage of substrates involved in maintenance of cytoskeletal and nuclear integrity, cell cycle progression, and DNA repair by caspase cysteine proteases results in the orderly demise of the cell. Mitochondria participate in the competent activation of caspases by releasing cytochrome c (the only soluble component of the respiratory chain) and additional apoptogenic factors (second mitochondrial activator of caspases, HtrA serine peptidase 2, endonuclease G and apoptosis inducing factor) from the mitochondrial intermembrane space into the cytosol. Cytochrome c in complex with Apaf -1 activates caspase-9 and other downstream caspases 4. This process is controlled by Bcl2 family proteins 10 and is accompanied by mitochondrial dysfunction and a distinct morphological derangement, mitochondrial fission 11.
The role and mechanism of mitochondrial dysfunction during programmed cell death has been extensively addressed in recent reviews. Here we will just remind the reader of two basic tenets of mitochondrial dysfunction that characterizes the initial stage of necrosis and late stages of apoptosis: Permeabilization of the inner mitochondrial membrane to protons, possibly as a consequence of the opening of the high conductance permeability transition pore (PTP); and blockage of electron flow along the respiratory chain as a consequence of cytochrome c dilution following its release into the cytoplasm or of feedback by activated caspases on individual complexes of the respiratory chain. The consequences of these events include decreased mitochondrial ATP production, loss of the driving force for Ca2+uptake, generation of reactive oxygen species, and ultimately, cell death. Thus, dysregulation of Ca2+signalling is one of the key events in the path to necrotic cell death. Additional evidence supports a role for Ca2+in the amplification of apoptosis signalling by a subset of death stimuli 12.
Regulation of mitochondrial morphology
An emerging and long neglected aspect of mitochondrial involvement in apoptotic death is the change in organelle shape, which has profound functional consequences on cell death progression. Mitochondria are morphologically complex. In certain cell types they are organized in networks of interconnected organelles, while in cardiac myocytes and other cells they exist primarily as individual entities 13 capable of undergoing dynamic fusion and fission reactions. From an ultrastructural perspective, mitochondria consist of an outer (OM) and an inner (IM) membrane that are further subdivided into an inner boundary portion and the cristae, bag -like folds of IM connected to OM via narrow tubular junctions 14. The ultrastructure and the reticular organization of the organelle are determined by mitochondria-shaping proteins that regulate the equilibrium between mitochondrial fusion and fission.
In mammalian cells, mitochondrial fission/division is regulated by Drp1 and Fis1 15–18. Drp1 is a cytosolic dynamin-related protein whose inhibition or downregulation result in a highly interconnected mitochondrial network. The same phenotype is caused by downregulation of Fis1 19, a 16 kDa integral protein of the outer mitochondrial membrane containing a single transmembrane domain and a tetratricopeptide repeat (TPR, involved in protein-protein interaction) domain facing the cytosol 19. Drp1 is recruited to mitochondria and directly or indirectly interacts with Fis1 to promote constriction of mitochondrial membranes 20(Figure 1). Translocation ofDrp1 to mitochondria occurs in response to cellular and mitochondrial cues, including mitochondrial dysfunction. Mitochondrial depolarization, associated or not with PTP opening, induces a sustained risein cytoplasmic Ca 2+that activates the phosphatase calcineurin, promoting dephosphorylation of conserved Ser637 of Drp1 21;Ser 637 dephosphorylated Drp1 translocates to mitochondria and promotes their fragmentation. Interestingly, Ser 637 is within a protein kinase A phosphorylation domain, functionally linking mitochondrial morphology to another crucial second messenger, cyclic AMP (cAMP) 22, 23. With mitochondrial dysfunction the phosphorylation status of Ser 637 is dominant over that of Ser 616 21, a target of cyclin dependent kinase 1 during mitosis 24, providing for distinct mechanisms by which the mitochondrial fission machinery can be regulated during cell stress and cell division.
Figure 1. Schematic depiction of the key proteins determining mitochondrial shapeby regulated fission and fusion.

Drp1, dynamin-related protein 1; Fis1, fission 1; Mfn1, mitofusin 1; Mfn2, mitofusin 2. MPTP = mitochondrial permeability transition pore.
The Ca2+-dependent kinase CAMKIa may also influence mitochondrial localization of Drp1 through phosphorylation of Ser 600 25, exerting a pro-fission effect similar to cyclin dependent kinase 1 phosphorylation of Ser 616. CAMKIa activation by Ca2+influx through voltage-dependent Ca2+channels functionally connects mitochondrial fission with transmembrane Ca2+cycling. Adrenergic stimulation by isoproterenol or physical activity is also associated with phosphorylation of Drp1 at Ser 600, linking Drp1 mitochondrial translocation to cardiac inotropy. However, mutants of Drp1 that mimic phosphorylation at Ser 600 do not constitutively localize to mitochondria, and Ca2+influx promotes their further mitochondrial localization, indicating that phosphorylation at other sites is more important in determining Drp1 translocation. Once Drp1 is localized at the mitochondria, it can be stabilized by SUMOylation 26, 27. In addition to Fis1 and Drp1, endophilin B1, a member of the endophilin family of fatty acid acyl transferases that participate in endocytosis, is involved in mitochondrial fission 28. The function of endophilin B1 may be similar to endophilin 1, i.e. lipid modification of dynamin I 29.
Fusion of mitochondrial OM is regulated by two dynamin -related GTPases –mitofusin (Mfn) 1 and 2 (Figure 1). These two highly homologous proteins share structures comprised of a terminal GTPase domain, two hydrophobic heptad repeats (HR), and two transmembrane domains that are inserted in to the mitochondrial OM 30. Notwithstanding their structural similarities, critical functional differences distinguish the two mitofusins: GTPase activity of Mfn1 is much higher, but affinity for GTP is lower 31; Mfn1 is responsible for mitochondria l-mitochondrial tethering through its anti-parallel interaction withHR2 of Mfn proteins from adjacent mitochondria 32; and in fibroblasts, Mfn1 (but not Mfn2)is required for fusion triggered by the inner membrane dynamin related protein Opa1 33.
The unique functional role of Mfn2 has been some what elusive to delineate. Mfn2can be retrieved in hetero-oligomers with Mfn1 and is suggested to participate in later steps of mitochondrial fusion 34. In addition, levels of Mfn2 correlate with oxidative metabolism of skeletal muscle 35 and with proliferation of vascular smooth muscle cells, where it sequester s the proto-oncogene Ras 36. Moreover, mutations in Mfn2 are associated with Charcot-Marie-Tooth type IIa peripheral neuropathy 37. Finally, as discussed in the final section below, Mfn2 exclusively controls the shape of ER(and in cardiac cells, presumably the sarcoplasmic reticulum)and tethers them to mitochondria 38. It is conceivable that the relative expression level of these mitochondrial fission and fusion proteins helps to dictate mitochondrial morphology in different cell types and during development, and contributes to the highly ordered mitochondrial organization seen in cardiomyocytes.
Mitochondrial morphology and cell death
During apoptosis mitochondria remodel their inner structure to allow the bulk of cytochrome c to be released from the cristae stores, a process christened “cristae remodeling” 39. Moreover, mitochondria undergo massive and reversible fragmentation prior to the release of cytochrome c 40, 41. Drp1 involvement in mitochondrial fragmentation was revealed by protection against cytochrome c release and cell death by a dominant negative Drp1 mutant 40. Likewise, Fis1 (the protein partner of Drp1 on mitochondrial membranes) overexpression induces cytochrome c release, while its ablation protects against cell death 17, 42. Importantly, mitochondrial fragmentation is the only known and essential involvement of mitochondria during developmental apoptosis of C. elegans 43.
Not only are mitochondrial fission proteins activated during apoptosis, but Mfn1-dependent fusion is impaired 44. This may occur either by functional inhibition of Mfn1 itself, or of its inner membrane partner Opa1 33. The latter possibility is supported by release of Opa1 together with cytochrome c early in the course of apoptosis 45. Also released from mitochondria is TIMMP8a, another intermembrane space protein involved in Drp1translocation from the cytosol to the organelle 46. Release of pro-fusion proteins and pro-fission cofactors maybe required to trigger mitochondrial fragmentation. However, it remains unclear as to why increased mitochondrial fission accelerates cell death. A unifying model implicates mitochondrial Ca2+uptake induced by the BH3 -only Bcl-2 family member BIK in cristae remodeling downstream of Drp1 activation47, 48. On the other hand, mitochondrial fission is not invariably associated with cell death. For example, Bax, Bak doubly deficient cells are resistant to apoptosis induced by stimuli that recruit the mitochondrial pathway, yet their mitochondria fragment following treatment with the same stimuli. In addition, a single conservative point mutation in the short inter-membrane space stretch of Fis1 dissociates its pro-fission from its pro-death activity 49. Conversely, fission by Drp1 can even protect from death induced by Ca2+-dependent apoptotic stimuli that require mitochondria to amplify deadly waves of this second messenger 50. Taken together, accumulated data show that excessive mitochondrial fission is almost always associated with cell death, but at this time the data are not sufficient to conclude that mitochondrial fission is essential for mitochondrial apoptosis.
Calcium, the link between cardiac contraction and the mitochondrial pathway of cardiomyocyte death
Ca2+is a versatile second messenger whose intracellular concentration impacts on a number of integrated cellular functions, including regulation of proliferation and gene transcription, stimulation of ATP production, and muscle contraction. It is therefore not surprising that persistent or very high elevations of intracellular Ca2+are detrimental, and that cells expend resources to regulate calcium(if they fail to do so cell death could be a just around the corner). Tight regulation of Ca2+may be even more important in the heart, where phasic high amplitude Ca2+transients are essential for normal minute -by-minute organ function (i.e. contraction). In heart failure, sarcoplasmic reticular (SR) calcium re-uptake is characteristically delayed and SR calcium stores are typically diminished. Although there may be a role for ryanodine receptor leakage in heart failure 51, abnormal calcium cycling is widely attributed to decreased SERCA expression or chronic SERCA inhibition by phospholamban 52. For this reason, a number of experimental and clinical efforts have aimed to improve cardiac function in heart failure by restoring SR calcium stores 53. Some mouse models of heart failure have been “rescued” by dis-inhibiting SERCA through genetic ablation of phospholamban 54. However, there is increasing evidence that enhancing SR calcium cycling and augmenting cardiac myocyte contraction in heart failure can also induce long-term increases in programmed cardiac myocyte death.
Song, et al 55 ablated phospholamban in two murine genetic cardiomyopathy models having calcium cycling abnormalities, the Gαq transgenic mouse 56 and the MYBP -C mutant mouse 57. As anticipated, SERCA function was improved by phospholamban ablation; cardiomyocyte calcium cycling and contraction were improved, but there was no corresponding improvement in the in vivo cardiomyopathy. In a recent study of similar design, Zhang et al used phospholamban ablation to correct SR calcium cycling defects in Ca++/calmodulin-dependent protein kinase (CaMK)II transgenic mice, but likewise found that the cardiomyopathy phenotype and signs of heart failure were made worse 58. There are a number of other reports that phospholamban ablation exacerbates murine heart failure and/or induces cardiac myocyte apoptosis 59–61, and it appears increasing SR calcium content by other mechanisms can have a similar detrimental effect on cardiac myocyte viability and heart failure 62–64. Taken together, the accumulated data support a direct relationship between SR calcium levels and programmed cardiac myocyte death, and suggest that increasing SR calcium beyond normal physiological limits can contribute to in vivo heart failure.
The mitochondrial permeability transition in heart failure
Cardiac mitochondria play a role in cardiac myocyte calcium dynamics by interacting with the SR to regulate beat-to-beat phasic calcium cycling 65 (reviewed in 2). However, the relative contribution of mitochondrial calcium to the phasic calcium transient is small, approximately 1% 66, and the dominant function of mitochondria is ATP production via oxidative phosphorylation. Indeed, mitochondria are the source of ~90% of ATP used for cardiac contraction. As noted above, however, mitochondria regulate critical aspects of cardiac function in addition to energy metabolism and calcium homeostasis: They are the source of reactive oxygen species (ROS) that can either stimulate cell signaling pathways or damage cardiac proteins, and are central regulators of cardiac myocyte apoptotic and necrotic death 67–70.
One of the key aspects of mitochondrial involvement in cell death is control of the PTP. The PTP is a non -selective pore in the mitochondrial inner membrane that opens in response to greatly increased calcium concentrations, initially described in 1979 by Haworth and Hunter 71. PTP opening permits transmembrane diffusion of molecules smaller than 1.5 kDa down their concentration gradients. Proton diffusion through PTP dissipates the normal pH gradient and membrane potential (Δψm) that are essential components of the proton motive force used by the ATP synthase for ATP production 72. Consequently, mitochondria try to compensate, maintaining the electrochemical gradient by reversing the ATP synthase. The net effect is that mitochondria begin consuming ATP. If a sufficient proportion of cardiac myocyte mitochondria undergo this permeability transition, the loss of ATP can be sufficient to initiate necrosis. On the other hand, if cytosolic ATP levels are sufficient to maintain minimal cell functions and to fuel apoptosis effectors, opening of the PTP can activate apoptosis signaling as a consequence of cytochrome c released from ruptured mitochondria and/or cristae remodeling.
Opening the PTP is only one of the Ca2+dependent mitochondrial events. Increased beat-to-beat sarcoplasmic calcium concentrations are reflected by phasic changes in mitochondrial calcium that activate enzymatic production of NADH for the electron transport chain, stimulating ADP production 1. In cardiac ischemia, calcium overloading combines with ROS production and increased phosphate concentration to induce PTP opening 73, although findings have even suggested that, in situ, the PTP is relatively calcium insensitive because of stabilization by cytosolic factors 74. Thus, although a genetic analysis has proven that calcium triggers apoptosis by opening the PTP 12, calcium may be only one of several factors that interact to initiate mitochondrial permeability transition.
The exact structural components of the PTP are not known at this time. For a detailed discussion of the current state of the field and how individual PTP components interact and relate to the heart, the interested reviewer is referred to two excellent recent reviews 75, 76. Briefly, the core PTP is believed to consist of a multi-membrane protein complex comprised of the voltage-dependent anion channel (VDAC)in the OM 77–79 and the adenine nucleotide translocator (ANT)in the IM 80–83, which are regulated by cyclophilin D (CyP-D)in the matrix. However, this model has been convincingly refuted by the means of genetic models and techniques that excluded a role for VDAC 84 and for ANT 85 as essential functional components of the PTP. On the other hand, they could both play regulatory effects, as ANT appears to function as phosphate- and calcium-sensitive PTP regulator.
CyP-D was first identified as an ANT-binding protein that mediated the inhibitory effect of cyclosporin A on PTP opening 86. It is encoded by the peptidyl-prolyl cis-trans isomerase (Ppif) gene, and three independent groups have reported that its genetic ablation is sufficient to eliminate the mitochondrial permeability transition, without affecting Bcl2 factor-mediated outer membrane permeabilization and intrinsic pathway apoptosis 87–89. Accordingly, genetic Ppif ablation has been used to demonstrate the role of PTP and death in mouse models of Alzheimer’s disease, muscular dystrophy, diabetes mellitus, and heart failure 90–93. Notwithstanding these compelling results, it has also been suggested that CyP-D is a PTP regulator, rather than an essential component, and that the striking effects observed with its ablation are simply the consequence of PTP inhibition by inorganic phosphate 94, 95.
Bcl-2 protein Nix as a mediator of SR-mitochondrial cross-talk in programmed cardiac myocyte death
As described above, mitochondria are central regulators, the so-called “gatekeepers”, of programmed cell death 96. In part, this is because of PTP opening, and in part because mitochondria are the targets for many actions of Bcl-2 family proteins that regulate apoptosis. Bcl-2 family proteins are classified according to their structural features and function in cell death 97. Briefly, the “multidomain” proapoptotic proteins, like Bax and Bak, are pore-forming proteins that permeabilize mitochondrial OM, leading to cytochrome c release. Pore-formation by Bax and Bak 98, 99 is facilitated by pro -apoptotic BH3 domain-only factors 100, including cardiac-expressed BNip3 and Nix. BH3-only factors can hetero-dimerize with anti-apoptotic factors like Bcl-2 and Bcl-XL, preventing OM pore formation by Bax and Bak. Dynamic regulation of Bax, Bak, pro-apoptotic, and anti-apoptotic Bcl-2 family proteins is characteristic of heart failure and has been linked with programmed cardiac myocyte death (reviewed in 101). Particularly detailed information is available for transcriptional upregulation of Nix in cardiac hypertrophy 102, 103. The mechanisms by which Nix-induced SR-mitochondrial cross-talk contributes to the progression from non-failing hypertrophy to dilated cardiomyopathy through the programmed apoptotic and necrotic loss of cardiac myocytes have only recently been fully elucidated, and point to a key role for Ca2+transfer between SR and mitochondria in Nix -dependent cell death.
Having found by microarray analysis that Nix transcripts are increased in cardiac hypertrophy 102, the Dorn group used transgenesis to determine the in vivo consequences of its upregulation, independent of hypertrophy per se or of any stimulus thereof. Forced cardiac myocyte Nix expression with the conventional α-myosin heavy chain (αMHC, MYH6) promoter produced mice that were normal at birth, but that died of rapidly progressive heart failure after one week. TUNEL staining showed massive cardiomyocyte apoptosis with apoptotic indices of 15–20% 102. A follow-up study using conditional cardiac-specific Nix overexpressing mice revealed synergy between Nix and surgical pressure overloading for inducing apoptotic heart failure 104, suggesting that Nix can coordinate transcriptional and physiological cues leading to programmed cardiomyocyte death. Accordingly, we hypothesized that elimination of Nix might retard the progression of cardiac hypertrophy to heart failure by preventing programmed cardiac myocyte death. Our approach was to create Nix gene knockout mice, subject them to pressure overload or Gq-mediated hypertrophy (in which Nix is normally transcriptionally upregulated), and compare cardiomyocyte apoptosis, ventricular remodeling, and cardiac function between surgically or genetically modeled mice with and without Nix.
Because germ-line Nix ablation produces a striking hematological phenotype that could potentially interfere with our cardiac studies 105–107, we employed a Nkx-2.5 Cre-lox strategy to generate mice in which Nix was deleted only in cardiac myocytes. The cardiac -specific Nix knockout mice then underwent surgical transverse aortic banding to create achronic pressure overload. Whereas pressure overloaded wild-type hearts developed the typical cardiomyocyte apoptosis, ventricular dilatation and cardiac failure, pressure overloaded cardiac Nix knockout mice exhibited only half as much cardiomyocyte apoptosis (TUNEL positivity), less late replacement fibrosis, almost no ventricular dilation and wall thinning, and had preserved systolic function 108. Germ-line Nix ablation provided a similar rescue for the apoptotic peripartum cardiomyopathy that is characteristic of mice with cardiomyocyte-specific overexpression of the alpha subunit of heterotrimeric Gq 108, 109. These findings established Nix as a critical inducible factor mediating programmed cardiac myocyte death in pressure overload hypertrophy, and linked programmed loss of cardiac myocytes with ventricular remodeling and progression to heart failure. Recently, we have better defined the mechanism for in vivo Nix-mediated cardiomyocyte death.
Like all pro-apoptotic Bcl-2 factors, Nix induces caspase-dependent apoptosis by stimulating mitochondrial OM permeabilization. Nix localizes to mitochondria, induces cytochrome c release, caspase activation and oligonucleosomal DNA degradation 102. However, we recently observed that only ~80% of transfected Nix localizes to mitochondria, and that the remainder is localized to ER and SR reticular structures, depending upon cell type 110. Furthermore, transcriptional upregulation of Nix in pressure overload hypertrophy preferential increases SR-(not mitochondrial -) associated Nix. We also observed that ER/SR-localized Nix increased cardiomyocyte SR calcium stores, as previously described for Bax 12, 111, while Nix ablation decreased SR calcium. The concordance between Nix ablation preventing Gq-mediated hypertrophy decompensation and cardiomyocyte apoptosis, 108 and also decreasing cardiac myocyte SR calcium stores 110 suggested a direct relationship between SR calcium levels and the programmed cardiomyocyte death that produced cardiomyopathy. We tested this possibility by concomitantly ablating phospholamban (PLB) and Nix in mice, superimposing the cardiac-specific Gq transgene, and then determining the effects on programmed cardiomyocyte death and cardiomyopathy development in the peripartum state. As anticipated, ablation of phospholamban (that inhibits SERCA -mediated SR calcium uptake) normalized cardiac myocyte SR calcium stores and improved excitation-contraction coupling. Whereas Nix ablation had protected hearts against apoptosis, thereby enhancing ventricular function and abrogating peripartum lethality in Gq mice 108, 110, Nix null/Gq transgenic mice in whom SR calcium stores were normalized through PLB ablation developed an exaggerated cardiomyopathy and increased mortality. These results established a causal link between SR calcium levels, programmed cardiac myocyte elimination, and in vivo cardiomyopathy mediated by pro-apoptotic Bcl-2 family member, Nix.
A number of recent studies have revealed pathophysiological involvement of SR -mitochondrial calcium transfer in heart failure not primarily caused by proapoptotic Bcl -2 family members. In the first such report, Nakayama et al 112 interrogated the mechanism for cardiac myocyte necrosis and heart failure induced by L -type calcium channel-mediated cardiac myocyte calcium overloading. Whereas overexpression of anti-apoptotic Bcl2 failed to rescue the cardiomyopathy of L-type calcium channel overexpression, ablating CyP-D (and thus preventing the mitochondrial permeability transition) normalized cardiac structure, function, and survival.
The second report came from the Heller Brown laboratory, which had previously described cardiomyopathic effects of activating (through overexpression) cardiomyocyte calcium/calmodulin kinase IIδc (CaMKIIδc) signaling 113. These investigators observed that SR calcium levels were decreased in the CaMKII transgenic mice, which they attributed to ryanodine receptor leak. In a follow-up study 58, the same group used the same phospholamban ablation approach described above as a means to restore SR calcium in the hopes that contractile function would be enhanced 54. Although PLB ablation normalized SR calcium levels in CaMKII transgenic mice, increased SR calcium was associated with worsening (rather than the anticipated improvement) of the cardiomyopathy and increased mortality from heart failure. Elegant cell-based studies demonstrated a link between increased SR calcium stores, increased SR calcium export independent of normal excitation-contraction coupling (SR calcium “sparks” and leakage), and increased mitochondrial calcium loading. PTP-induced cell death was linked to SR-mitochondrial calcium transport by in vivo studies where CaMKII transgenic/PLB knockout cardiac myocytes were rescued from programmed death by cyclosporin A (CyP-D and PTP inhibitor) or RU-360 (mitochondrial calcium uniporter inhibitor).
These studies and other data indicated that calcium transport from sarcoplasmic reticulum to mitochondria through junctional “calcium hot-spots” 114 can be a potent stimulus for programmed cardiac myocyte death. We hypothesized that reticular-mitochondrial cross-talk stimulated by ER/SR-localized Nix might be inducing the necrotic pathway to programmed cell death. If this were the case, then cardiomyocyte “apoptosis” we and others had reported based on evidence of cytochrome c release, caspase activation, and TUNEL positivity might in part be a collateral effect of mitochondrial rupture after PTP opening. To test this notion we created and recombinantly expressed in HEK293 cells 110 or Nix null embryonic fibroblasts 93 mitochondria -specific and ER/SR-specific Nix mutants. Mitochondrial-directed Nix produced cell death associated with caspase activation, but with no net decrease in Δψm, i.e. apoptosis. In contrast, reticular-directed Nix produced cell death preceded by PTP opening, but associated with caspase activation that we attributed to cytochrome c release after outer mitochondrial membrane rupture. Furthermore, we found that pharmacological (cyclosporin A) or genetic (CyP-D, Ppif ablation) inhibition of the PTP prevented cell death induced by reticular Nix, but not by mitochondrial Nix 93. Finally, while death mediated by mitochondrial -directed Nix required Bax or Bak, reticular-directed Nix induced cell death independently of the multidomain pro-apoptotics. We interpreted these results as evidence that mitochondrial-directed Nix stimulates conventional intrinsic pathway apoptosis, whereas reticular-directed Nix induces programmed cell necrosis by increasing reticular calcium concentration and delivery to mitochondria, thereby promoting the mitochondrial permeability pore transition(Figure 2).
Figure 2. Activation of cell apoptosis and necrosis by Nix, and role of organelle localization.
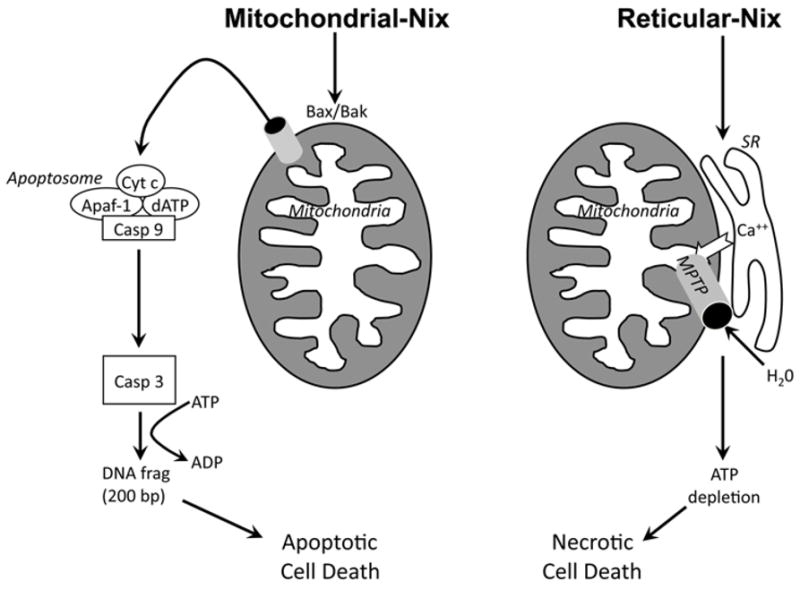
On left is Bax/Bak-dependent apoptosis mediated through mitochondrial Nix; right is mitochondrial permeability transition pore (MPTP)-dependent necrosis mediated through reticular Nix.
Recently, we established the in vivo relevance of Nix -mediated activation of dual apoptotic and necrotic programmed cell death pathways to in vivo cardiomyocyte death and ventricular remodeling 93. Conditional, cardiac-specific transgenic expression of wild-type, mitochondrial-directed, or SR-directed Nix induced similar dilated cardiomyopathy phenotypes associated with similar levels of programmed cardiac myocyte death. However, in vivo cardiac myocyte necrosis visualized by anti-complement 9 staining of the membrane attack complex occurred only in mouse hearts expressing Nix that was localized all or in part to the SR(i.e. wild-type Nix or its SR-directed mutant), whereas TUNEL labeling occurred with mitochondrial- or SR-directed Nix. Likewise, ultrastructural evidence for cardiomyocyte PTP opening (mitochondrial swelling, matrix degeneration, and outer membrane disruption) was found only in hearts expressing an SR –localizing Nix. W e demonstrated causality for PTP opening in SR -directed Nix-mediated cardiac myocyte death through concomitant ablation of Ppif(encoding CyP-D) with overexpression of Nix or its organelle-directed mutants. CyP-D ablation rescued the cardiomyopathy and cardiomyocyte death only in SR-directed Nix expressing mice, completely eliminated complement 9 staining, and normalized mitochondrial ultrastructure. These findings show that an important aspect of Nix-mediated cell death is programmed necrosis mediated by SR -mitochondrial crosstalk that is a consequence of SR-localized Nix. Since we had previously observed that Nix which is endogenously upregulated during cardiac hypertrophy preferentially localizes to the SR 110, we concluded that MTPT opening stimulated by SR-mitochondrial calcium cross-talk may play a greater role than previously suspected in hypertrophy decompensation and the progression to overt heart failure. Consistent with this notion are a number of recent reports that otherwise implicate cardiac myocyte or SR calcium levels and PTP opening in cardiac injury and heart failure progression 115–117.
Mitofusins in SR -mitochondrial calcium signaling
The implication of SR-mitochondrial calcium transfer through putative high calcium microdomains by Nix and other factors, 110 and observations that a rigidly defined cardiac myocyte subcellular architecture and “mitochondrial packing” are essential to cardiac contractility, 118 suppor t a specific requirement for physical interactions between cardiac SR and mitochondria, suggesting a specific mechanism for mitochondrial-mitochondrial and mitochondrial-ER/SR tethering.
Interactions between organelles are key to spatial organization of cell signaling. The example of mitochondria and ER/SR is prototypical, and is determined by the biophysical properties of the mitochondrial Ca2+uniporter that is responsible for Ca2+uptake in the organelle. This mitochondrial IM channel has a very low affinity for Ca2+, and therefore requires high concentrations of the ion that are not normally achieved in the bulk of cytoplasm following release of Ca2+from the RyR or inositol phosphate receptor (IP3R) 119. However, seminal studies by Rizzuto and Pozzan revealed that following release of Ca2+by the IP3R, mitochondria do take it up 120, leading to elaboration of the theory that Ca2+microdomains, hot spots of high [Ca 2+], are present at the interface between ER and the mitochondria 121. This theory was corroborated by observations that the two organelles are in close proximity in a variety of cell types, 122 and that release of Ca2+from ER triggers activation of mitochondrial dehydrogenases 123 that prolong ATP production 124. Additional functions of the ER-mitochondria juxtaposition include the transfer of lipids between the former and the latter, where most biosynthethic pathways are lacking 125. Accumating evidencs suggests that lipid trafficking betweem ER and mitochondria may have a role in PTP-independent cell death 126, generation of autophagosomal membranes 127 and in the above noted pathways to cell death that require inter-oraganelle Ca2+transfer 12. Artificial zippers between mitochondria and ER further substantiated that cells require this physical interaction for ATP production and death by selected stimuli 128, 129.
Despite the importance of ER/SR-mitochondria connection in cellular pathophysiology, the nature of the physical tether was only recently elucidated. Earlier reports had uncovered roles for the sorting protein PACS2 130 and for VDAC and IP3 receptor together with the chaperone grp75 131 in the interaction. However, these proteins did not appear to constitute the physical bridge between the organelles, rather being crucial regulators of the interaction (Figure 3). Trans-organellar bridges are formed by another protein whose primary role lies in the modulation of mitochondrial fusion, Mfn2 38. Cytochemical and biochemical analyses revealed that the bulk of Mfn2 is retrieved in mitochondria-associated membranes (MAMs), patches of ER attached to the mitochondrial OM. In addition, a relevant (approx7% of the total) fraction of Mfn2 resides in the endoplasmic reticulum, and Mfn2 ablation alters the structure of this organelle, causing its fragmentation, as substantiated by fluorescence recovery after photobleaching experiments. Selective reconstitution of the endoplasmic reticulum pool of Mfn2 in Mfn2−/−cells completely restored the reticular nature of the organelle, suggesting a role for t he ER-Mfn2 in the modulation of its shape. Finally, cells lacking Mfn2 display an increased average distance between ER and mitochondria, consistent with the localization of the protein in the MAMs. Selective correction of ER and mitochondria using targeted chimeras of Mfn2 and Mfn1 in cells lacking both Mfn1 and Mfn2 supports a model in which ER-Mfn2 engages in homo or hetero interactions with mitochondrial Mfn1 and/or Mfn2 to tether the two organelles (Figure 3). Further support to this model is given by an in vitro assay showing the requirement of ER Mfn2 for co-sedimentation of ER and mitochondria. In fact, cross-linkable, trans complexes of ER-Mfn2 with mitochondrial Mfn1 and Mfn2 exist and are further supported by co-immunoprecipitation assays. The lack of interaction between the two organelles has a major impact on Ca2+transfer between them, and cells without Mfn2 are perfect tools to verify the Ca 2+ microdomains theory. Release of Ca2+from the ER is coupled with a reduced mitochondrial Ca 2+ uptake, whose rate is considerably slower in Mfn2−/− cells, compared to wild -type. This is not the consequence of impaired mitochondrial Ca2+uptake, which is unaffected by Mfn2 ablation, but reflects increased distance between the organelles and the resulting limited generation of Ca2+ microdomains. Thus, Mfn2−/− cells are lack of function models that, after 15 years, have provided experimental proof for Ca2+microdomains postulated by Rizzuto and Pozzan. Further evidence supports a role for Mfn2 in tissues where the ER-mitochondrial coupling is crucial, like the heart. For example, in cardiomyocytes, during oxidative stress induced apoptosis, upregulation of Mfn2 levels seem to play a crucial role, in a way independent from the effect of Mfn2 on mitochondrial dynamics 132. During hypertrophy induced by pressure overload, Mfn2 seems to be conversely downregulated in what looks like a compensatory mechanism orchestrated by PPARδ and PGC1β 133, 134.
Figure 3.
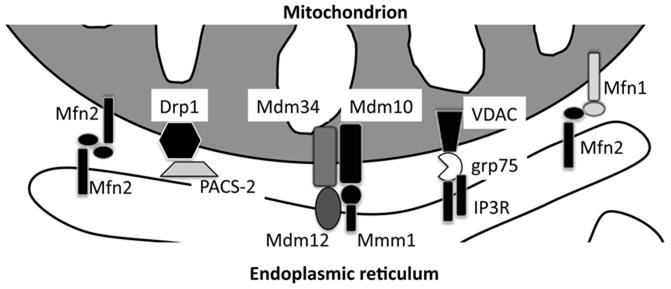
Schematic depiction of proteins identified to date in yeast and mammals that regulate the juxtaposition between mitochondria and ER.
Recently, a multiprotein complex has been identified using a genetic screen in yeast as responsible for ER-mitochondrial tether. The yeast homologue of Mfn, Fzo1p, was not part of this multiprotein complex that comprised two mitochondrial OM integral proteins, Mdm10 and Mdm34, Mdm12, a cytosolic protein, and Mmm1, which can be retrieved in mitochondria or in the ER (Figure 3) 135. The deletion strains of these components display growth defects that can be reconstituted using the artificial ER-mitochondrial tether invented by Hajnockzy and colleagues, pointing to a crucial role for these proteins in the establishment of ER-mitochondria interaction. In yeast, this is likely to impact only on phospholipid transfer between the two organelles, since ER is not the main Ca2+stores and yeast mitochondria do not uptake Ca 2+, as they lack the uniporter. That this complex is a specialized yeast feature would be confirmed by the lack of higher eukaryotes orthologues for these proteins, mitigated by the retrieval of conserved sequences similar to syanptotagmin in at least two components of this complex 136. Further research will uncover the orthologues of this tethering complex identified in yeast, extending its importance to Ca2+signaling and apoptosis.
In conclusion, the crosstalk between ER and mitochondria is a key feature of the spatial organization of cell signaling. This strict relationship is key to insure proper mitochondrial responses to Ca2+release from the ER, but can also lead to the amplification of cardiac myocyte death by a plethora of different stimuli.
Acknowledgments
Sources of Funding: This work was supported by NIH/NHLBI, SNF, Telethon Italy, and Oncosuisse.
Non-standard Abbreviations and Acronyms
- LCC
L-type calcium channel
- ER
Endoplasmic reticulum
- SR
Sarcoplasmic reticulum
- SERCA
Sarcoplasmic reticular calcium ATPase
- ATP
Adenosine tri-phosphate
- PTP
Permeability transition pore
- TPR
Tetratricopeptide repeat
- OM
Mitochondrial outer membrane
- IM
Mitochondrial inner membrane
- CAMK
Ca++/calmodulin-dependent protein kinase
- HR
Heptad repeats
- mfn
Mitofusin
- Opa1
Optic atrophy 1
- Drp1
Dynamin related protein 1
- CyP-D
Cyclophilin D
- VDAC
voltage-dependentanion channel
- ANT
adenine nucleotide translocator
- αMHCMYH6
α-myosin heavy chain
- PLN
Phospholamban
- Δψm
Mitochondrial inner membrane electrical potential
- MAM
Mitochondrial-associated membrane
- RyR
Ryanodine receptor
- IP3R
Inositol-tri-phosphate receptor
Footnotes
Disclosures: none.
References
- 1.Balaban RS. The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta. 2009;1787:1334–1341. doi: 10.1016/j.bbabio.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murgia M, Giorgi C, Pinton P, Rizzuto R. Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. J Mol Cell Cardiol. 2009;46:781–788. doi: 10.1016/j.yjmcc.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukyanenko V, Chikando A, Lederer WJ. Mitochondria in cardiomyocyte Ca2+ signaling. Int J Biochem Cell Biol. 2009;41:1957–1971. doi: 10.1016/j.biocel.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 5.Leist M, Single B, Castoldi AF, KÅhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: A switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- 7.Zamaraeva MV, Sabirov RZ, Maeno E, Ando-Akatsuka Y, Bessonova SV, Okada Y. Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ. 2005;12:1390–1397. doi: 10.1038/sj.cdd.4401661. [DOI] [PubMed] [Google Scholar]
- 8.Dimmer KS, Scorrano L. (De)constructing mitochondria: what for? Physiology (Bethesda) 2006;21:233–241. doi: 10.1152/physiol.00010.2006. [DOI] [PubMed] [Google Scholar]
- 9.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 10.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 11.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 12.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 13.Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- 14.Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–324. doi: 10.1016/s0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- 15.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 17.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 18.Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 24.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 25.Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–1039. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt A, Wolde M, Thiele C, Fest W, Kratzin H, Podtelejnikov AV, Witke W, Huttner WB, Soling HD. Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature. 1999;401:133–141. doi: 10.1038/43613. [DOI] [PubMed] [Google Scholar]
- 30.Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115:1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 31.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117:6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- 32.Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 33.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eura Y, Ishihara N, Yokota S, Mihara K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem. 2003;134:333–344. doi: 10.1093/jb/mvg150. [DOI] [PubMed] [Google Scholar]
- 35.Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacin M, Vidal H, Rivera F, Brand M, Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 36.Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 37.Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 38.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 39.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 40.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 41.Martinou I, Desagher S, Eskes R, Antonsson B, Andre E, Fakan S, Martinou JC. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 44.Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol. 2004;164:493–499. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem. 2005;280:35742–35750. doi: 10.1074/jbc.M505970200. [DOI] [PubMed] [Google Scholar]
- 46.Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 47.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160:1115–1127. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alirol E, James D, Huber D, Marchetto A, Vergani L, Martinou JC, Scorrano L. The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell. 2006;17:4593–4605. doi: 10.1091/mbc.E06-05-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 51.Bellinger AM, Mongillo M, Marks AR. Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J Clin Invest. 2008;118:445–453. doi: 10.1172/JCI34006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- 53.Hoshijima M, Knoll R, Pashmforoush M, Chien KR. Reversal of calcium cycling defects in advanced heart failure toward molecular therapy. J Am Coll Cardiol. 2006;48:A15–23. doi: 10.1016/j.jacc.2006.06.070. [DOI] [PubMed] [Google Scholar]
- 54.Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J, Jr, Kranias EG, Giles WR, Chien KR. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–322. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 55.Song Q, Schmidt AG, Hahn HS, Carr AN, Frank B, Pater L, Gerst M, Young K, Hoit BD, McConnell BK, Haghighi K, Seidman CE, Seidman JG, Dorn GW, 2nd, Kranias EG. Rescue of cardiomyocyte dysfunction by phospholamban ablation does not prevent ventricular failure in genetic hypertrophy. J Clin Invest. 2003;111:859–867. doi: 10.1172/JCI16738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.D’Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., 2nd Transgenic G-alphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM, Brown JH. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res. 2010;106:354–362. doi: 10.1161/CIRCRESAHA.109.207423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delling U, Sussman MA, Molkentin JD. Re-evaluating sarcoplasmic reticulum function in heart failure. Nat Med. 2000;6:942–943. doi: 10.1038/79592. [DOI] [PubMed] [Google Scholar]
- 60.Cross HR, Kranias EG, Murphy E, Steenbergen C. Ablation of PLB exacerbates ischemic injury to a lesser extent in female than male mice: protective role of NO. Am J Physiol Heart Circ Physiol. 2003;284:H683–690. doi: 10.1152/ajpheart.00567.2002. [DOI] [PubMed] [Google Scholar]
- 61.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP, Anderson ME. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. 2006;291:H3065–3075. doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- 62.Wu G, Long X, Marin-Garcia J. Adenoviral SERCA1 overexpression triggers an apoptotic response in cultured neonatal but not in adult rat cardiomyocytes. Mol Cell Biochem. 2004;267:123–132. doi: 10.1023/b:mcbi.0000049361.89265.2b. [DOI] [PubMed] [Google Scholar]
- 63.Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD, Houser SR. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ Res. 2005;97:1009–1017. doi: 10.1161/01.RES.0000189270.72915.D1. [DOI] [PubMed] [Google Scholar]
- 64.Miyamoto S, Howes AL, Adams JW, Dorn GW, 2nd, Brown JH. Ca2+ dysregulation induces mitochondrial depolarization and apoptosis: role of Na+/Ca2+ exchanger and AKT. J Biol Chem. 2005;280:38505–38512. doi: 10.1074/jbc.M505223200. [DOI] [PubMed] [Google Scholar]
- 65.Seguchi H, Ritter M, Shizukuishi M, Ishida H, Chokoh G, Nakazawa H, Spitzer KW, Barry WH. Propagation of Ca2+ release in cardiac myocytes: role of mitochondria. Cell Calcium. 2005;38:1–9. doi: 10.1016/j.ceca.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 66.Andrienko TN, Picht E, Bers DM. Mitochondrial free calcium regulation during sarcoplasmic reticulum calcium release in rat cardiac myocytes. J Mol Cell Cardiol. 2009;46:1027–1036. doi: 10.1016/j.yjmcc.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res. 2008;103:1363–1369. doi: 10.1161/CIRCRESAHA.108.186551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dorn GW., 2nd Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc Res. 2009;81:465–473. doi: 10.1093/cvr/cvn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest. 2005;115:565–571. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haworth RA, Hunter DR. The Ca 2+-induced membrane transition in mitochondria. II. Nature of the Ca 2+ trigger site. Arch Biochem Biophys. 1979;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 72.Krasnikov BF, Zorov DB, Antonenko YN, Zaspa AA, Kulikov IV, Kristal BS, Cooper AJ, Brown AM. Comparative kinetic analysis reveals that inducer-specific ion release precedes the mitochondrial permeability transition. Biochim Biophys Acta. 2005;1708:375–392. doi: 10.1016/j.bbabio.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 73.Crompton M, Costi A. A heart mitochondrial Ca2(+)-dependent pore of possible relevance to re-perfusion-induced injury. Evidence that ADP facilitates pore interconversion between the closed and open states. Biochem J. 1990;266:33–39. doi: 10.1042/bj2660033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 76.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharm Exp Ther. 2009;330:670–678. doi: 10.1124/jpet.109.153213. [DOI] [PubMed] [Google Scholar]
- 77.Szabo I, De Pinto V, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett. 1993;330:206–210. doi: 10.1016/0014-5793(93)80274-x. [DOI] [PubMed] [Google Scholar]
- 78.Beutner G, Ruck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7. [DOI] [PubMed] [Google Scholar]
- 79.Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Remy R, Xie ZH, Reed JC, Kroemer G. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J Exp Med. 1998;187:1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tikhonova IM, Andreyev A, Antonenko Yu N, Kaulen AD, Komrakov A, Skulachev VP. Ion permeability induced in artificial membranes by the ATP/ADP antiporter. FEBS Lett. 1994;337:231–234. doi: 10.1016/0014-5793(94)80197-5. [DOI] [PubMed] [Google Scholar]
- 81.Brenner C, Cadiou H, Vieira HL, Zamzami N, Marzo I, Xie Z, Leber B, Andrews D, Duclohier H, Reed JC, Kroemer G. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19:329–336. doi: 10.1038/sj.onc.1203298. [DOI] [PubMed] [Google Scholar]
- 82.Jacotot E, Ferri KF, El Hamel C, Brenner C, Druillennec S, Hoebeke J, Rustin P, Metivier D, Lenoir C, Geuskens M, Vieira HL, Loeffler M, Belzacq AS, Briand JP, Zamzami N, Edelman L, Xie ZH, Reed JC, Roques BP, Kroemer G. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J Exp Med. 2001;193:509–519. doi: 10.1084/jem.193.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baines CP, Molkentin JD. Adenine nucleotide translocase-1 induces cardiomyocyte death through upregulation of the pro-apoptotic protein Bax. J Mol Cell Cardiol. 2009;46:969–977. doi: 10.1016/j.yjmcc.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocatoris not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, 2nd, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 88.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 89.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fujimoto K, Chen Y, Polonsky KS, Dorn GW., 2nd Targeting cyclophilin D and the mitochondrial permeability transition enhances β-cell survival and prevents diabetes in Pdx1 deficiency. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.0914209107. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW., 2nd Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA. 2010;107:9035–9042. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 95.Basso E, Petronilli V, Forte MA, Bernardi P. Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J Biol Chem. 2008;283:26307–26311. doi: 10.1074/jbc.C800132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 98.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro -apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 99.Zhou L, Chang DC. Dynamics and structure of the Bax-Bak complex responsible for releasing mitochondrial proteins during apoptosis. J Cell Sci. 2008;121:2186–2196. doi: 10.1242/jcs.024703. [DOI] [PubMed] [Google Scholar]
- 100.Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 101.Dorn GW, 2nd, Kirshenbaum LA. Cardiac reanimation: targeting cardiomyocyte death by BNIP3 and NIX/BNIP3L. Oncogene. 2008;27 (Suppl 1):S158–S167. doi: 10.1038/onc.2009.53. [DOI] [PubMed] [Google Scholar]
- 102.Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN, Dorn GW., 2nd Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med. 2002;8:725–730. doi: 10.1038/nm719. [DOI] [PubMed] [Google Scholar]
- 103.Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirshenbaum LA, Dorn GW., 2nd Distinct Pathways Regulate Proapoptotic Nix and BNip3 in Cardiac Stress. J Biol Chem. 2006;281:1442–1448. doi: 10.1074/jbc.M509056200. [DOI] [PubMed] [Google Scholar]
- 104.Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA, Marreez Y, Sanbe A, Robbins J, Dorn GW., 2nd Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ Res. 2004;95:1200–1206. doi: 10.1161/01.RES.0000150366.08972.7f. [DOI] [PubMed] [Google Scholar]
- 105.Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, Daria D, Jegga AG, Geiger H, Aronow BJ, Molkentin JD, Macleod KF, Kalfa TA, Dorn GW., 2nd Unrestrained erythroblast development in Nix−/−mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA. 2007;104:6794–6799. doi: 10.1073/pnas.0610666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW., 2nd Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation. 2008;117:396–404. doi: 10.1161/CIRCULATIONAHA.107.727073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., 2nd Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, Molkentin JD, Kranias EG, Dorn GW., 2nd Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest. 2009;119:203–212. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nutt LK, Pataer A, Pahler J, Fang B, Roth J, McConkey DJ, Swisher SG. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca 2+ stores. J Biol Chem. 2002;277:9219–9225. doi: 10.1074/jbc.M106817200. [DOI] [PubMed] [Google Scholar]
- 112.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+-and mitochondrial -dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 114.Rizzuto R, Pozzan T. Microdomains of intracellular Ca 2+: Molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 115.de Jesus Garcia-Rivas G, Guerrero-Hernandez A, Guerrero-Serna G, Rodriguez-Zavala JS, Zazueta C. Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart. FEBS J. 2005;272:3477–3488. doi: 10.1111/j.1742-4658.2005.04771.x. [DOI] [PubMed] [Google Scholar]
- 116.Ruiz-Meana M, Abellan A, Miro-Casas E, Agullo E, Garcia-Dorado D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol. 2009;297:H1281–1289. doi: 10.1152/ajpheart.00435.2009. [DOI] [PubMed] [Google Scholar]
- 117.Odagiri K, Katoh H, Kawashima H, Tanaka T, Ohtani H, Saotome M, Urushida T, Satoh H, Hayashi H. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol. 2009;46:989–997. doi: 10.1016/j.yjmcc.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 118.Wilding JR, Joubert F, de Araujo C, Fortin D, Novotova M, Veksler V, Ventura-Clapier R. Altered energy transfer from mitochondria to sarcoplasmic reticulum after cytoarchitectural perturbations in mice hearts. J Physiol. 2006;575:191–200. doi: 10.1113/jphysiol.2006.114116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529(Pt 1):37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- 121.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 122.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 123.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 124.Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vance JE, Vance DE. Phospholipid biosynthesis in mammalian cells. Biochem Cell Biol. 2004;82:113–128. doi: 10.1139/o03-073. [DOI] [PubMed] [Google Scholar]
- 126.Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, Forte M, d’Azzo A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol Cell. 2009;36:500–511. doi: 10.1016/j.molcel.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Garcia-Perez C, Hajnoczky G, Csordas G. Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J Biol Chem. 2008;283:32771–32780. doi: 10.1074/jbc.M803385200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005;24:717–729. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. 2007;282:23354–23361. doi: 10.1074/jbc.M702657200. [DOI] [PubMed] [Google Scholar]
- 133.Fang L, Moore XL, Gao XM, Dart AM, Lim YL, Du XJ. Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci. 2007;80:2154–2160. doi: 10.1016/j.lfs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 134.Li Y, Yin R, Liu J, Wang P, Wu S, Luo J, Zhelyabovska O, Yang Q. Peroxisome proliferator-activated receptor delta regulates mitofusin 2 expression in the heart. J Mol Cell Cardiol. 2009;46:876–882. doi: 10.1016/j.yjmcc.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kornmann B, Walter P. ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J Cell Sci. 2010;123:1389–1393. doi: 10.1242/jcs.058636. [DOI] [PMC free article] [PubMed] [Google Scholar]